

ABSTRACT
Activation of naïve CD8+ T cells stimulates proliferation and differentiation into cytotoxic T-lymphocytes (CTLs). Adoptive T Cell Therapy (ACT) involves multiple rounds of ex vivo activation to generate enough CTLs for reinfusion into patients, but this drives differentiation into terminal effector T cells. Less differentiated CTL populations, such as stem cell memory T cells, are more ideal candidates for ACT because of increased self-renewal and persistent properties. Ex vivo targeting of T cell differentiation with epigenetic modifiers is a potential strategy to improve cytotoxic T-lymphocyte (CTL) generation for ACT. We established a pipeline to assess the effects of epigenetic modifiers on CD8+ T cell proliferation, differentiation, and efficacy in a preclinical melanoma model. Single treatment with epigenetic modifiers inhibited T cell proliferation in vitro, producing CD44hiCD62Lhi effector-like T cells rather than a stem cell memory T cell phenotype. Most epigenetic modifying agents had no significant effect on ACT efficacy with the notable exception of the bromodomain and extraterminal (BET)-inhibitor JQ1 which was associated with a decrease in efficacy compared to unmodified T cells. These findings reveal the complexity of epigenetic targeting of T cell differentiation, highlighting the need to precisely define the epigenetic targeting strategies to improve CTL generation for ACT.
KEYWORDS: Cancer immunotherapy, adoptive cell therapy, T cell differentiation, melanoma, cytotoxic T lymphocytes, BET inhibition
Introduction
Adoptive cell therapy (ACT) involves the ex vivo expansion and reinfusion of tumour-specific CD8+ T cells as a potential treatment for different malignancies [1,2]. ACT can induce durable responses in a subset of patients with melanoma [3,4], but overall complete response rates remain relatively low. The persistence and survival of transferred CD8+ T cells highly correlates with therapy success [5] and appears to be intrinsically associated with the T cell differentiation status. ACT with less differentiated memory CD8+ T cell subsets results in superior persistence and anti-tumour immunity as compared to more differentiated effector T cell subsets [6]. However, to achieve the large numbers required for ACT, T cells undergo multiple rounds of expansion, which inevitably promotes T cell differentiation, resulting in a loss of proliferative potential, survival, and multipotency [7].
It has been proposed previously that skewing the expansion of specific subsets of CD8+ T cells during expansion may enhance ACT efficacy. For example, Gattinoni and colleagues have successfully altered CD8+ T cell differentiation in mice using a Wnt-signalling inhibitor TWS119, resulting in T cells persisting in a less differentiated subgroup [8]. These less differentiated CD8+ T cells, defined as stem cell memory T cells (Tscm), have increased self-renewal and multipotency capabilities. Importantly, CD8+ Tscm cells were qualitatively superior compared to central memory (Tcm) and effector memory (Tem) CD8+ T cells when used in ACT [8,9].
As an alternative approach to manipulating T-cell phenotype by cell-signalling inhibition, the epigenetic features of T-cells could also be targeted during ex vivo activation and expansion. Epigenetic modifications (marks), such as DNA methylation and histone modifications at gene-regulatory regions, are associated with the expression of key transcription factors controlling T cell differentiation and proliferation [10]. Importantly, different epigenetic marks can be manipulated by drugs that inhibit enzymes which covalently modify histones or DNA and are potential therapeutic targets to improve ACT. The effects of epigenetic modifiers on tumour cells have been extensively studied. Histone deacetylase inhibitors (HDACis), such as vorinostat and panobinostat, are approved for the treatment of cutaneous T-cell lymphoma and multiple myeloma, respectively [11,12]. HDACis can enhance tumour immunity by upregulating MHC-I on tumour cells and are potentially synergistic with ACT [13]. Histone methyltransferase inhibitors (such as DOT1L, GSK343, and GSKJ4) inhibit proliferation and growth of leukemia, and glioblastoma cell lines [14]. Bromodomain and extraterminal (BET)-domain readers of acetyl marks in histone tails can be also be targeted (for example, by BET-inhibitors, JQ1, IBET, and GSK2801), and has recently been demonstrated to improve the efficacy of ACT [15]. Whilst the effects of targeting different epigenetic marks on cancer cell proliferation have been reported [16], the effects of direct epigenetic modification on CD8+ T cell proliferation and differentiation are not completely understood. CD8+ T cell differentiation is tightly regulated by epigenetic events [10,17,18], and epigenetic targeting to reprogram T cells to overcome terminally differentiated cells used for ACT is a possible way of improving therapy.
Here we have further investigated the hypothesis that ex vivo treatment of activated CD8+ T cells may reprogram T cells into a Tscm phenotype and improve ACT. The effects of single epigenetic modifiers, or the WNT-signalling inhibitor TWS119 on CD8+ T cell activation, were investigated in a preclinical murine model of ACT. We found that most epigenetic modifiers promoted CD44hiCD62Lhi surface phenotype, but not into a Tscm like CD44loCD62Lhi phenotype. Surprisingly, cells treated with JQ1, the prototype BET-inhibitor, at high doses caused impaired proliferation and reduced treatment efficacy. We also report that we were unable to replicate changes to the Tscm phenotype by treatment with TWS119 during ex vivo T-cell expansion in our pre-clinical models.
Results
Epigenetic modifiers inhibited CD8+ T cell proliferation but did not alter the expression of surface differentiation markers
We utilized an in vitro CD8+ T cell activation assay to assess the effects of epigenetic modifiers on T cell proliferation and differentiation. gBT-I are well characterized CD8+ T cell receptor (TCR) transgenic mice specific for a H-2Kb-restricted Herpes Simplex Virus (HSV) glycoprotein B (gB) epitope, gB498-505 [19]. We stimulated CFSE labelled gBT-I splenocytes with gB498-505 peptide-pulsed C57BL/6 splenocytes at a 1:1 ratio for 72 h. In the absence of epigenetic modifiers, >95% of gBT-I CD8+ T cells proliferated in response to cognate antigen (Figure 1(a)). In vitro activation of CD8+ T cells leads to division dependent cell differentiation, and changes in expression of activation markers such as CD44 and CD62L [20]. Inactivated, naïve gBT-I CD8+ T cells are CD44loCD62Lhi [21], and differentiate into CD44hiCD62Lhi cells upon activation. We postulated that addition of epigenetic modifiers would inhibit T cell proliferation and alter the expression of CD44 and CD62L.
Figure 1.
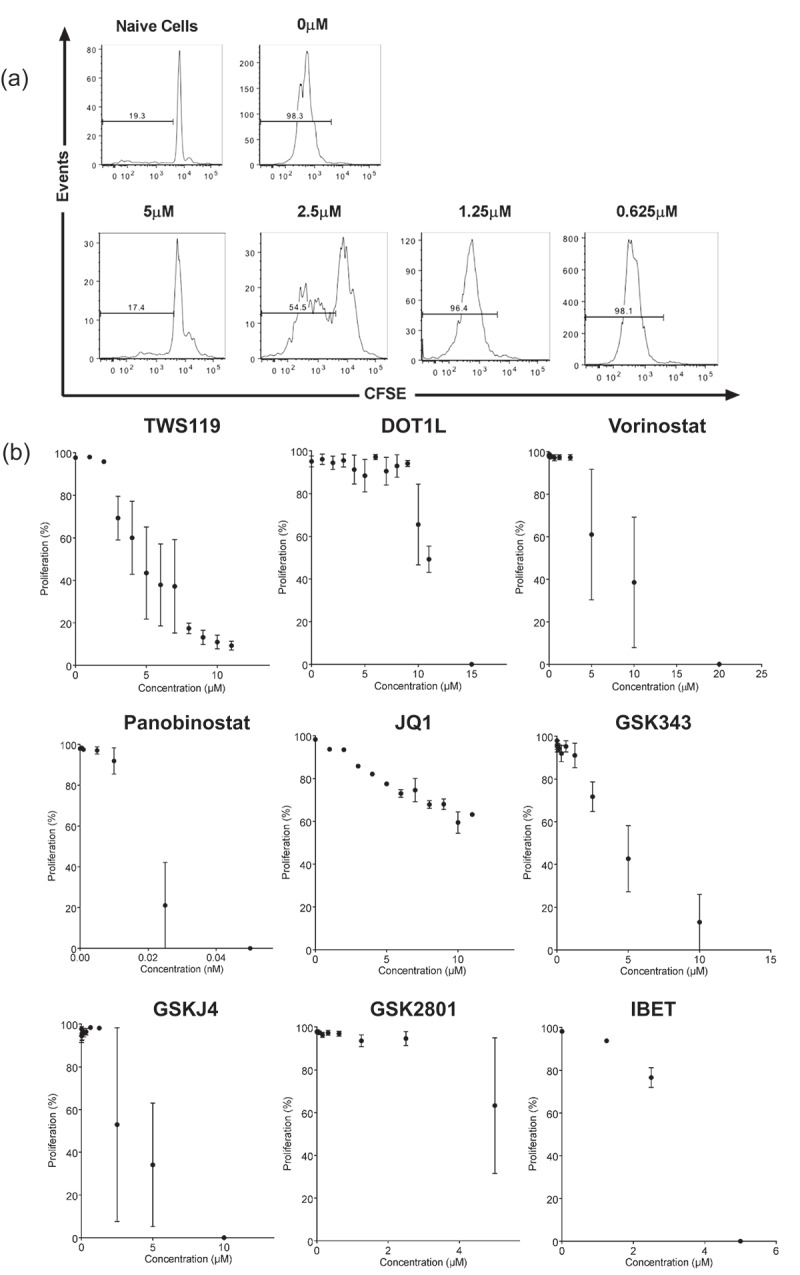
T cell proliferation inhibition by epigenetic modifiers in vitro. (a) Representative FACS diagram depicting gBT-I T cell proliferation measured by CFSE dilution 72-h post-activation, in the presence of titrated concentrations of TWS119. Plots are gated on live, CD8+ cells. (b) Dot plots representing the percentage of proliferating (CFSElo) gBT-I T cells in the presence of titrated concentration of epigenetic modifiers, 72 h after activation in a 384-well plate. Each graph represents treatment with one drug. Each datapoint represents the mean ± SD proliferation of three separate experiments.
We first screened the effects of eight individual epigenetic modifiers on T cell proliferation (Table 1). TWS119, a known inhibitor of T cell proliferation, was also tested [8]. The concentration of each drug that would reduce proliferation (determined by % CFSE dilution) to approximately 50%, but not affect cell viability was determined. The optimum range for TWS119 was between 3 and 6 μM (Figure 1(b)). With the exception of Panobinostat and GSK2801, all epigenetic modifiers inhibited T cell proliferation. Panobinostat and GSK2801 were highly toxic to T cells and were not tested further. The screen was repeated in 384 and 6 well plates, and the final optimal concentrations for each epigenetic modifier were determined (Table 2). Although there were varying standard deviations, measurements of T cell proliferation were a clear indicator of cellular changes and an effective way to ensure cells were still undergoing differentiation in the presence of epigenetic modifiers.
Table 1.
Epigenetic modifiers selected for project.
| DOT1L inhibitor SGC0946 (DOT1-like histone H3K79 methyltransferase inhibitor) |
| Vorinostat (Histone deacetylase inhibitor) |
| Panobinostat (Histone deacetylase inhibitor) |
| GSK2801 (selective inhibitor of bromodomains BAZ2A/B) |
| GSKJ4 (histone demethylase inhibitor) |
| GSK343 (EZH2 selective inhibitor) |
| IBET (selective bromodomain inhibitor for BRD4) |
| JQ1 (selective bromodomain inhibitor for BRD2, BRD3, BRD4) |
Table 2.
Optimal dose of epigenetic modifiers used.
| Drug name | Optimal concentration |
|---|---|
| TWS119 | 3.5 µM |
| DOT1L inhibitor SGC0946 | 2.5 µM |
| Vorinostat | 2.5 µM |
| Panobinostat | – |
| JQ1 | 8 µM |
| GSK343 | 6 µM |
| GSKJ4 | 2.5 µM |
| GSK2801 | – |
| IBET | 3 µM |
T cell differentiation was assessed by the expression of CD44 and CD62L [20]. TWS119-inhibited T cell differentiation, as 67.17 ± 0.85% of CD8+ T cells were CD44loCD62Lhi 72-h post-activation, consistent with previously published data [8] (Figure 2(a)). With the exception of vorinostat, activated gBT-I CD8+ T cells treated with all epigenetic modifiers differentiated into CD44hiCD62Lhi T cells, similar to untreated cells. Activated gBT-I CD8+ T cells exposed to vorinostat expressed a unique CD44loCD62Llo phenotype (Figure 2(a,b)). In summary, we did not observe differentiation into a different surface phenotype with most of our epigenetic modifiers, with the exception of vorinostat.
Figure 2.
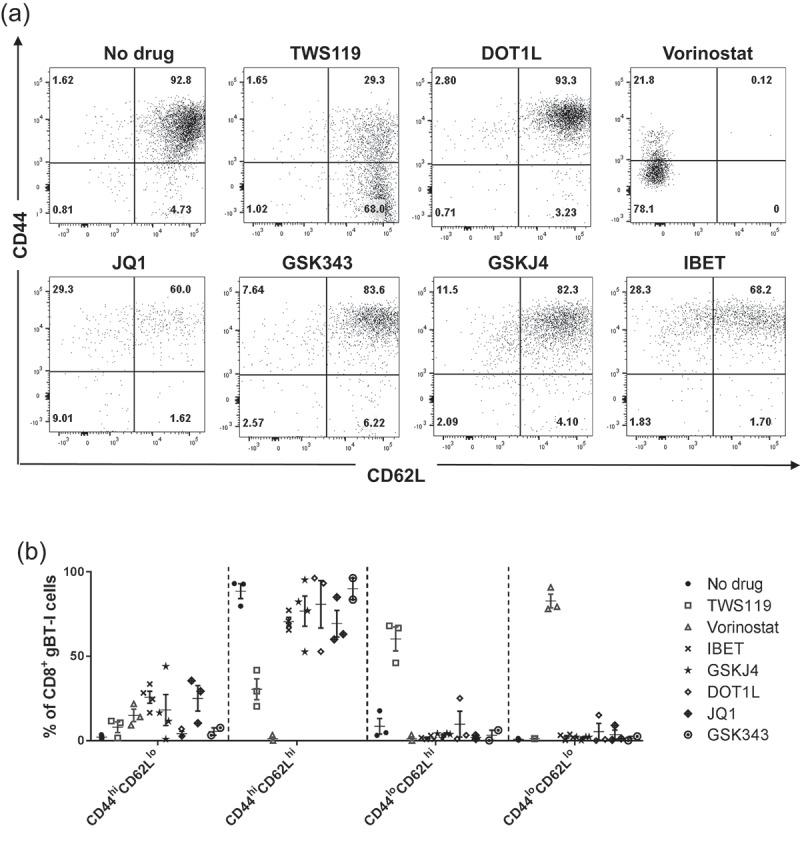
T cell phenotype of activated T cells in the presence of epigenetic modifiers. (a) Representative FACS diagram depicting expression of CD44 and CD62L on activated gBT-I T cells, in the presence or absence of different drugs. Plots are gated on live, CD8+ cells, and are representative of three to four separate experiments. (b) CD44 and CD62L expression represented at dot plots.
Inhibition of WNT/β-catenin signalling pathway did not form T stem cell memory in gBT-I TCR transgenic cells
It has been demonstrated previously that activation of CD8+ T cells in the presence of TWS119 reduces proliferation and differentiates T cells towards a Tscm phenotype [8]. We sought to replicate this in our model, and similarly found that 3.5 μM TWS119 inhibited gBT-I CD8+ T cell proliferation (66.4% compared to 97.3% in untreated cells) (Figure 3(a)).
Figure 3.
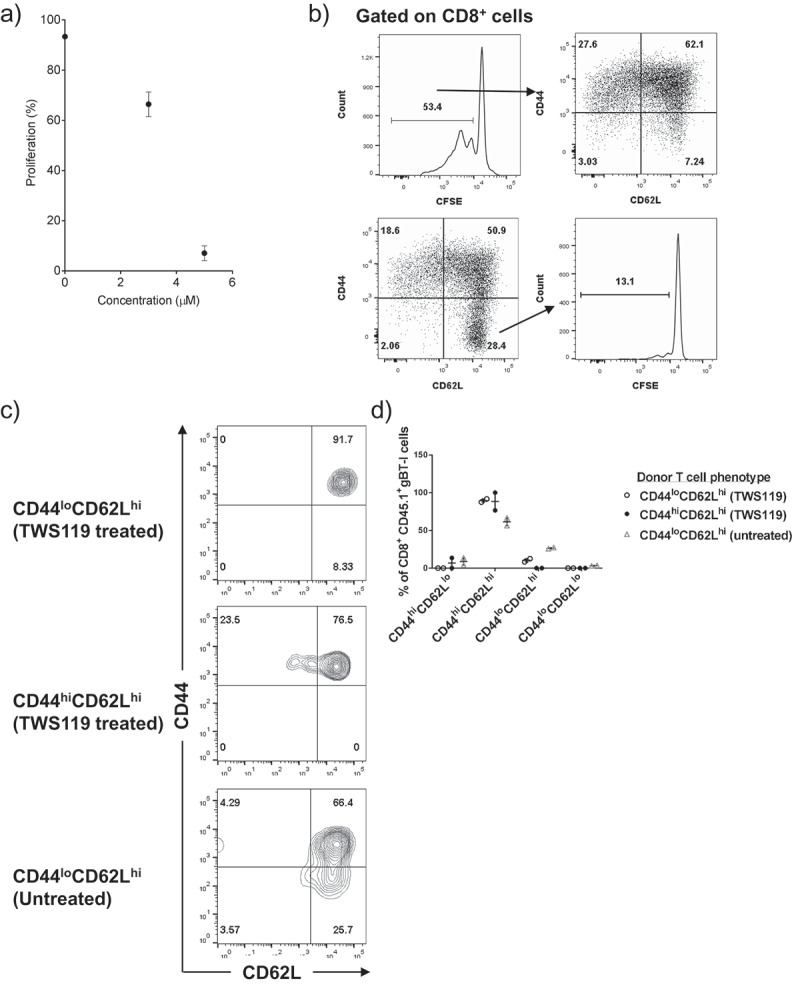
TWS119 treatment failed to generate a Tscm phenotype on gBT-I T cells. (a) Dot plot representing CFSElo proliferating gBT-I T cells in the presence of TWS119, 72 h after activation in a 6-well plate. Each datapoint represents the mean ± SD proliferation. (b) CD44 and CD62L expression of CFSElo T cells post-activation (top row). CFSE profile of CD44loCD62Lhi cells demonstrates that most cells in this population remain naïve (bottom row). Plots are representative for three separate experiments. (c) TWS119-treated cells were sorted into CD44loCD62Lhi, CD44hiCD62Lhi populations, and untreated CD44loCD62Lhi populations (1 x 10^5 cells) were transferred into a lymphodepleted animal. CD44 and CD62L expression on transferred (CD45.1) cells 4 weeks post-transfer are represented as FACS diagrams, and in (d) a dot plot.
Tscm cells are proliferating (CFSElo) cells that uniquely retain the surface phenotype similar to a naïve T cell (CD44loCD62Lhi). However, we found that the majority of CFSElo proliferating gBT-I CD8+ T cells had a CD44hiCD62Llo or CD44hiCD62Lhi surface phenotype (Figure 3(b)). Less than 10% of proliferating cells displayed the reported Tscm (CD44loCD62Lhi) phenotype (Figure 3(b)), compared to 30% of proliferating cells as reported in the pMEL transgenic model [8]. The majority of the CD44loCD62Lhi cells post TWS119 treatment remained CFSEhi (Figure 3(b)), suggesting that TWS119 inhibited gBT-I proliferation, with the majority of cells retaining a Tnaive phenotype. We further tested TWS119 in vitro with two other CD8+ TCR transgenic models (CL4 and OT-I) and were also unable to generate percentage of Tscm cells comparable to previously described literature (data not shown).
Even though we failed to observe changes in surface phenotype post TWS119 treatment, we tested if treated cells would recapitulate its self-renewal and stem-like phenotype in vivo. We sorted CD44loCD62Lhi and CD44hiCD62Lhi gBT-I CD8+ T cells post TWS119 treatment and transferred them into lympho-depleted mice. However, we did not observe any evidences of self-renewal or stemness, as most transferred cells differentiated into CD44hiCD62Lhi T cells after 4 weeks, similar to untreated Tnaive cells (Figure 3(c,d)). Taken together, our data suggested that although we were able to inhibit proliferation with TWS119, we did not observe any reprogramming of memory T cell phenotype.
ACT with JQ1 modified T cells reduced treatment efficacy in a preclinical cancer model
Although we did not observe changes to gBT-I CD8+ T cells surface phenotype in vitro, we postulated that epigenetic modifying agents could have altered T cell function. We tested this with an established model of adoptive cell therapy whereby tumour-specific CD8+ T cells are the key mediators of tumour cell death. We sought to determine if modifying gBT-I CD8+ T cells in vitro would affect their anti-tumour effect in vivo. gBT-I CD8+ T cells were activated in vitro in the presence of different epigenetic modifiers and administered to mice harbouring a subcutaneous B16 melanoma expressing the gB antigen (B16-gB). B16-gB tumours grew rapidly in untreated mice (no ACT), with all tumours forming large palpable masses by 22 d. Administration of untreated, activated gBT-I T cells resulted in rapid tumour regression in the first 3 d, and tumour growth was suppressed for the next 7 d (Figure 4(a)). However, tumours eventually escape this control and relapse, growing exponentially. Mice within this group survive an average for 35 d, and there was a significant difference in survival between animals treated with unmodified T cells and untreated animals (p < 0.001). We examined the effect of modified gBT-I CD8+ T cells on tumour growth and survival.
Figure 4.
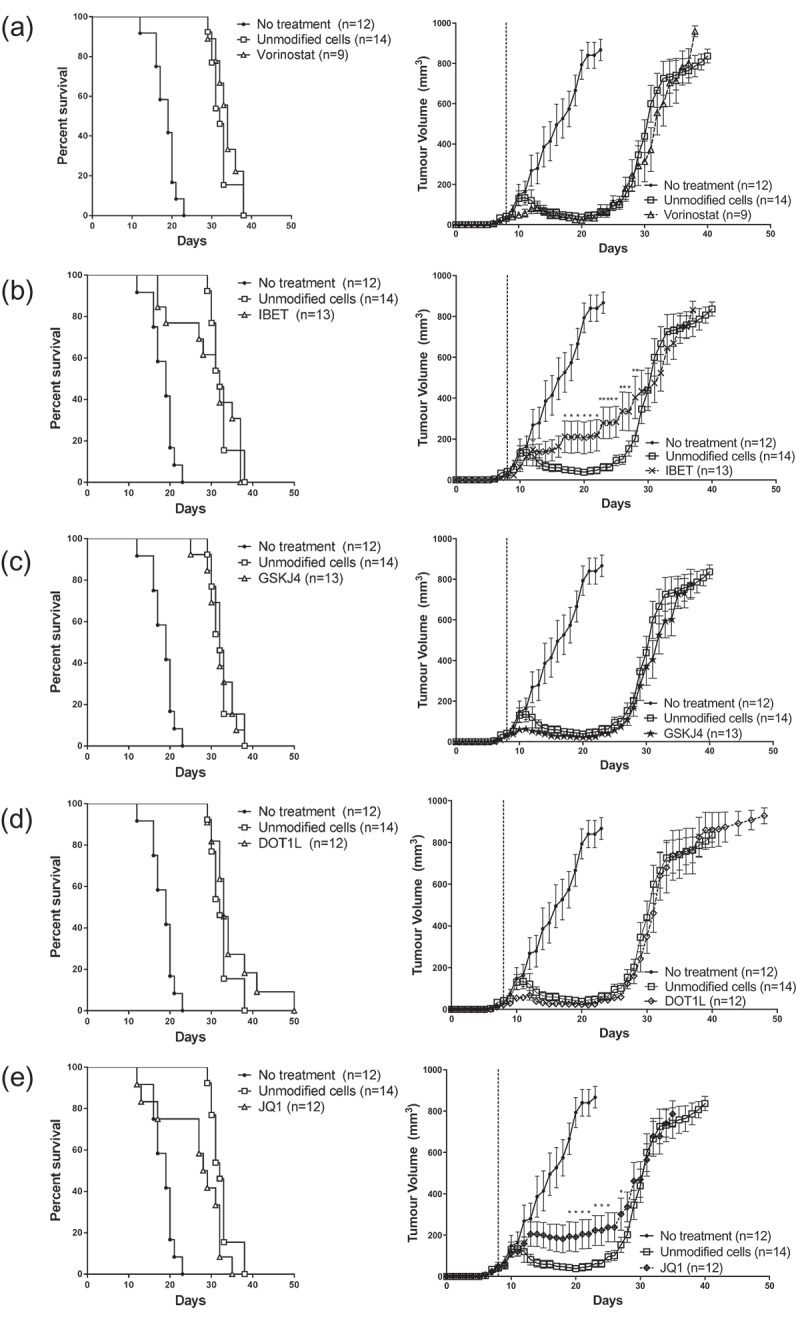
ACT with epigenetic modified gBT-I T cells did not improve tumour regression and survival. Tumour growth and survival curves of mice inoculated subcutaneously with B16.gB prior to receiving ACT consisting of activated gBT-I T cells treated with (a) vorinostat, (b) IBET, (c) GSKJ4, (d) DOT1L and (e) JQ1. Graphs represent three separate experiments. Tumour growth between unmodified group and each modified group were compared with a Mixed Model ANOVA. Time points with significant differences in mean tumour volumes are represented with an asterisk (*<0.05, **<0.01). Survival curves were compared with a Log-Rank (Mantel-Cox) test and there was a significant difference between unmodified T cells and untreated animals (p < 0.001). There was no significant difference between each treatment and unmodified T cells, with the exception of JQ1 treated cells. (JQ1 vs unmodified, p = 0.0237).
Treatment with gBT-I CD8+ T cells modified with vorinostat, DOT1L inhibitor and GSKJ4 did not alter tumour growth differently from unmodified T cells (Figure 4(a–d). Interestingly, treatment with IBET and JQ1 modified T cells abrogated tumour regression in a proportion of animals, suggesting that CD8+ T cell function was impaired (Figure 4(b,e)). Tumours were maintained at a mean volume of approximately 200mm3 – 400mm3 for 20 d after treatment with IBET or JQ1 modified T cells before relapsing, and were significantly different from mice treated with unmodified T cells over time periods indicated in Figure 4(b,e) (IBET: days 17–28; JQ1: days 19–25,27: *p < 0.05, **p < 0.01). There was no significant difference in overall survival between each treatment and unmodified T cells, with the exception of JQ1-treated T cells (p = 0.0237) (Figure 4(e)). Although the effect was modest, modification with JQ1 and IBET inhibited tumour regression in our models of ACT.
Discussion
ACT can be effective in some individuals with advanced melanoma. However, a major hurdle for improving ACT efficacy is the development of T cell exhaustion during ex vivo expansion, which is necessary to generate sufficient numbers of cells for autologous infusion. In this study, we sought to manipulate CD8+ T cell reprogramming by targeting epigenetic enzymes during the ex vivo T cell expansion step. Whilst multiple epigenetic modifiers demonstrated the capacity to inhibit proliferation in vitro, none showed evidence of differentiating cells into a Tscm surface phenotype or enhancing the efficacy of ACT when the treated cells were administered.
We successfully established an in vitro screen to examine the effects of epigenetic modifiers on proliferation and differentiation of TCR transgenic cells during antigen-specific activation. T cell proliferation was inhibited to different extents by different epigenetic modifiers. High toxicity was observed when T cells were treated with low concentrations of GSK2801 and panobinostat, which is consistent with published observations where human lymphocytes are exposed to panobinostat long term (>10 h) [22,23]. Although T cell proliferation is closely linked to its differentiation status, we did not observe measurable differences in differentiation, even though T cell proliferation was altered to varying extents by the epigenetic inhibitors. With the exception of vorinostat, all proliferating T cells differentiated into a CD44hiCD62Lhi phenotype either with or without treatment. In contrast, proliferating T cells exposed to vorinostat exhibited an unusual CD44lo surface phenotype post activation. Vorinostat can interfere with TCR signalling pathways and might be indirectly affecting the expression of activation molecules [24]. Another possible mechanism is that the HDAC1/miR-34a axis regulates CD44 expression in some tumour cell lines and inhibiting HDAC1 with vorinostat might have affected CD44 expression in this instance [25]. We were unable to confirm this finding with the other HDAC inhibitor, panobinostat due to its toxicity.
There are limited reports investigating the pharmacological manipulation of epigenetic mechanisms with respect to T cell differentiation in clinically relevant settings. A recent study has reported that T cells treated with the BET-inhibitor JQ1 in a xenograft model [15], demonstrated greater cell persistence, proliferation, enhanced formation of Tcm, Tscm populations, and improved ACT efficacy. However, in the present study, JQ1 treatment during ex vivo expansion formed T cells with a Tcm like surface phenotype, but JQ1-treated T cell grafts demonstrated reduced efficacy in vivo. The disparity in findings could be attributed to differences in expansion protocols in both studies. Kagoya et al. treated their cells at a significantly lower concentration of JQ1 (0.15 µM) over a longer period of time (14 d) compared to our study (8 µM for 3 d). Furthermore, human studies utilizes non-specific, polyclonal T cell activation to generate T cell grafts for ACT, whereas we investigated antigen-specific CD8+ T cell activation in our study. As TCR affinity affects the ability of CD8+ T cells to differentiate into Tscm cells, our results might be influenced by our TCR transgenic model [26]. Lastly, JQ1 improved both CD8+ and CD4+ T cell function and engraftment [15], whereas our model is limited to the study of CD8+ T cells. CD4+ T cells play a crucial role in the maintenance of CD8+ memory T cell function [27] and is another possible contribution to the disparity in findings.
In our study, we examined the expression of CD44 and CD62L as readouts for CD8+ T cell in vitro differentiation. However, expression of these two markers did not correlate with observed functional impairment in vivo of JQ1- and iBET-treated T cells. In depth analysis of other T cell differentiation surface markers (such as CD25, CCR7, KLRG1, and transcription factors such as TCF-1 and EOMES) or transcriptome-wide surveys of ex vivo expanded T cells may provide additional insight into how broad-based epigenetic targeting alters T cells differentiation and function in the context of ACT.
While we were unable to identify compounds that promote the generation of Tscm cells ex vivo, we note distinctive features of our model system which serve as important caveats for interpreting the results reported herein. We included the Wnt/β-catenin pathway inhibitor TWS119 as a positive control to promote Tscm ex vivo as reported previously [8]. However, despite the expected reduction in proliferation, we did not observe a coincident increase in Tscm cells in our TCR transgenic models. This inconsistency has been reported previously in a mouse model in which polyclonal T cells were activated and treated with TWS119 [28] resulting in reduced proliferation, but no measurable change to the Tscm phenotype. Effects of Wnt/β-catenin signalling on ex vivo human T cell differentiation were investigated previously with either a combination of IL-7 and IL-15 [9], or TWS119 and IL-21 [29], and it therefore cannot be ruled out that differing protocols are associated with discrepant effects on Tscm phenotype. In addition, we note limitations of our model system that may impact upon the outcomes of modulating ACT efficacy. We recently reported that resistance to ACT in B16-OVA melanoma cells arises through multiple processes including transcriptional silencing of the targeted antigen genes [30]. In the present study, we observed down-regulation of GFP in relapsing tumour cell explants (data not shown) suggesting that this evolutionary process was likely operative. Therefore, enhanced ACT efficacy is expected to promote initial tumour regression, however, the emergence of resistance in this model might not necessarily be delayed, for instance, if relapsing tumour cells are selected from pre-existing clonal outgrowths. Our data show that none of the ex vivo T-cell epigenetic treatments promoted survival and did not cause significant shrinkage to tumour volume arguing against these protocols as enhancing ACT. In contrast, both BET-inhibitors tested (JQ1 and IBET) appeared to impair shrinkage of tumour volume, with JQ1-treated T cells also inducing significantly reduced survival.
Conclusions
Our study demonstrates that single epigenetic modifiers can alter T cell proliferation; however, at these doses, we do not consistently observe measurable effects on differentiation. The epigenetic patterns regulating T cell differentiation are complex, with different epigenetic marks on multiple genes changing over the course of T cell activation [10]. Most studies currently expose T cells ex vivo with epigenetic modifiers from the start of T cell activation. Further studies are required to identify how altering the timing, and combination of such modifiers can improve ACT and conversely to document protocols that inhibit efficacy.
Materials and methods
Mice
gBT-I on a C57BL/6.SJL-PtprcaPep3b/BoyJ background (CD45.1) [19] were bred and maintained at Telethon Kids Institute (Perth, Australia). gBT-I are T cell receptor (TCR) transgenic mice specific for a H-2Kb-restricted Herpes Simplex Virus (HSV) glycoprotein B (gB) epitope, gB498-505. Wild type C57BL/6 mice were obtained from Animal Resources Centre WA (Perth, Australia). The Telethon Kids Institute Animal Ethics Committee approved all experimental procedures.
In vitro T cell activation assay
gBT-I splenocytes were labelled with 2.5 μM of carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes) as per manufacturer’s instructions. C57BL/6 splenocytes were pulsed with 1 μM of gB498-505 peptide (Mimotopes) at 37°C for 1 h. CFSE labelled gBT-I splenocytes were incubated with gB498-505 pulsed splenocytes at a 1:1 ratio, in the presence of varying concentrations of epigenetic modifiers. Concentrations were selected at doses that inhibited proliferation but caused minimal cell death thereby maintaining a viable pool of cells for infusion. Cells were diluted to a final concentration of 8.0 × 106 cells/ml and transferred into a 384-well plate or 6-well plate. Cells were incubated in cRPMI media (RPMI media supplemented with 10% FCS), 50 μM 2-mercaptoethanol, 2mM L-glutamine, 100U/ml penicillin, 100 μg/ml streptomycin (Life Technologies), 3μg/ml LPS (Sigma, MO, USA) and 10U/ml IL-2 (Peprotech) in a 5% CO2 incubator at 37°C for 72 h. Drugs used in this study are available from Selleckchem including SGC0946 (DOT1L-inhibitor; Catalogue No.S7079), Vorinostat (SAHA, MK0683; Catalogue No.S1047), Panobinostat (LBH589; Catalogue No.S1030), GSK2801 (Catalogue No.S7231), GSKJ4 (Catalogue No.S7070), GSK343 (Catalogue No.S7164), IBET151 (GSK1210151A; Catalogue No.S2780) and JQ1 (Catalogue No.S7110).
Flow cytometry
Cell proliferation was analysed by flow cytometry. Activated cells were stained with anti-mouse CD45.1-V450 (clone A20), anti-mouse CD8-APC (clone 53–6.7), anti-mouse Va2-PE antibody (clone B20.1), anti-mouse CD44 (clone IM7) and anti-mouse CD62L (clone MEL-14) (BD Biosciences). Prior to acquisition cells were stained with propidium iodide (PI; Sigma) to exclude dead cells. Flow cytometry was performed on a BD LSR Fortessa (BD Biosciences). Flow cytometry analysis was performed using FlowJo (Treestar).
Tumour cell lines and inoculation
B16.F10 melanoma cells were purchased from the American Type Culture Collection (ATCC). B16 lines were transduced to express gB498-505 and eGFP as previously described [31]. Transduction was confirmed by eGFP expression and eGFP+ cells were sorted by flow cytometry to establish purified cell lines. B16.gB tumour cell lines were passaged routinely at 70–80% confluency and cultured in cRPMI media. 5 × 105 tumour cells were inoculated subcutaneously under the dermis of the left-hand flank, and tumour growth was monitored daily with callipers.
Adoptive T cell therapy (ACT)
gBT-I T cells were activated in vitro as described above, in the presence and absence of epigenetic drugs. Eight days after B16.gB inoculation, mice with palpable tumours were exposed to 550 Rad gamma irradiation, and received 1.0 × 107 gBT-I T cells intravenously on the same day.
Statistics
All graphs and statistical analysis were performed using GraphPad Prism (GraphPad). Tumour growth curves were compared using Mixed Model Type 3 Analysis of Variance (ANOVA), with Kenward-Roger Approximation using Estimated Marginal Means for multiple comparisons [32]. (http://doi.org/10.5281/zenodo.3341720)
Funding Statement
This work was supported by a project grant and Fellowship from the Children’s Leukaemia and Cancer Research Foundation, Western Australia (to MNC). JC was supported by a Cancer Council Western Australia Suzanne Cavanagh Early Career Investigator Award, and a US Department of Defense Peer Reviewed Cancer Program Cancer Horizon Award. JW was supported by fellowships from the NHMRC and Cancer Council WA.
Acknowledgments
The authors thank the Telethon Kids Institute Bioresources team for their excellent animal care and the Telethon Kids Flow Cytometry Facility for assistance in sorting experiments.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Rosenberg SA, Restifo NP.. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015. April 3;348(6230):62–68. PubMed PMID: 25838374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Verdegaal EM. Adoptive cell therapy: a highly successful individualized therapy for melanoma with great potential for other malignancies. Curr Opin Immunol. 2016. April;39:90–95. PubMed PMID: 26829458. [DOI] [PubMed] [Google Scholar]
- [3].Khammari A, Labarriere N, Vignard V, et al. Treatment of metastatic melanoma with autologous Melan-A/MART-1-specific cytotoxic T lymphocyte clones. J Invest Dermatol. 2009. December;129(12):2835–2842. PubMed PMID: 19554023. [DOI] [PubMed] [Google Scholar]
- [4].Tran E, Robbins PF, Lu YC, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med. 2016. December 8;375(23):2255–2262. PubMed PMID: 27959684; PubMed Central PMCID: PMCPMC5178827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Robbins PF, Dudley ME, Wunderlich J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Iimmunol. 2004. December 15;173(12):7125–7130. PubMed PMID: 15585832; PubMed Central PMCID: PMCPMC2175171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Klebanoff CA, Gattinoni L, Restifo NP. Sorting through subsets: which T-cell populations mediate highly effective adoptive immunotherapy? J Immunother. 2012. Nov-Dec;35(9):651–660. PubMed PMID: 23090074; PubMed Central PMCID: PMCPMC3501135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012. October;12(10):671–684. PubMed PMID: 22996603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Gattinoni L, Zhong XS, Palmer DC, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009. July;15(7):808–813. PubMed PMID: 19525962; PubMed Central PMCID: PMCPMC2707501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gattinoni L, Lugli E, Ji Y, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011. September 18;17(10):1290–1297. PubMed PMID: 21926977; PubMed Central PMCID: PMCPMC3192229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Henning AN, Roychoudhuri R, Restifo NP. Epigenetic control of CD8(+) T cell differentiation. Nat Rev Immunol. 2018. May;18(5):340–356. PubMed PMID: 29379213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mann BS, Johnson JR, Cohen MH, et al. FDA approval summary: vorinostat for treatment of advanced primary cutaneous T-cell lymphoma. Oncologist. 2007. October;12(10):1247–1252. PubMed PMID: 17962618. [DOI] [PubMed] [Google Scholar]
- [12].Laubach JP, Moreau P, San-Miguel JF, et al. Panobinostat for the treatment of multiple myeloma. Clin Cancer Res off J Am Assoc Cancer Res. 2015. November 1;21(21):4767–4773. 10.1158/1078-0432.CCR-15-0530. PubMed PMID: 26362997 [DOI] [PubMed] [Google Scholar]
- [13].Lisiero DN, Soto H, Everson RG, et al. The histone deacetylase inhibitor, LBH589, promotes the systemic cytokine and effector responses of adoptively transferred CD8+ T cells. J Immunother Cancer. 2014;2:8. PubMed PMID: 25054063; PubMed Central PMCID: PMCPMC4105687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yu T, Wang Y, Hu Q, et al. The EZH2 inhibitor GSK343 suppresses cancer stem-like phenotypes and reverses mesenchymal transition in glioma cells. Oncotarget. 2017. November 17;8(58):98348–98359. PubMed PMID: 29228694; PubMed Central PMCID: PMCPMC5716734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kagoya Y, Nakatsugawa M, Yamashita Y, et al. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest. 2016. September 1;126(9):3479–3494. PubMed PMID: 27548527; PubMed Central PMCID: PMCPMC5004946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011. September 16;146(6):904–917. PubMed PMID: 21889194; PubMed Central PMCID: PMCPMC3187920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011. July 12;20(1):53–65. PubMed PMID: 21741596; PubMed Central PMCID: PMCPMC4046888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Duvic M, Talpur R, Ni X, et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood. 2007. January 1;109(1):31–39. PubMed PMID: 16960145; PubMed Central PMCID: PMCPMC1785068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mueller SN, Heath W, McLain JD, et al. Characterization of two TCR transgenic mouse lines specific for herpes simplex virus. Immunol Cell Biol. 2002. April;80(2):156–163. PubMed PMID: 11940116. [DOI] [PubMed] [Google Scholar]
- [20].Oehen S, Brduscha-Riem K. Differentiation of naive CTL to effector and memory CTL: correlation of effector function with phenotype and cell division. J Iimmunol. 1998. November 15;161(10):5338–5346. PubMed PMID: 9820507 [PubMed] [Google Scholar]
- [21].Lang A, Nikolich-Zugich J. Development and migration of protective CD8+ T cells into the nervous system following ocular herpes simplex virus-1 infection. J Iimmunol. 2005. March 1;174(5):2919–2925. PubMed PMID: 15728503 [DOI] [PubMed] [Google Scholar]
- [22].Wong DJ, Rao A, Avramis E, et al. Exposure to a histone deacetylase inhibitor has detrimental effects on human lymphocyte viability and function. Cancer Immunol Res. 2014. May;2(5):459–468. PubMed PMID: 24795358; PubMed Central PMCID: PMCPMC4010948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Clutton G, Xu Y, Baldoni PL, et al. The differential short- and long-term effects of HIV-1 latency-reversing agents on T cell function. Sci Rep. 2016. August;2(6):30749. PubMed PMID: 27480951; PubMed Central PMCID: PMCPMC4969750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wozniak MB, Villuendas R, Bischoff JR, et al. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica. 2010. April;95(4):613–621. PubMed PMID: 20133897; PubMed Central PMCID: PMCPMC2857191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lin L, Jiang H, Huang M, et al. Depletion of histone deacetylase 1 inhibits metastatic abilities of gastric cancer cells by regulating the miR-34a/CD44 pathway. Oncol Rep. 2015. August;34(2):663–672. PubMed PMID: 26035691. [DOI] [PubMed] [Google Scholar]
- [26].Wu S, Zhu W, Peng Y, et al. The antitumor effects of vaccine-activated CD8(+) T cells associate with weak TCR signaling and induction of stem-like memory T cells. Cancer Immunol Res. 2017. October;5(10):908–919. PubMed PMID: 28851693; PubMed Central PMCID: PMCPMC5626646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gao FG, Khammanivong V, Liu WJ, et al. Antigen-specific CD4+ T-cell help is required to activate a memory CD8+ T cell to a fully functional tumor killer cell. Cancer Res. 2002. November 15;62(22):6438–6441. PubMed PMID: 12438231 [PubMed] [Google Scholar]
- [28].Prlic M, Bevan MJ. Cutting edge: beta-catenin is dispensable for T cell effector differentiation, memory formation, and recall responses. J Iimmunol. 2011. August 15;187(4):1542–1546. PubMed PMID: 21724993; PubMed Central PMCID: PMCPMC3150307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sabatino M, Hu J, Sommariva M, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016. July 28;128(4):519–528. PubMed PMID: 27226436; PubMed Central PMCID: PMCPMC4965906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wylie B, Chee J, Forbes CA, et al. Acquired resistance during adoptive cell therapy by transcriptional silencing of immunogenic antigens. OncoImmunology. 2019;8(8):1609874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wylie B, Seppanen E, Xiao K, et al. Cross-presentation of cutaneous melanoma antigen by migratory XCR1+CD103- and XCR1+CD103+ dendritic cells. Oncoimmunology. 2015. August;4(8):e1019198. PubMed PMID: 26405572; PubMed Central PMCID: PMCPMC4570138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wilson W. Varix/TumourGrowthAnalysis: tumour growth kinetics analysis. Zenodo. 2019. DOI: 10.5281/zenodo.3341720 [DOI] [Google Scholar]