

ABSTRACT
In our recent study, we demonstrated that oncogenic RAS (rat sarcoma)-mediated transformation and tumorigenesis are supported by transcriptional induction of a crucial antioxidant component, SLC7A11 (solute carrier family 7 member 11), otherwise known as XCT, a gene encoding the cystine/glutamate transporter. Our data highlight that this promotes the biosynthesis of glutathione, in turn allowing RAS transformed cells to mitigate tumorigenesis-linked oxidative stress.
Oncogenic mutations in RAS (rat sarcoma) are present in approximately 20% of all human cancers. The role of intracellular redox balance in governing oncogenic RAS-mediated cancer initiation and progression has thus far remained unclear. Some evidence supports that RAS promotes reactive oxygen species (ROS) to drive transformation,1 while more recent studies have shown that RAS enhances the antioxidant response to support tumorigenicity.2 In our recent study, we addressed this controversy by investigating how redox components contribute to RAS oncogenic properties.
Our data showed that oncogenic RAS protects fibroblasts against oxidative stress by enhancing intracellular glutathione (GSH) levels. We discovered that this property arises, at least in part, through the transcriptional activation of SLC7A11 (solute carrier family 7 member 11), otherwise known as XCT, a gene encoding the cystine/glutamate transporter that supports GSH biosynthesis.3 Mechanistically, we elucidated that the promoter of XCT is transactivated by two distinct transcriptional arms acting in synergy: on the one hand, the transcription factor ETS-1 (E26 transformation-specific 1), downstream of RAS-ERK (extracellular signal-regulated kinase) signaling, and on the other hand ATF4 (activating transcription factor 4), a critical regulator of the oxidative stress response (Figure 1).3 Notably, our findings demonstrate that oncogenic RAS upregulates XCT not merely as an adaptive response to oxidative stress, but also as an intrinsic mechanism supporting cellular transformation. Indeed, we uncovered that genetic or pharmacological inhibition of XCT severely impaired the transformation and tumorigenic potential of oncogenic RAS-expressing cells, as demonstrated in vitro and in vivo using tumor xenografts in immunocompromised mice.3 Thus, our study reveals that oncogenic RAS enhances intracellular antioxidant capacity through the upregulation of XCT to support transformation, which highlights XCT as a form of non-oncogene addiction.
Figure 1.
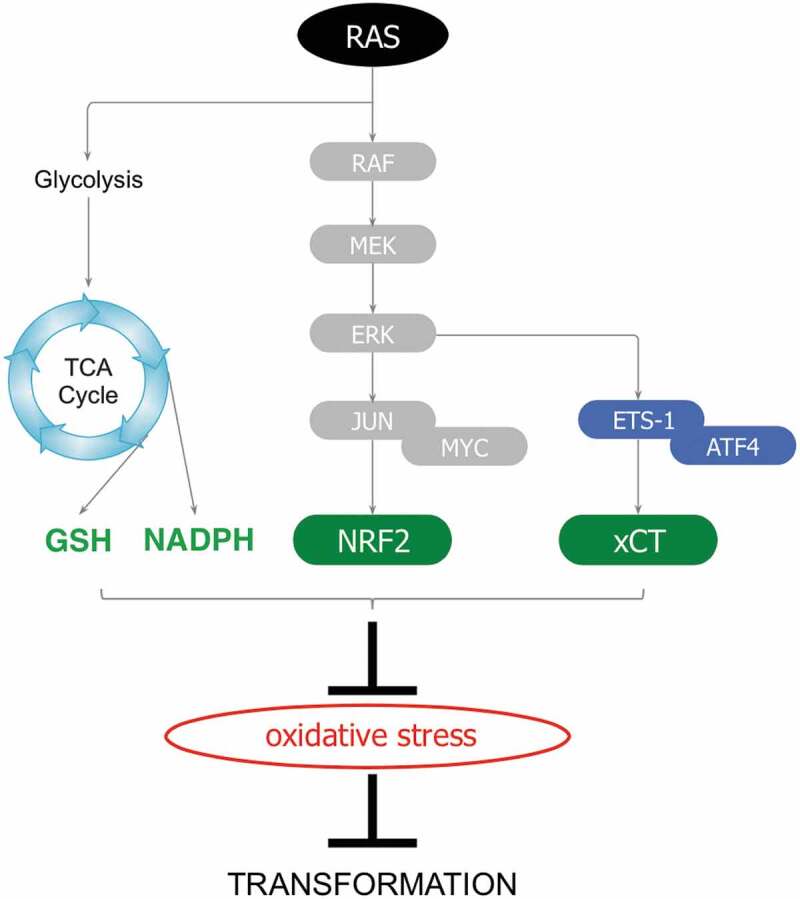
Antioxidant pathways facilitating RAS-driven oncogenesis. Downstream signaling or metabolic pathways that support oncogenic rat sarcoma (RAS) transformation by activating antioxidant programs to mitigate the deleterious effects of oxidative stress (TCA cycle: tricarboxylic acid cycle; GSH: reduced glutathione; NADPH: Nicotinamide adenine dinucleotide phosphate; RAF: rapidly accelerated fibrosarcoma; MEK: mitogen-activated protein kinase/extracellular signal-regulated kinase; ERK: extracellular signal-regulated kinase; JUN: jun-nana; MYC: myelocytomatosis proto-oncogene; NRF2: NFE2L2/nuclear factor, erythroid 2 like 2; ETS1: E26 transformation-specific 1; ATF4: activating transcription factor 4; XCT: System xc, cysteine/glutamate transporter).
Overall, our findings carry important potential clinical relevance, as the targeting of oncogenic RAS remains an elusive challenge in cancer research, while patients harboring RAS mutations are still characterized by dismal prognoses. Until recently, mutant RAS has been considered as undruggable, as numerous drug resistance mechanisms or toxic drug side effects limit the utilization of many candidate oncogenic RAS-targeting compounds. It has thus been proposed that targeting the downstream dependencies or vulnerabilities of mutant RAS may carry potential utility, instead of targeting RAS itself. In this regard, blocking XCT may be an efficacious therapeutic strategy in treating RAS-driven tumors for several reasons. First of all, XCT function is not essential for normal cells or normal tissues, as XCT deficiency (XCT−/-) in mice is not lethal,4 therefore predicting that targeting XCT should have little to no toxic effects. Second, pharmacological inhibitors against XCT, such as Erastin and Sulfasalazine, have been developed and their usage has led to successful impeding of tumor development in several tumor models in vivo. Interestingly, Erastin was originally characterized as a Ras-selective lethal drug (RSL).5 Our findings potentially explain this selectivity as we determined that mutant RAS drives XCT expression to support oncogenic transformation. XCT inhibitors have been primarily used in combination with traditional chemotherapeutic agents. The reasoning is that since a number of these drugs increase ROS, reducing the cellular antioxidant capacity by blocking XCT function in cancer cells will lead to unresolved oxidative stress. Since our data highlight that XCT supports RAS transformation, and not only RAS-mediated protection against exogenous oxidants, the use of XCT inhibitors as a single agent may also prove to be effective.3 Notably, this strategy can be exploited not only in a wide spectrum of cancers that harbor RAS mutations, such as pancreatic, colon, and lung cancer but also in those that exhibit hyperactive RAS signaling pathway. This further encompasses various cancer types such as glioma, melanoma, acute myeloid leukemia, and rhabdomyosarcoma among others characterized by genetic alterations of EGFR (epidermal growth factor receptor), ERBB2 (avian erythroblastosis oncogene-B2 receptor tyrosine kinase 2), BRAF (v-rapidly accelerated fibrosarcoma murine sarcoma viral oncogene homolog B1) or NF1 (neurofibromatosis type 1). Therefore, targeting a non-oncogene addiction, i.e. XCT, holds promise for selectively inhibiting RAS-driven tumors.
Together with previous work, our study further supports a model whereby oncogenic transformation relies on the induction of antioxidant programs. Indeed, it was reported that mutant RAS altered metabolism toward increasing GSH biosynthesis to facilitate tumorigenesis in vivo.6 It was also shown that oncogenic RAS, as well as mutant BRAF and MYC (myelocytomatosis proto-oncogene) induce Nfe2l2 (nuclear factor, erythroid 2 like 2), better known as Nrf2, which encodes the master regulator of the intracellular antioxidant response, to support tumor progression.2 Additionally, the oncogene eIF4E (eukaryotic translation initiation factor 4E) was reported to support oncogenicity by selectively promoting the translation of mRNAs involved in the antioxidant response.7 Therefore, targeting the specific dependency of tumors on antioxidant programs may be a generalizable therapeutic strategy, in particular with oncogene-driven or difficult-to-treat cancers. Previous attempts to inhibit antioxidant capacities in tumors relied, in part, on using agents such as BSO (1-buthionine-S, R-sulfoximine), a classical drug known to inhibit GCL (γ–glutamyl-cysteine ligase), and this is still undergoing clinical testing.8 However, one study elegantly illustrates that at later stages of tumor progression, GSH is dispensable due to compensation from the thioredoxin pathway, and thus combined inhibition of both redox pathways may be required for a synergistic effect on tumors.9 Another potential therapeutic approach to cripple antioxidant capacities is treatment with Vitamin C, which selectively targets RAS-driven tumors in vivo by reducing the ratio of reduced to oxidized GSH.10 With this growing understanding of the mechanisms by which tumors modulate antioxidant response programs, we expect to see continued improvements of such redox-based therapeutic approaches, which will undoubtedly lead to better clinical outcomes. In this light, our study thus uncovers a novel vulnerability of oncogenic RAS-driven tumors, namely the downstream XCT antioxidant program.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Mitsushita J, Lambeth JD, Kamata T.. The superoxide-generating oxidase Nox1 is functionally required for Ras oncogene transformation. Cancer Res. 2004;64(10):3580–3585. doi: 10.1158/0008-5472.CAN-03-3909. [DOI] [PubMed] [Google Scholar]
- 2.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475(7354):106–109. doi: 10.1038/nature10189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lim JKM, Delaidelli A, Minaker SW, Zhang H-F, Colovic M, Yang H, Negri GL, von Karstedt S, Lockwood WW, Schaffer P, et al. Cystine/glutamate antiporter xCT (SLC7A11) facilitates oncogenic RAS transformation by preserving intracellular redox balance. Proc. National Acad. Sci. U.S. Am. 2019;116(19):9433–9442. doi: 10.1073/pnas.1821323116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, Makino N, Sugiyama F, Yagami K-I, Moriguchi T, et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem. 2005;280(45):37423–37429. doi: 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
- 5.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerr EM, Gaude E, Turrell FK, Frezza C, Martins CP.. Mutant Kras copy number defines metabolic reprogramming and therapeutic susceptibilities. Nature. 2016;531(7592):110–113. doi: 10.1038/nature16967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Truitt ML, Conn CS, Shi Z, Pang X, Tokuyasu T, Coady AM, Seo Y, Barna M, Ruggero D. Differential requirements for eIF4E dose in normal development and cancer. Cell. 2015;162(1):59–71. doi: 10.1016/j.cell.2015.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villablanca JG, Volchenboum SL, Cho H, Kang MH, Cohn SL, Anderson CP, Marachelian A, Groshen S, Tsao-Wei D, Matthay KK, et al. A phase I new approaches to neuroblastoma therapy study of buthionine sulfoximine and melphalan with autologous stem cells for recurrent/refractory high-risk neuroblastoma. Pediatr Blood Cancer. 2016;63(8):1349–1356. doi: 10.1002/pbc.25994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell. 2015;27(2):211–222. doi: 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- 10.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio IIC, Giannopoulou EG, Rago C, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350(6266):1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]