

Abstract
Newly formed (NF) B cells that emerge from the bone marrow to the periphery have often been referred to as immature or transitional B cells. However, NF B cells have several striking characteristics, including a distinct BCR repertoire, high expression of activation-induced cytidine deaminase, high sensitivity to PAMPs, and the ability to produce cytokines. A number of findings do not support their designation as immature since NF B cells have the potential to become Ab-producing cells and to undergo class switch recombination. Here, we provide a fresh perspective on NF B cell functions and describe some of the signals driving their activation. We summarize growing evidence supporting a role for NF B cells in protection against infections and as a potential source of auto-Ab-producing cells in autoimmune diseases such as systemic lupus erythematosus.
Introduction
B cell subsets can be named based on where they reside. The B cells in follicles are follicular (FO) B cells. B cells in marginal zones are marginal zone (MZ) B cells. B cells in bone marrow (BM) are BM B cells, and the B cells in GALT are GALT B cells. Some B cells simply don’t stay put and recirculate throughout the body. Many of the recirculating B cells are newly minted and have just left the BM. These newly formed (NF) naïve B cells in some respects are like teenagers leaving home for the first time, young adults but not experienced adults. They haven’t yet gone through all of life’s checkpoints to obtain a final specificity. They can be selected against and die young. Or in the presence of inflammatory or other signals, they can proliferate, secrete Abs or produce cytokines. In healthy humans, about 40% of the Abs made by what Wardemann et al. termed ‘immature B cells’ are autoreactive (1, 2). NF B cells are present in the peripheral B-cell pool throughout life, but are the most abundant peripheral B cell subset in neonates, before the naïve B cell pool is established. NF B cells are also the main peripheral B cell population in patients undergoing B cell-depletion therapy (3) and in some patients with immunodeficiency (4).
One widely accepted classification of B cells newly arriving to the spleen has been to define them as immature in contrast to mature FO or MZ B cells. The immature B cells in mice are surface IgM (sIgM)++ and surface IgD (IgD)+ while the mature B cells are sIgM+sIgD++ (5, 6). A number of differences were identified between immature and mature B cells (6). Neonatal and immature B cells are particularly sensitive to clonal deletion or tolerance induction (7). Given the importance of defining how autoreactive B cells and Abs are selected against, the field has tended to focus on how NF B cells are altered or selected to become FO or MZ B cells, rather than on the possible functions of the NF B cells per se.
This review summarizes findings on NF B cell selection and functions and the current understating of what factors are important for NF B cell activation (Fig. 1). We have chosen the term ‘NF B cell’ as opposed to ‘immature B cell’ since it more accurately denotes their status as recent BM emigrants, naïve yet competent B cells.
Fig.1. The fate of newly-formed (NF) B cells in the periphery.
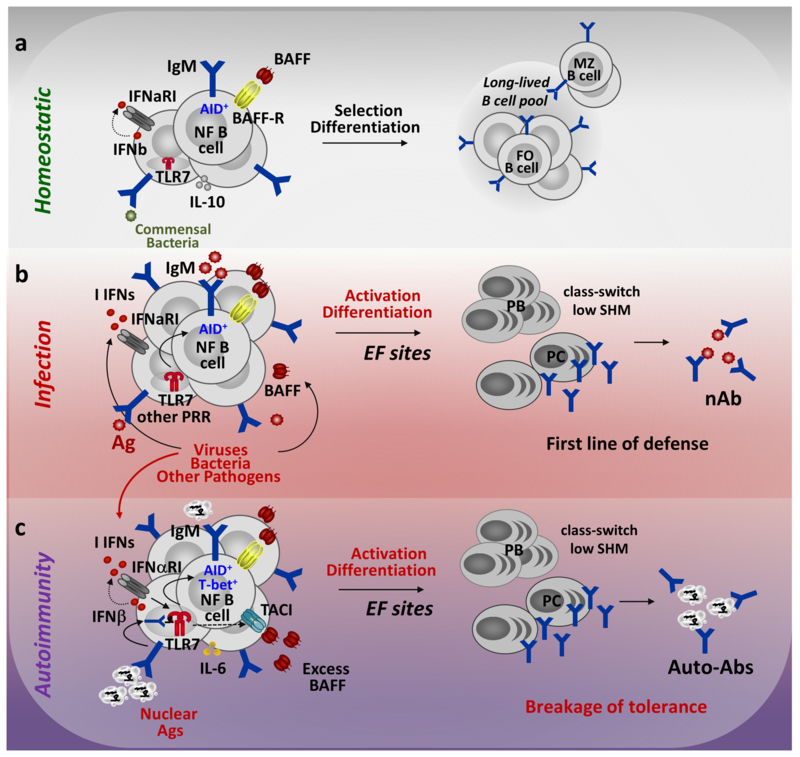
NF B cells migrating to the periphery from the bone marrow express AID, have a unique BCR repertoire, and 25-45% of NF B cells are autoreactive. Under homeostatic conditions (a) NF B cells undergo selection and become follicular (FO) or marginal zone (MZ) B cells. Their selection depends on signals received via the BCR, BAFFR, commensal bacteria, and other receptors. (b) Infections by e.g., viruses or bacteria lead to increases in type I IFN (I IFN) production and BAFF. NF B cells recognize PAMPs and pathogen-associated Ags, become activated and differentiate into plasmablasts (PBs) or IgG-secreting plasma cells (PCs), which produce neutralizing Abs. This provides a first line of defense against pathogens. (c) In autoimmune conditions NF B cell recognize self-Ags via the BCR, which combined with signals through TLR7, IFNαRI and TACI (in the excess of BAFF) can drive direct activation of NF B cells and the production of class-switched auto-Abs.
1. NF B cell development and selection in the periphery
After expressing a functional BCR on their surface, developing B cells in the BM may undergo rounds of selection through receptor editing or clonal deletion. Pre-B cells that escape negative selection in the BM develop into NF B cells which enter the periphery and circulate through the blood or enter the splenic red pulp (RP) (5, 8). In mice, NF B cells represent about 15–20% of blood B cells and about 10–15% of splenic B cells (9), although these percentage decrease with age. In humans, NF B cells represent about 4-10 % of CD19+ B cells in adult peripheral blood (10). The NF B population in both mice and humans is heterogeneous. Several different classifications have been proposed to delineate different subsets of NF B or transitional B cells (Table 1). In general, mouse NF B cells express high levels of sIgM, heat-stable Ag (CD24) and CD93. When they differentiate towards becoming FO or MZ B cells, they upregulate sIgD, CD23 and CD21 (8, 10-12). Human NF B cells express sIgM and sIgD and are defined as CD24hi, CD38hi, CD10+, and CD27−. As they become mature naïve B cell they downregulate sIgM, CD24, CD38 and CD10 and upregulate sIgD, CD23 and CD21 (3, 10, 14, 15).
Table 1.
Characteristics of newly-formed (NF) B cells in comparison to other naïve B cell subsets in the periphery
| Cell Subsets |
Surface Phenotype |
Lifespan and Location |
BCR Repertoire /AID expression |
Possible Functions |
|---|---|---|---|---|
| NF B cells |
Human* sIgMhi, sIgD+/lo, CD10+, CD27−, CD24hi, CD38hi, CD21−/lo (T1) sIgM+, sIgD+, CD10+, CD27− CD24+/hi, CD38+/hi, CD21+ (T2) Mouse** sIgMhi, sIgD−/lo, CD93hi, CD24hi, CD21−/lo, CD23−/lo (T1) sIgMhi, sIgD+, CD93+, CD24hi, CD21+, CD23+ (T2) |
Short lifespan Circulation Splenic Red Pulp |
High Autoreactivity AID+ (constitutive) |
Precursors to Mature Naïve B cells and short lived PCs Rapid, short lived Ab responses (T cell independent) High sensitivity to PAMPs Production of polyreactive neutralizing Ab, Auto-Abs Cytokine production Ag-presentation to T cells |
| Mature Naïve B cells |
Human* sIgM+/lo, sIgD+, CD27−, CD10− CD24+/lo, CD38+/lo, CD21+, CD23+ Mouse** CD93−, sIgM+, sIgDhi, CD21/CD35lo, CD24lo, CD23hi (FO) or CD93−, sIgMhi, sIgDlo, CD21/CD35hi, CD24hi, CD23−/lo, CD1dhi (MZ) |
Long-lived Follicles (FO), Spleen or LNs Marginal zone (MZ), Spleen Circulation |
Reduced Autoreactivity AID−(inducible) |
Precursors for GC, memory B cells and short or long lived PCs Delayed and sustained Ab responses (T cell-dependent or independent). Production of hi-affinity protective Abs, Auto-Abs Cytokine production Ag-presentation to T cells |
Note: All cells in question are * CD19+, CD20+ and ** CD19+, B220+
NF B cells in mice have rapid turnover and are subjected to both negative and positive selection (16, 17). Early studies suggested that NF B cells cannot proliferate and undergo apoptosis upon BCR engagement. However, most of this work used strong BCR crosslinking as an Ag surrogate (7, 18, 19) without taking into consideration innate signals and the functions of NF B cells per se, which, as discussed below, strongly depend on the settings where NF B cells encounter Ags.
Under homeostatic conditions, some NF B cells die, and others join the peripheral long-lived B cell pool (Fig. 1a). Just how and where NF B cells are selected to become MZ and FO B cells is not entirely clear, even after decades of study (reviewed in more detail elsewhere (16, 17, 20)). A current model suggests that NF B cells are selected to become other B cell subsets or undergo apoptosis depending on the strength of the BCR signal or ‘BCR signaling threshold’, which is established based on the crosstalk between signals received through the BCR and other secondary signals (20). An example of how other signals can affect NF B cell fate is the crosstalk between BCR and B cell-activating factor receptor (BAFFR). Although BAFF promotes mainly mature B cell survival, signaling via BAFFR also plays a significant role in NF B cell differentiation and survival (21-23). BCR and BAFFR signaling engage in complex crosstalk (24-27). In NF B cells, BCR engagement drives the production of p100, which in turn is used by BAFFR signaling to promote cell survival (25). The expression of BAFFR on NF B cells appears to require a tonic BCR signal (28). The absence of BAFF or BAFFR results in a reduction of peripheral B cells and a failure in B cell differentiation passed the NF B cell stage (22, 28). BAFF transgenic (Tg) mice that overexpress BAFF, on the other hand, have expanded peripheral B cells and develop systemic autoimmunity similar to human systemic lupus erythematosus (SLE) and Sjögren’s syndrome (29). This might be due to the rescue of autoreactive NF B cells from negative selection (30, 31) or be associated with the activation of NF B cells capable of class switch recombination (CSR) and producing IgG auto-Abs (32, 33). Since BAFF levels can become elevated during infections, and in some patients with autoimmune diseases, the effects of BAFF on NF B cells are relevant to human disease. In addition to BCR/BAFFR crosstalk, NF B cell selection and survival may depend on CD40 signaling (34, 35) or signals to endosomal TLRs (as discussed in more details below).
Unlike FO and MZ B cells, NF B cells constitutively express activation-induced deaminase (AID) (32, 36-38). This suggests they may rapidly respond to Ags in vivo and undergo CSR or even somatic hypermutation (SHM) (36). Alternatively, Kuraoka et al. (39) and others (40) have found that AID must be expressed in NF B cells for developing autoreactive B cells to be removed. Just how AID mediates this effect is not known. However, several groups have reported that TLR signals can upregulate AID in NF B cells (32, 41).
Wardemann et al. cloned Abs from single B cells derived from the BM and blood of healthy donors and tested them for reactivity against nuclear and cytoplasmic Ags. About 40% of newly emigrated blood B cells (i.e., NF B cells) react with more than one self-Ag (e.g., are autoreactive/polyreactive) (1, 2). Martin et al. (42) isolated pre-B cells and B cells (sIgM+ CD38+) from human BM and from the same donors, peripheral blood NF/transitional B cells (sIgD+CD10+CD27−) and naïve B cells (sIgD+CD10−CD27−). Using long-read, high-throughput sequencing, they obtained and analyzed heavy and light chain gene sequences from these subsets. NF B cells have a BCR repertoire different from the other B cell subsets, and importantly, are not simply precursors to naïve B cells. This is pertinent in light of a recent report suggesting that the NF B cell BCR repertoire may be shaped by signals and/or microbial symbiont Ags (43). The distinct BCR repertoire of NF B cells may contribute to protection against pathogens. However, since NF B cells are highly enriched for autoreactive/polyreactive specificities, they may also be a source of potentially harmful clones expressing auto-Abs.
Remarkably, the studies of Fink and her colleagues suggest that there is a T cell equivalent to the NF B cell: recent thymic emigrants (RTEs). CD8 RTEs can rapidly respond to low-affinity ligands better than mature T cells and can quickly become effectors (44, 45). As with NF B cells, the downside of RTE responses is that they can elicit autoimmunity. Like some NF B cells, RTEs are tolerized without secondary inflammatory signals and express an anergy-associated gene signature (46).
2. NF-B cells in the protection against infections
Since NF B cells have a distinct BCR repertoire, they are a potential unique source of Abs specific for pathogens. NF B cells in humans and mice expand after viral or bacterial infections, suggesting they respond to pathogens and their products (Fig. 1b). Notably, patients with viral infections, including HIV and hepatitis C virus (HCV), have increased frequencies of circulating NF B cells (4, 10, 47-50). A few in vivo studies assessing the role of NF B cells during infections suggest they play an active role in protective immunity.
2.1. NF B cells increase after viral infections
The frequency of NF B cells increases during some viral infections including HIV and HCV and after influenza vaccine administration in children. HIV-infected patients with active disease have a significantly increased frequency of peripheral blood CD21loCD27−CD10+ NF B cells (47, 51). HIV immune responses are also characterized by the rapid expansion of atypical CD21loCD27+ IgG-producing (PCs) and a reduction of memory B cells (47, 52). Since NF B cells can class-switch, it is possible that the rapid forming IgG-producing cells in these patients develop directly from NF B cells.
Furthermore, using a germline targeting strategy, Jardine et al. showed that a population of human naive B cells that includes NF B cells could be induced to undergo affinity maturation and produce broadly-neutralizing Abs (bnAb) against HIV (53). Given that NF B cells have a distinct repertoire from naïve B cells (42) and apparently respond to commensal bacteria (43), they may have the potential to respond in a distinct way to pathogens like HIV. Whether germline BCRs found within the naïve/NF B cell repertoire can be cloned and modified to produce hi-affinity, bnAbs against HIV and other viruses is worth studying (53-56). The production of bnAb might be induced during polyclonal B cell activation, which occurs early after HIV and acute Ebola virus infection (57, 58).
The signals driving the expansion of NF B cells in HIV patients are not known; however, HIV ssRNA activates TLR7 that can drive the expansion and activation of autoreactive NF B cells (discussed below). HIV-infected individuals with advanced disease produce increased levels of auto-Abs (1, 59, 60). Since NF B cells are enriched in autoreactivity, this raises the possibility that during HIV infection, NF B cells become activated and produce Abs (47).
Chronic HCV infection is another example where polyclonal B cell activation is observed coupled with the expansion of NF B cells. HCV+ patients have increased frequencies of NF B cells, which correlates with increases in serum BAFF levels (61). NF B cells from HCV-infected patients compared to NF B cells from healthy controls have a more proliferative phenotype, i.e., higher expression of the Ki67 marker. An increase in NF B cells is also detected in children who have received influenza vaccinations (48). NF B cell increases and activation might contribute to early and robust nAb responses and increased number of plasmablasts (PBs), particularly when a trivalent inactivated influenza vaccine is used (48). Thus, several studies report changes in NF B cells after vaccination or infection with viruses known to induce early polyclonal B cell responses. NF B cells respond to viral infection and presumably may play an active role in immune responses to viruses.
2.2. NF B cells can provide protection against viral infection
Not many in vivo studies have assessed the role of NF B cells during viral infections. NF B cells can contribute to protective immunity against West Nile virus (WNV) in mice (62). Using a CD180-based immunization method that targets Ags directly to B cells, we induced strong and persistent humoral immune responses in BAFFR-deficient mice that have NF B cells, but lack FO and MZ B cells. After immunization, these mice develop nAbs and are protected from an otherwise lethal WNV infection. After WNV infection NF B cells in BAFFR−/− mice produce both WNV-specific IgG Abs and nAbs in the absence of germinal center (GC) formation. Furthermore, immunization of BAFFR−/− mice with the hapten NP attached to anti-CD180 induces the expansion of NP-specific NF B cells and increases in NP-specific IgG Ab secreting cells. Thus, NF B cells can be induced to respond to specific Ags and can go on to make Ag specific-Abs in vivo.
Ag targeting to B cell subsets is a strategy to improve vaccine efficiency. After Ag is targeted to the CD180 receptor, which is expressed on all B cell subsets as well as other Ag presenting cells (APCs), strong Ab and T cell responses are induced (63, 64). In mice inoculated with Ag conjugated to an anti-CD180 Ab (Ag-αCD180), NF B cells, MZ B cells and FO B cells process and present Ag and stimulate Ag-specific CD4+ T follicular helper cells (64). Within 24-48 hours after immunization Ag-specific B cells are detectable at T-B borders in the spleen, after which Ag-specific NF B and FO B cells proliferate in a T cell-dependent manner. In contrast, Ag targeting to CD40 fails to activate NF B cells and other B cells to become APCs, even though Ag-presenting dendritic cells (DCs) are activated. These results are consistent with previous findings that the consequences of Ag presentation by B cells are multifaceted and distinct from those of DCs. For example, the peptide/MHC II complexes derived from BCR-associated Ag acquisition are predominately of the rare M1 MHC II conformation, defined by the Ia.2 epitope (65). This MHC epitope is associated with the robust activation of CD4+ T cells (66). Thus, the CD4+ T cells activated by B cell APCs, including NF B cells, may be qualitatively different than those stimulated by DCs. Furthermore, while with some Ags, initial CD4+ T cell activation and expansion may rely on DCs, B cell APCs may be required for memory T cell formation (67). In some conditions, naïve CD4+ T cells can be activated directly by Ag-presenting B cells but not by DCs, such as following virus-like-particle vaccination (68). The fact that NF B cells can be induced to become APCs suggests that under some circumstances, NF B cells contribute to protective T cell-dependent immunity. Further studies are required to define fully how and when NF B cells are activated to become APCs.
2.3. NF B cells respond to commensal bacteria
Chen et al. (43) reported that microbial symbionts influence host immunity by enriching frequencies of antibacterial specificities within the pre-immune B cell repertoires early in life and in a T-independent manner. Mice colonized with microbiota for 21 days after weaning have increased frequencies of progenitor and splenic NF B cells compared to their germ-free littermates. Importantly, changes in Ig repertoire (VH preference) induced by symbiotic microbiota evident in the naïve B cell compartment are present in NF B cells as early as seven days after colonization. The exposure to microbial symbionts enriches the frequencies of antibacterial B cell specificities in the intestine and spleen (43). These results are in line with earlier findings by the Wesemann group (69) showing that primary B-cell development in the intestine includes mucosal BCR editing and contributes to differential diversification of pre-immune repertoires in the lamina propria and BM. The exposure to bacteria apparently enhances the diversity of the pre-immune Ig repertoire of developing NF B cells. This broadening of the NF B cell BCR repertoire may enable hosts to respond to microbes rapidly and provide protection early in life. Commensal microbes influence the IgA repertoire to provide protection against bacterial infections (70-72). Willmore et al. showed that serum IgA and IgA secreting PCs in the BM elicited by a variety of commensal bacteria colonizing the gut protect against polymicrobial sepsis (73). Vossenkamper et al. suggest that NF B cell can migrate from the BM to GALT and differentiate into IgA-producing cells, providing protection against bacterial infections (74).
2.4. NF B cells as a first line of defense against bacteria and parasites
NF B cells can respond to bacterial stimuli. Capolunghi et al. (75) showed that cord blood NF B cells respond to bacterial DNA through TLR9, express AID and Blimp-1, undergo SHM and CSR and can produce anti-pneumococcal Abs. The ability of NF B cells to respond to bacterial DNA may provide an important first line of defense early in life against pathogenic bacteria. NF B cells and naïve B cells transiently decrease after immunization of healthy individuals with the pneumococcal polysaccharide-based vaccine (64). NF B cells and naïve B cells also decrease early after vaccination, suggesting they may respond to bacterial products and turn into PCs. NF B cells may also contribute to protection against neonatal sepsis (76). Elevated levels of circulating CD19+CD38hiCD24hi NF B cells correlated with protection. NF B cells were significantly increased in survivors compared with healthy controls. In contrast, naïve B cells (CD19+CD38int CD24int) significantly decreased in all septic patients during the first seven days post-onset compared with healthy neonates, and their levels did not correlate with protection. The authors propose that NF B cells may play a protective role in neonatal sepsis by releasing IL-10.
NF B cells may also contribute to parasite-specific immune responses. Plasmodium chabaudi induces AID expression in NF B cells and MZ B cells during acute infection in mice (77). NF B cells differentiate into IgM- and IgG-secreting cells in response to P. chabaudi infection in vivo, and in vitro in response to CpG. However, whether or not NF B cells contribute to protection against malaria infection is still unclear. A recent study (78) showed that after acute infection of P. vivax, peripheral B cell subsets skew toward NF B cells (CD20+CD21+CD10+CD27−), PCs and atypical memory B cells. The increase in these B cell subsets in the acute phase of P. vivax infection reverts to baseline levels by day 30 when parasites have been cleared. P. vivax also induces a decrease in naïve B cells and no changes in classical memory B cells. Both the NF and naïve B cells appear to be activated. These changes in B cells after malaria infection result in parasite-specific and autoreactive IgM responses.
Children in malaria hyperendemic regions have increased levels of NF B cells, together with increased levels of serum BAFF and inflammatory cytokines (79, 80). A recent study examined Ab repertoire development and diversification after malaria infection in infants and discovered unexpectedly high levels of SHM in blood B cells from infected infants as young as three months old (81). Although infants have almost entirely naïve and NF B cells, after malaria infection, they have a degree of repertoire diversification comparable to that of B cells from adults after acute malaria infection. This study analyzed the repertoire of populations containing both NF and non-NF B cells. However, since NF B cells express AID, it is possible that malaria infection induces SHM within the NF-B cell population.
Clonal selection of naïve B cells rather than affinity maturation drives protective B cell responses to Plasmodium falciparum (82). With complex Ags, the efficiency of affinity maturation may not determine the quality of Ag-specific B cell responses, but rather the frequency of Ag-reactive precursors and their activation (82). Tan et al. identified broadly reactive Abs that all carried an insertion of the LIAR1 domain and were common in malaria-endemic regions (83, 84). It will be interesting to investigate if NF B cells are a source of these broadly reactive Abs and contribute to protective immunity against malaria.
3. NF B cells in autoimmunity
Increases in circulating NF B cells are found in SLE and other autoimmune diseases, including type 1 diabetes and juvenile dermatomyositis (JDM) (49, 50). NF B cells appear to play a key role in autoimmunity, and both TLR7 and type I IFN signaling are major drivers of their activation (85, 86) (Fig. 1c). Since NF B cells are both enriched in autoreactive specificities and express AID, they may be an important source of pathogenic auto-Abs.
3.1. NF B cell activation driven by signals to TLR7 and TLR9
B cells that recognize nuclear self-Ags receive second signals through the endosomal TLRs, TLR7 and TLR9 that recognize RNA and DNA motifs (87-89). Ligation of TLR9 with CpG drives AID expression and the differentiation of NF B cells into IgM+ memory B cells and natural IgM-secreting PCs (75). TLR9 may also drive NF B cell differentiation towards auto-Ab producing MZ B cells (90). The role of TLR9 in B cell selection and activation remains unclear since the removal of TLR9 in lupus-prone mice results in worsen disease, which might be linked to the role of TLR9 in the regulation of TLR7-mediated B cell responses (86, 91). Consistent with this model, SLE patients have impaired TLR9 responses in NF B cells (92, 93), whereas patients with severe disease have expanded NF B cells, associated with high expression of TLR7 (94).
In mice, an increase in Tlr7 dosage drives the development of lupus-like disease (85, 95-97) by promoting type I IFN production by myeloid cells and DCs, and by driving autoreactive B cell activation. While all peripheral B cells express TLR7, NF B cells seem to be especially sensitive to signals via TLR7. H564Igi transgenic (Tg) mice (98) have rearranged Ig heavy and light chain genes specific for RNA/RNP-associated Ags. Some B cells in H564Igi mice undergo receptor editing and lose autoreactivity; the remaining BCR Tg (Id+) B cells express markers suggesting they are developmentally arrested at the NF B cell stage. Despite a presumed immature phenotype, Id+ B cells in these mice produce class-switched (IgG2a and IgG2b) Abs (98). The development of auto-Abs in H564Igi mice requires TLR7 and AID expression in NF B cells but is independent of T-cell help (98, 99). NF Id+ Tg B cells can also be induced in vitro to produce high levels of IgG Abs (32). Thus, data from this model suggest that NF B cells contribute to TLR7-mediated auto-Ab production.
Tlr7Tg mice with 8-16 extra copies of Tlr7 gene (95) have a massive expansion and activation of NF B cells in the splenic RP. Furthermore, TLR7 overexpression leads to selective and rapid BrdU uptake in NF B cells but not in FO B cells (32). The Tlr7Tg NF B cells are hyperresponsive to TLR7 stimulation. The hyper-proliferative phenotype of NF B cells is associated with a further increase in AID expression and upregulation of T-box transcription factor (T-bet). Upon in vitro stimulation with a TLR7 ligand, NF B cells produce high levels of IgG2b and IgG2c, including anti-RNA IgG. Although the effects of Tlr7 overexpression on NF B cell activation does not require IFN signals, the Tlr7Tg B cells we evaluated already expressed high levels of TLR7 (32). Other studies found that IFN signaling upregulates TLR7 expression in B cells including NF B cells, and is required for B cell responses to TLR7 stimulation (94, 100-103). The activation of NF B cell in Tlr7Tg mice does not rule out the possibility that Ab-secreting cells in these mice also develop after FO B cell activation and GC formation, especially since Tlr7Tg mice have increased FO and GC B cells (32, 104). However, Tlr7Tg NF B cells produce considerably higher levels of IgG Abs in response to TLR7 stimulation in vitro compared to Tlr7Tg FO B cells (32). The CD93+ NF B cells in Tlr7Tg mice are present in splenic RP. Thus, NF B cells normally may become activated at extrafollicular (EF) sites. The relative contribution of NF B cells as pathogenic auto-Ab producers and whether or not activated NF B cell interact with T cells in vivo needs further exploration.
3.2. NF B cell dysregulation in human SLE and other autoimmune diseases
Studies in human SLE support a link between copy number variations (CNV) and single-gene polymorphisms (SNP) in the TLR7 gene locus and SLE susceptibility (105). CD10+CD24hiCD38hi NF B cells are significantly expanded in TLR7hi SLE patients, of which the majority are TLR7 rs3853839 G risk allele-carriers (94). Similar to the NF B cells in the Tlr7Tg mice, NF B cells obtained from TLR7hi SLE patients produce anti-nuclear IgG in response to TLR7 ligation in vitro, suggesting that human NF B cells may be a direct source of auto-Abs. Elevated expression of TLR7 in SLE patients is also associated with an increase in IFNα, which likely further contributes to NF B cell activation and the overall disease activity in these patients. The TLR7hi SLE patients developed a wide range of auto-Ab specificities, which may well be due to increased survival and differentiation of autoreactive NF B cells.
Other studies have reported increases in circulating NF B cells in SLE patients (10, 106, 107). Wu et al. found that newly-diagnosed SLE patients have increased PTEN expression in B cells associated with the expansion of NF B cells (108). Chang et al. showed that IFNα stimulation of NF B cells from SLE patients induces increased Syk kinase activation after BCR stimulation (109). The expansion of NF B cells has also been linked to increases in IFNα levels and BAFF/APRIL production by neutrophils (110). Liu et al. also explored the effects of IFNα on NF B cell-survival, which leads to increased NF-κB activation and reduced expression of the pro-apoptotic molecule Bax (106). These changes in IFN responses and increases in NF B cells may be driven by increased TLR7 expression or signaling (94). Since overall B cell numbers may be decreased in SLE patients, the increase frequencies of NF B cells could also be due to a loss of other B cell subsets, and not necessarily reflect actual NF B cell expansion. Nevertheless, newly-diagnosed SLE patients and TLR7hi SLE patients show an increase in NF B cell frequencies and numbers (94, 108).
NF B cells (again defined as CD10+CD24hiCD38hi cells) are expanded in other autoimmune diseases, such as T1D (49). NF B cells from pediatric patients with JDM actively divide and their frequencies correlate with disease activity. NF B cells in JDM patients showed high IFNα and TLR7-signatures and increase in IL-6 production (50).
NF B cells are also increased in patients with X-linked lymphoproliferative immunodeficiency, some common variable immune deficiency patients, and patients recovering from hematopoietic stem cell transplantation (4, 111, 112). Immunodeficiencies are frequently associated with systemic autoimmunity and the production of auto-Abs (113). While this might be linked to defects in B cell selection in the bone marrow, another possibility is the abundance of poly/autoreactive NF B cells which in the absence of other B cell subsets may ‘fill the space’ in the peripheral B cell compartment as reviewed more elsewhere (112). A study from Kolhatkar et al. showed an increase in NF B cell activation and defects in NF B cell selection in Wiskott-Aldrich syndrome patients associated with dysregulation of BCR and TLR signals (114).
3.3. Cytokine production and other effector functions of NF B cells, related to autoimmunity.
Another important function of B cells is cytokine production (115). NF B cells in healthy individuals have been reported to mainly produce IL-10 and regulate T cells responses, consistent with Breg functions (116). However, NF B cells in patients with SLE, rheumatoid arthritis and JDM fail to suppress T cell responses, and compared to NF B cells from healthy controls, produce less IL-10 and more IL-6 (50, 106, 116). This alteration of cytokine production by NF B cells may be driven by type I IFN signals (94, 106), suggesting that under certain conditions NF B can produce pro-inflammatory cytokines such as IL-6, and drive the activation of other immune cells.
Recent studies revealed another property of NF B cells – an ability to produce type I IFNs (117, 118). Using a single-cell RNA seq approach, Hamilton et al. (103) showed that mouse splenic NF(T1) B cells constitutively express high levels of IFNβ. Furthermore, NF B cells from BXD2 lupus-prone mice overexpress IFNβ compared to WT NF B cells. In BM chimera experiments, NF B cells lacking endogenous IFNβ express low levels of Ifna genes and Tlr7 and have reduced survival and activation in vivo compared to IFNβ-sufficient cells. These results suggest that IFNβ is required for the optimal survival of and TLR7-driven responses of NF B cells. Based on these data, the authors proposed a model in which endogenous IFNβ, via an IFNβ-TLR7-IFNα loop, regulates NF B cell development. Increased production of IFNβ production by NF B cells may drive the induction of TLR7 and other type I IFNs during NF B cell development, resulting in the loss of tolerance and increases in auto-Ab production. Circulating NF B cells and naïve B cells in SLE patients express elevated levels of IFN-β, which correlates with disease severity (117). These new findings establish NF B cells not only as a target of IFNs but also as producers of type I IFNs. However, they raise further questions as to what the signals are that regulate endogenous IFNβ expression in NF B cells.
Since NF B cells can function as APCs, it is possible they may present auto-Ags and promote autoreactive T cell responses. Further studies are required to assess this.
3.4. NF B cell activation driven by BAFF signaling
BAFF-Tg mice develop an autoimmune phenotype associated with the production of IgG2c and IgG2b autoAbs. Although BAFF-Tg mice develop GCs, Groom et al. (119) showed that they can produce class-switched Abs even in the absence of T cells, suggesting that the activation of autoreactive B cells in these mice may occur in EF sites. NF B cells were only thought to express BAFFR; however, a recent study showed that NF B cells in BAFF-Tg mice express detectable levels of another BAFF receptor, the transmembrane activator and CAML interactor (TACI) (33). Notably, the TACI+ NF B cells are increased in BAFF-Tg mice. TACI+ NF B cells are enriched in self-reactive BCR specificities, express AID and T-bet, and when cultured in vitro spontaneously produced class-switched IgG Abs including anti-nuclear auto-Abs (33). TACI+ NF B cells also produce greater levels of class-switched IgG autoantibodies ex vivo, as compared to MZ and/or FO B cells, suggesting that they may be a dominant source of autoAb production in BAFF-Tg mice. The exact mechanisms driving TACI expression in BAFF-Tg NF B cells and how TACI-signaling promotes auto-Ab production are not clear. Jacobs et al. propose that self-nucleic acids internalized via self-reactive BCRs may drive TACI expression. Since TACI shares signaling components (MyD88, TRAF5, and TRAF6) with the TLR pathway, TACI may crosstalk with TLRs (120, 121). BAFF signaling through TACI upregulates TLR7 expression in B cells (122). However, Du et al. showed that the lack of TLR signaling does not affect the production of autoAbs significantly and/or the expression of TACI on NF B cells in the BAFF-Tg mice (123). The overall role of TACI in SLE remains somewhat controversial since some studies suggest that the expression of TACI inhibits B cell activation (124, 125). The studies of Jacobs et al. and Du et al. (33, 123), however, suggest that TACI may play a different role in NF B cells and help drive the formation of Ab producing cells, particularly when BAFF levels are elevated.
Serum levels of BAFF are increased in patients with SLE, RA, SjS, or systemic sclerosis (126). However, the potential link between BAFF, TACI and NF B cells in humans requires further exploration. The deletion of TACI on B cells protects against BAFF-mediated autoimmunity in mice (122), suggesting a selective TACI blockade may be of therapeutic value in human SLE.
Conclusions and Future Perspectives
The collective properties of NF B cells underscore the plasticity of this remarkable B cell subset (Table 1). Clearly, NF B cells can undergo selection in the periphery and contribute to an expanded B cell repertoire and protective immunity. A growing set of findings suggest they have important stand-alone functions in their own right and may play key roles in defenses against infections. NF B cells not only express AID, have a distinctive BCR repertoire that can be shaped by commensal bacteria, and can undergo CSR, but also are highly sensitive to programming by type I IFN and TLR7 signaling. They can respond not only to commensals but also to foreign Ags and can become APCs. We propose that NF B cells are an important innate B cell subset that contributes to rapid first line responses to infections (Fig 1b).
EF B cell responses are responsible for the rapid Ab production after infection and are associated with little or no hypermutation (127). During EF responses, AID is expressed and developing PBs can undergo isotype switch independently of T cells (127-132). It is tempting to speculate that NF B cells contribute to EF Ab responses not only because of the properties noted above but also because they are found in the RP. They may be key participants in EF B cell responses to infections and in the EF responses described in autoimmune mouse models and more recently in human SLE (133, 134). Infection by an attenuated strain of Salmonella typhimurium or by S. enterica leads to EF B cell activation associated with SHM and the production of Salmonella Ag-binding Abs (135-137). These studies did not determine which B cell subsets are involved in such responses. Further studies are required to assess if and when rapid EF responses and protective immune responses are driven by NF B cells.
Tipton et al. showed that a major fraction of Ab-secreting cell clones during SLE flares arise from naïve B cells, rather than from GC or memory B cells (107, 134). While this study excluded NF B cells, Wang et al. (94) reported that NF B cells derived from SLE patients could become activated and produce anti-nuclear Abs upon TLR7 ligand stimulation. Data from Tlr7Tg, H564Igi, and BAFF Tg autoimmune prone mice further support EF activation of NF B cells in the splenic RP (32, 33).
There are important implications if this remarkably understudied B cell subset can in fact be harnessed and programmed to contribute to immunity. First, possible roles for NF B cells in immunocompromised patients may have been overlooked. Infants, the elderly and immunocompromised individuals are at greater risk for severe viral infections, but also are the most unresponsive populations to vaccination (138, 139). Thus, vaccination strategies that recruit NF B cell and other B cell APCs, such as targeting Ags to CD180, may be particularly beneficial for these groups. The importance of NF B cells in autoimmunity is emphasized by the fact that NF B cells are enriched in autoreactive specificities, and can become activated, particularly in response to innate signals (IFNs and TLRs), known to be major drivers of disease pathology, particularly in SLE (Fig 1c). The fact that RTEs have efficient trafficking to the periphery and a metabolism different from mature T cells (45) also suggests avenues for exploration of their B cell counterparts. Clearly, further studies are needed to define how recently minted lymphocytes can be programmed best to contribute to protective immunity. Perhaps newly formed T cells and B cells work together during initial immune responses.
Acknowledgments
We would like to thank Kevin Draves, Kelsey Roe, Ting Wang, and John Marken members of the Clark and Giltiay laboratories whose work has contributed to the findings described in this review.
This work was supported by:
NIH/NIAID 2R01AI044257 (Clark EA, Giltiay NV), NIH/NIAID 2R01AI052203 (Clark EA); Lupus Research Alliance grant 519408 (Giltiay NV), and UL1 TR002319 (ITHS Catalyst Award) from the National Center for Advancing Translational Sciences, NIH (Giltiay NV).
Abbreviations
- AID
activation-induced cytidine deaminase
- APC
Ag presenting cell
- BAFF
B cell-activating factor
- BAFFR
B cell-activating factor receptor
- BM
bone marrow
- bnAb
broadly neutralizing Ab
- CSR
class switch recombination
- EF
extrafollicular
- FO
follicular
- GC
germinal center
- HCV
hepatitis C virus
- JDM
juvenile dermatomyositis
- MZ
marginal zone
- nAb
neutralizing Ab
- NF
newly formed
- PB
plasmablast
- PC
plasma cell
- RP
red pulp
- RTE
recent thymic emigrant
- SHM
somatic hypermutation
- sIgD
surface IgD
- sIgM
surface IgM
- SLE
systemic lupus erythematosus
- TACI
transmembrane activator and CAML interactor
- Tg
transgenic
- WNV
West Nile virus
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, and Nussenzweig MC. 2003. Predominant Autoantibody Production by Early Human B Cell Precursors. Science 301: 1374–1377. [DOI] [PubMed] [Google Scholar]
- 2.Meffre E, and Wardemann H. 2008. B-cell tolerance checkpoints in health and autoimmunity. Current Opinion in Immunology 20: 632–638. [DOI] [PubMed] [Google Scholar]
- 3.Palanichamy A, Barnard J, Zheng B, Owen T, Quach T, Wei C, Looney RJ, Sanz I, and Anolik JH. 2009. Novel Human Transitional B Cell Populations Revealed by B Cell Depletion Therapy. The Journal of Immunology 182: 5982–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cuss AK, Avery DT, Cannons JL, Yu LJ, Nichols KE, Shaw PJ, and Tangye SG. 2006. Expansion of Functionally Immature Transitional B Cells Is Associated with Human-Immunodeficient States Characterized by Impaired Humoral Immunity. The Journal of Immunology 176: 1506–1516. [DOI] [PubMed] [Google Scholar]
- 5.Hardy RR, and Hayakawa K. 2001. B Cell Development Pathways. Annual Review of Immunology 19: 595–621. [DOI] [PubMed] [Google Scholar]
- 6.Monroe JG 2004. B-cell positive selection and peripheral homeostasis. Immunological Reviews 197: 5–9. [DOI] [PubMed] [Google Scholar]
- 7.Carsetti R, Köhler G, and Lamers MC. 1995. Transitional B cells are the target of negative selection in the B cell compartment. Journal of Experimental Medicine 181: 2129–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Agrawal S, Smith S. a. B. C., Tangye SG, and Sewell WA. 2013. Transitional B cell subsets in human bone marrow. Clinical & Experimental Immunology 174: 53–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loder BF, Mutschler B, Ray RJ, Paige CJ, Sideras P, Torres R, Lamers MC, and Carsetti R. 1999. B Cell Development in the Spleen Takes Place in Discrete Steps and Is Determined by the Quality of B Cell Receptor–Derived Signals. Journal of Experimental Medicine 190: 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sims GP, Ettinger R, Shirota Y, Yarboro CH, Illei GG, and Lipsky PE. 2005. Identification and characterization of circulating human transitional B cells. Blood 105: 4390–4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chung JB, Sater RA, Fields ML, Erikson J, and Monroe JG. 2002. CD23 defines two distinct subsets of immature B cells which differ in their responses to T cell help signals. Int Immunol 14: 157–166. [DOI] [PubMed] [Google Scholar]
- 12.Su TT, and Rawlings DJ. 2002. Transitional B Lymphocyte Subsets Operate as Distinct Checkpoints in Murine Splenic B Cell Development. The Journal of Immunology 168: 2101–2110. [DOI] [PubMed] [Google Scholar]
- 13.Chung JB, Silverman M, and Monroe JG. 2003. Transitional B cells: step by step towards immune competence. Trends in Immunology 24: 342–348. [DOI] [PubMed] [Google Scholar]
- 14.Kaminski DA, Wei C, Qian Y, Rosenberg AF, and Sanz I. 2012. Advances in Human B Cell Phenotypic Profiling. Front. Immunol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suryani S, Fulcher DA, Santner-Nanan B, Nanan R, Wong M, Shaw PJ, Gibson J, Williams A, and Tangye SG. 2010. Differential expression of CD21 identifies developmentally and functionally distinct subsets of human transitional B cells. Blood 115: 519–529. [DOI] [PubMed] [Google Scholar]
- 16.Carsetti R, Rosado MM, and Wardmann H. 2004. Peripheral development of B cells in mouse and man. Immunological Reviews 197: 179–191. [DOI] [PubMed] [Google Scholar]
- 17.Metzler G, Kolhatkar NS, and Rawlings DJ. 2015. BCR and co-receptor crosstalk facilitate the positive selection of self-reactive transitional B cells. Current Opinion in Immunology 37: 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norvell A, Mandik L, and Monroe JG. 1995. Engagement of the antigen-receptor on immature murine B lymphocytes results in death by apoptosis. The Journal of Immunology 154: 4404–4413. [PubMed] [Google Scholar]
- 19.Monroe JG 1996. Tolerance sensitivity of immature-stage B cells: can developmentally regulated B cell antigen receptor (BCR) signal transduction play a role? The Journal of Immunology 156: 2657–2660. [PubMed] [Google Scholar]
- 20.Pillai S, and Cariappa A. 2009. The follicular versus marginal zone B lymphocyte cell fate decision. Nature Reviews Immunology 9: 767–777. [DOI] [PubMed] [Google Scholar]
- 21.Mackay F, and Schneider P. 2009. Cracking the BAFF code. Nature Reviews Immunology 9: 491–502. [DOI] [PubMed] [Google Scholar]
- 22.Rickert RC, Jellusova J, and Miletic AV. 2011. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunological Reviews 244: 115–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smulski CR, and Eibel H. 2018. BAFF and BAFF-Receptor in B Cell Selection and Survival. Front. Immunol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Craxton A, Draves KE, Gruppi A, and Clark EA. 2005. BAFF regulates B cell survival by downregulating the BH3-only family member Bim via the ERK pathway. Journal of Experimental Medicine 202: 1363–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn Iii WJ, Brezski RJ, Treml LS, Jordan KA, Monroe JG, Sen R, and Cancro MP. 2008. Tonic B cell antigen receptor signals supply an NF-κB substrate for prosurvival BLyS signaling. Nature Immunology 9: 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, and Tybulewicz VLJ. 2013. The BAFF Receptor Transduces Survival Signals by Co-opting the B Cell Receptor Signaling Pathway. Immunity 38: 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rawlings DJ, Metzler G, Wray-Dutra M, and Jackson SW. 2017. Altered B cell signalling in autoimmunity. Nature Reviews Immunology 17: 421–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rowland SL, Leahy KF, Halverson R, Torres RM, and Pelanda R. 2010. BAFF Receptor Signaling Aids the Differentiation of Immature B Cells into Transitional B Cells following Tonic BCR Signaling. The Journal of Immunology 185: 4570–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, Tschopp J, and Browning JL. 1999. Mice Transgenic for Baff Develop Lymphocytic Disorders along with Autoimmune Manifestations. Journal of Experimental Medicine 190: 1697–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lesley R, Xu Y, Kalled SL, Hess DM, Schwab SR, Shu H-B, and Cyster JG. 2004. Reduced Competitiveness of Autoantigen-Engaged B Cells due to Increased Dependence on BAFF. Immunity 20: 441–453. [DOI] [PubMed] [Google Scholar]
- 31.Thien M, Phan TG, Gardam S, Amesbury M, Basten A, Mackay F, and Brink R. 2004. Excess BAFF Rescues Self-Reactive B Cells from Peripheral Deletion and Allows Them to Enter Forbidden Follicular and Marginal Zone Niches. Immunity 20: 785–798. [DOI] [PubMed] [Google Scholar]
- 32.Giltiay NV, Chappell CP, Sun X, Kolhatkar N, Teal TH, Wiedeman AE, Kim J, Tanaka L, Buechler MB, Hamerman JA, Imanishi-Kari T, Clark EA, and Elkon KB. 2013. Overexpression of TLR7 promotes cell-intrinsic expansion and autoantibody production by transitional T1 B cells. Journal of Experimental Medicine 210: 2773–2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jacobs HM, Thouvenel CD, Leach S, Arkatkar T, Metzler G, Scharping NE, Kolhatkar NS, Rawlings DJ, and Jackson SW. 2016. Cutting Edge: BAFF Promotes Autoantibody Production via TACI-Dependent Activation of Transitional B Cells. The Journal of Immunology 196: 3525–3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lesley R, Kelly LM, Xu Y, and Cyster JG. 2006. Naive CD4 T cells constitutively express CD40L and augment autoreactive B cell survival. PNAS 103: 10717–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz MA, Kolhatkar NS, Thouvenel C, Khim S, and Rawlings DJ. 2014. CD4+ T Cells and CD40 Participate in Selection and Homeostasis of Peripheral B Cells. The Journal of Immunology 193: 3492–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mao C, Jiang L, Melo-Jorge M, Puthenveetil M, Zhang X, Carroll MC, and Imanishi-Kari T. 2004. T Cell-Independent Somatic Hypermutation in Murine B Cells with an Immature Phenotype. Immunity 20: 133–144. [DOI] [PubMed] [Google Scholar]
- 37.Ueda Y, Liao D, Yang K, Patel A, and Kelsoe G. 2007. T-Independent Activation-Induced Cytidine Deaminase Expression, Class-Switch Recombination, and Antibody Production by Immature/Transitional 1 B Cells. The Journal of Immunology 178: 3593–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han J-H, Akira S, Calame K, Beutler B, Selsing E, and Imanishi-Kari T. 2007. Class Switch Recombination and Somatic Hypermutation in Early Mouse B Cells Are Mediated by B Cell and Toll-like Receptors. Immunity 27: 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuraoka M, Meffre E, and Kelsoe G. 2018. Chapter Two - The First B-Cell Tolerance Checkpoint in Mice and Humans: Control by AID In Advances in Immunology vol. 139 Alt F, ed. Academic Press; 51–92. [DOI] [PubMed] [Google Scholar]
- 40.Cantaert T, Schickel J-N, Bannock JM, Ng Y-S, Massad C, Oe T, Wu R, Lavoie A, Walter JE, Notarangelo LD, Al-Herz W, Kilic SS, Ochs HD, Nonoyama S, Durandy A, and Meffre E. 2015. Activation-Induced Cytidine Deaminase Expression in Human B Cell Precursors Is Essential for Central B Cell Tolerance. Immunity 43: 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuraoka M, Snowden PB, Nojima T, Verkoczy L, Haynes BF, Kitamura D, and Kelsoe G. 2017. BCR and Endosomal TLR Signals Synergize to Increase AID Expression and Establish Central B Cell Tolerance. Cell Reports 18: 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martin VG, Wu Y-CB, Townsend CL, Lu GHC, O’Hare JS, Mozeika A, Coolen ACC, Kipling D, Fraternali F, and Dunn-Walters DK. 2016. Transitional B Cells in Early Human B Cell Development – Time to Revisit the Paradigm? Front. Immunol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen Y, Chaudhary N, Yang N, Granato A, Turner JA, Howard SL, Devereaux C, Zuo T, Shrestha A, Goel RR, Neuberg D, and Wesemann DR. 2018. Microbial symbionts regulate the primary Ig repertoire. Journal of Experimental Medicine 215: 1397–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berkley AM, and Fink PJ. 2014. Cutting Edge: CD8+ Recent Thymic Emigrants Exhibit Increased Responses to Low-Affinity Ligands and Improved Access to Peripheral Sites of Inflammation. The Journal of Immunology 193: 3262–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cunningham CA, Helm EY, and Fink PJ. 2018. Reinterpreting recent thymic emigrant function: defective or adaptive? Current Opinion in Immunology 51: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Friesen TJ, Ji Q, and Fink PJ. 2016. Recent thymic emigrants are tolerized in the absence of inflammation. Journal of Experimental Medicine 213: 913–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Malaspina A, Moir S, Ho J, Wang W, Howell ML, O’Shea MA, Roby GA, Rehm CA, Mican JM, Chun T-W, and Fauci AS. 2006. Appearance of immature/transitional B cells in HIV-infected individuals with advanced disease: Correlation with increased IL-7. PNAS 103: 2262–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cao RG, Suarez NM, Obermoser G, Lopez SMC, Flano E, Mertz SE, Albrecht RA, García-Sastre A, Mejias A, Xu H, Qin H, Blankenship D, Palucka K, Pascual V, and Ramilo O. 2014. Differences in Antibody Responses Between Trivalent Inactivated Influenza Vaccine and Live Attenuated Influenza Vaccine Correlate With the Kinetics and Magnitude of Interferon Signaling in Children. J Infect Dis 210: 224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Habib T, Funk A, Rieck M, Brahmandam A, Dai X, Panigrahi AK, Prak ETL, Meyer-Bahlburg A, Sanda S, Greenbaum C, Rawlings DJ, and Buckner JH. 2012. Altered B Cell Homeostasis Is Associated with Type I Diabetes and Carriers of the PTPN22 Allelic Variant. The Journal of Immunology 188: 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piper CJM, Wilkinson MGL, Deakin CT, Otto GW, Dowle S, Duurland CL, Adams S, Marasco E, Rosser EC, Radziszewska A, Carsetti R, Ioannou Y, Beales PL, Kelberman D, Isenberg DA, Mauri C, Nistala K, and Wedderburn LR. 2018. CD19+CD24hiCD38hi B Cells Are Expanded in Juvenile Dermatomyositis and Exhibit a Pro-Inflammatory Phenotype After Activation Through Toll-Like Receptor 7 and Interferon-α. Front. Immunol 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Amu S, Fievez V, Nozza S, Lopalco L, and Chiodi F. 2016. Dysfunctions in the migratory phenotype and properties of circulating immature transitional B cells during HIV-1 infection. AIDS 30: 2169. [DOI] [PubMed] [Google Scholar]
- 52.Moir S, Malaspina A, Ogwaro KM, Donoghue ET, Hallahan CW, Ehler LA, Liu S, Adelsberger J, Lapointe R, Hwu P, Baseler M, Orenstein JM, Chun T-W, Mican JAM, and Fauci AS. 2001. HIV-1 induces phenotypic and functional perturbations of B cells in chronically infected individuals. PNAS 98: 10362–10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jardine JG, Kulp DW, Havenar-Daughton C, Sarkar A, Briney B, Sok D, Sesterhenn F, Ereño-Orbea J, Kalyuzhniy O, Deresa I, Hu X, Spencer S, Jones M, Georgeson E, Adachi Y, Kubitz M, deCamp AC, Julien J-P, Wilson IA, Burton DR, Crotty S, and Schief WR. 2016. HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351: 1458–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kelsoe G, and Haynes BF. 2017. Host controls of HIV broadly neutralizing antibody development. Immunological Reviews 275: 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crowe JE 2019. Influenza Virus–Specific Human Antibody Repertoire Studies. The Journal of Immunology 202: 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Havenar-Daughton C, Abbott RK, Schief WR, and Crotty S. 2018. When designing vaccines, consider the starting material: the human B cell repertoire. Current Opinion in Immunology 53: 209–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levesque MC, Moody MA, Hwang K-K, Marshall DJ, Whitesides JF, Amos JD, Gurley TC, Allgood S, Haynes BB, Vandergrift NA, Plonk S, Parker DC, Cohen MS, Tomaras GD, Goepfert PA, Shaw GM, Schmitz JE, Eron JJ, Shaheen NJ, Hicks CB, Liao H-X, Markowitz M, Kelsoe G, Margolis DM, and Haynes BF. 2009. Polyclonal B Cell Differentiation and Loss of Gastrointestinal Tract Germinal Centers in the Earliest Stages of HIV-1 Infection. PLOS Medicine 6: e1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McElroy AK, Akondy RS, Davis CW, Ellebedy AH, Mehta AK, Kraft CS, Lyon GM, Ribner BS, Varkey J, Sidney J, Sette A, Campbell S, Ströher U, Damon I, Nichol ST, Spiropoulou CF, and Ahmed R. 2015. Human Ebola virus infection results in substantial immune activation. PNAS 112: 4719–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Susal C, Daniel V, Oberg HH, Terness P, Huth-Kuhne A, Zimmerman R, and Opelz G. 1992. Striking inverse association of IgG-anti-Fab gamma antibodies and CD4 cell counts in patients with acquired immunodeficiency syndrome (AIDS)/AIDS-related complex. Blood 79: 954–957. [PubMed] [Google Scholar]
- 60.Massabki PS, Accetturi C, Nishie IA, da Silva NP, Sato EI, and Andrade L. E. c. 1997. Clinical implications of autoantibodies in Hiv infection. Aids 11: 1845–1850. [DOI] [PubMed] [Google Scholar]
- 61.Sugalski JM, Rodriguez B, Moir S, and Anthony DD. 2010. Peripheral Blood B Cell Subset Skewing Is Associated with Altered Cell Cycling and Intrinsic Resistance to Apoptosis and Reflects a State of Immune Activation in Chronic Hepatitis C Virus Infection. The Journal of Immunology 185: 3019–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Giordano D, Draves KE, Young LB, Roe K, Bryan MA, Dresch C, Richner JM, Diamond MS, G. M Jr, and Clark EA. 2017. Protection of mice deficient in mature B cells from West Nile virus infection by passive and active immunization. PLOS Pathogens 13: e1006743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chaplin JW, Chappell CP, and Clark EA. 2013. Targeting antigens to CD180 rapidly induces antigen-specific IgG, affinity maturation, and immunological memory. Journal of Experimental Medicine 210: 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roe K, Shu GL, Draves KE, Giordano D, Pepper M, and Clark EA. 2019. Targeting Antigens to CD180 but Not CD40 Programs Immature and Mature B Cell Subsets to Become Efficient APCs. The Journal of Immunology ji1900549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barroso M, Tucker H, Drake L, Nichol K, and Drake JR. 2015. Antigen-B Cell Receptor Complexes Associate with Intracellular MHC Class II Molecules. J. Biol. Chem jbc.M115.649582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Busman-Sahay K, Sargent E, Harton JA, and Drake JR. 2011. The Ia.2 Epitope Defines a Subset of Lipid Raft-Resident MHC Class II Molecules Crucial to Effective Antigen Presentation. The Journal of Immunology 186: 6710–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Misumi I, and Whitmire JK. 2014. B Cell Depletion Curtails CD4+ T Cell Memory and Reduces Protection against Disseminating Virus Infection. The Journal of Immunology 192: 1597–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hong S, Zhang Z, Liu H, Tian M, Zhu X, Zhang Z, Wang W, Zhou X, Zhang F, Ge Q, Zhu B, Tang H, Hua Z, and Hou B. 2018. B Cells Are the Dominant Antigen-Presenting Cells that Activate Naive CD4+ T Cells upon Immunization with a Virus-Derived Nanoparticle Antigen. Immunity 49: 695–708.e4. [DOI] [PubMed] [Google Scholar]
- 69.Wesemann DR, Portuguese AJ, Meyers RM, Gallagher MP, Cluff-Jones K, Magee JM, Panchakshari RA, Rodig SJ, Kepler TB, and Alt FW. 2013. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature 501: 112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Slack E, Balmer ML, and Macpherson AJ. 2014. B cells as a critical node in the microbiota–host immune system network. Immunological Reviews 260: 50–66. [DOI] [PubMed] [Google Scholar]
- 71.McCoy KD, Ronchi F, and Geuking MB. 2017. Host-microbiota interactions and adaptive immunity. Immunological Reviews 279: 63–69. [DOI] [PubMed] [Google Scholar]
- 72.Zhao Q, and Elson CO. 2018. Adaptive immune education by gut microbiota antigens. Immunology 154: 28–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wilmore JR, Gaudette BT, Gomez Atria D, Hashemi T, Jones DD, Gardner CA, Cole SD, Misic AM, Beiting DP, and Allman D. 2018. Commensal Microbes Induce Serum IgA Responses that Protect against Polymicrobial Sepsis. Cell Host & Microbe 23: 302–311.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vossenkämper A, Blair PA, Safinia N, Fraser LD, Das L, Sanders TJ, Stagg AJ, Sanderson JD, Taylor K, Chang F, Choong LM, D’Cruz DP, MacDonald TT, Lombardi G, and Spencer J. 2013. A role for gut-associated lymphoid tissue in shaping the human B cell repertoire. Journal of Experimental Medicine 210: 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Capolunghi F, Cascioli S, Giorda E, Rosado MM, Plebani A, Auriti C, Seganti G, Zuntini R, Ferrari S, Cagliuso M, Quinti I, and Carsetti R. 2008. CpG Drives Human Transitional B Cells to Terminal Differentiation and Production of Natural Antibodies. The Journal of Immunology 180: 800–808. [DOI] [PubMed] [Google Scholar]
- 76.Li S, Ma F, Hao H, Wang D, Gao Y, Zhou J, Li F, Lin H-C, Xiao X, and Zeng Q. 2018. Marked elevation of circulating CD19 + CD38 hi CD24 hi transitional B cells give protection against neonatal sepsis. Pediatrics & Neonatology 59: 296–304. [DOI] [PubMed] [Google Scholar]
- 77.Wilmore JR, Maue AC, and Rochford R. 2016. Plasmodium chabaudi infection induces AID expression in transitional and marginal zone B cells. Immunity, Inflammation and Disease 4: 497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patgaonkar M, Herbert F, Powale K, Gandhe P, Gogtay N, Thatte U, Pied S, Sharma S, and Pathak S. 2018. Vivax infection alters peripheral B-cell profile and induces persistent serum IgM. Parasite Immunology 40: e12580. [DOI] [PubMed] [Google Scholar]
- 79.Asito AS, Moormann AM, Kiprotich C, Ng’ang’a ZW, Ploutz-Snyder R, and Rochford R. 2008. Alterations on peripheral B cell subsets following an acute uncomplicated clinical malaria infection in children. Malaria Journal 7: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nduati E, Gwela A, Karanja H, Mugyenyi C, Langhorne J, Marsh K, and Urban BC. 2011. The Plasma Concentration of the B Cell Activating Factor Is Increased in Children With Acute Malaria. J Infect Dis 204: 962–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wendel BS, He C, Qu M, Wu D, Hernandez SM, Ma K-Y, Liu EW, Xiao J, Crompton PD, Pierce SK, Ren P, Chen K, and Jiang N. 2017. Accurate immune repertoire sequencing reveals malaria infection driven antibody lineage diversification in young children. Nature Communications 8: 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murugan R, Buchauer L, Triller G, Kreschel C, Costa G, Martí GP, Imkeller K, Busse CE, Chakravarty S, Sim BKL, Hoffman SL, Levashina EA, Kremsner PG, Mordmüller B, Höfer T, and Wardemann H. 2018. Clonal selection drives protective memory B cell responses in controlled human malaria infection. Science Immunology 3: eaap8029. [DOI] [PubMed] [Google Scholar]
- 83.Lanzavecchia A 2018. Dissecting human antibody responses: useful, basic and surprising findings. EMBO Molecular Medicine 10: e8879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tan J, Pieper K, Piccoli L, Abdi A, Foglierini M, Geiger R, Maria Tully C, Jarrossay D, Maina Ndungu F, Wambua J, Bejon P, Silacci Fregni C, Fernandez-Rodriguez B, Barbieri S, Bianchi S, Marsh K, Thathy V, Corti D, Sallusto F, Bull P, and Lanzavecchia A. 2016. A LAIR1 insertion generates broadly reactive antibodies against malaria variant antigens. Nature 529: 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, and Bolland S. 2006. Autoreactive B Cell Responses to RNA-Related Antigens Due to TLR7 Gene Duplication. Science 312: 1669–1672. [DOI] [PubMed] [Google Scholar]
- 86.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, and Shlomchik MJ. 2006. Toll-like Receptor 7 and TLR9 Dictate Autoantibody Specificity and Have Opposing Inflammatory and Regulatory Roles in a Murine Model of Lupus. Immunity 25: 417–428. [DOI] [PubMed] [Google Scholar]
- 87.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, and Marshak-Rothstein A. 2005. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. Journal of Experimental Medicine 202: 1171–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, and Marshak-Rothstein A. 2002. Chromatin–IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416: 603. [DOI] [PubMed] [Google Scholar]
- 89.Green NM, and Marshak-Rothstein A. 2011. Toll-like receptor driven B cell activation in the induction of systemic autoimmunity. Seminars in Immunology 23: 106–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guerrier T, Youinou P, Pers J-O, and Jamin C. 2012. TLR9 drives the development of transitional B cells towards the marginal zone pathway and promotes autoimmunity. Journal of Autoimmunity 39: 173–179. [DOI] [PubMed] [Google Scholar]
- 91.Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, and Shlomchik MJ. 2010. TLR9 Regulates TLR7- and MyD88-Dependent Autoantibody Production and Disease in a Murine Model of Lupus. The Journal of Immunology 184: 1840–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gies V, Schickel J-N, Jung S, Joublin A, Glauzy S, Knapp A-M, Soley A, Poindron V, Guffroy A, Choi J-Y, Gottenberg J-E, Anolik JH, Martin T, Soulas-Sprauel P, Meffre E, and Korganow A-S. 2018. Impaired TLR9 responses in B cells from patients with systemic lupus erythematosus. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dieudonné Y, Gies V, Guffroy A, Keime C, Bird AK, Liesveld J, Barnas JL, Poindron V, Douiri N, Soulas-Sprauel P, Martin T, Meffre E, Anolik JH, and Korganow A-S. 2019. Transitional B cells in quiescent SLE: An early checkpoint imprinted by IFN. Journal of Autoimmunity 102: 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang T, Marken J, Chen J, Tran VB, Li Q-Z, Li M, Cerosaletti K, Elkon KB, Zeng X, and Giltiay NV. 2019. High TLR7 Expression Drives the Expansion of CD19+CD24hiCD38hi Transitional B Cells and Autoantibody Production in SLE Patients. Front. Immunol 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bolland S, Yim Y-S, Tus K, Wakeland EK, and Ravetch JV. 2002. Genetic Modifiers of Systemic Lupus Erythematosus in FcγRIIB−/− Mice. Journal of Experimental Medicine 195: 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Subramanian S, Tus K, Li Q-Z, Wang A, Tian X-H, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, Schultz RA, and Wakeland EK. 2006. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A 103: 9970–9975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, and Bolland S. 2007. Control of Toll-like Receptor 7 Expression Is Essential to Restrict Autoimmunity and Dendritic Cell Proliferation. Immunity 27: 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Berland R, Fernandez L, Kari E, Han J-H, Lomakin I, Akira S, Wortis HH, Kearney JF, Ucci AA, and Imanishi-Kari T. 2006. Toll-like Receptor 7-Dependent Loss of B Cell Tolerance in Pathogenic Autoantibody Knockin Mice. Immunity 25: 429–440. [DOI] [PubMed] [Google Scholar]
- 99.Umiker BR, McDonald G, Larbi A, Medina CO, Hobeika E, Reth M, and Imanishi-Kari T. 2014. Production of IgG autoantibody requires expression of activation-induced deaminase in early-developing B cells in a mouse model of SLE. European Journal of Immunology 44: 3093–3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Green NM, Laws A, Kiefer K, Busconi L, Kim Y-M, Brinkmann MM, Trail EH, Yasuda K, Christensen SR, Shlomchik MJ, Vogel S, Connor JH, Ploegh H, Eilat D, Rifkin IR, van Seventer JM, and Marshak-Rothstein A. 2009. Murine B Cell Response to TLR7 Ligands Depends on an IFN-β Feedback Loop. The Journal of Immunology 183: 1569–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, and Hartmann G. 2005. Plasmacytoid Dendritic Cells Control TLR7 Sensitivity of Naive B Cells via Type I IFN. The Journal of Immunology 174: 4043–4050. [DOI] [PubMed] [Google Scholar]
- 102.Kiefer K, Oropallo MA, Cancro MP, and Marshak-Rothstein A. 2012. Role of type I interferons in the activation of autoreactive B cells. Immunology & Cell Biology 90: 498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hamilton JA, Wu Q, Yang P, Luo B, Liu S, Hong H, Li J, Walter MR, Fish EN, Hsu H-C, and Mountz JD. 2017. Cutting Edge: Endogenous IFN-β Regulates Survival and Development of Transitional B Cells. The Journal of Immunology 199: 2618–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Walsh ER, Pisitkun P, Voynova E, Deane JA, Scott BL, Caspi RR, and Bolland S. 2012. Dual signaling by innate and adaptive immune receptors is required for TLR7-induced B-cell–mediated autoimmunity. PNAS 109: 16276–16281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shen N, Fu Q, Deng Y, Qian X, Zhao J, Kaufman KM, Wu YL, Yu CY, Tang Y, Chen J-Y, Yang W, Wong M, Kawasaki A, Tsuchiya N, Sumida T, Kawaguchi Y, Howe HS, Mok MY, Bang S-Y, Liu F-L, Chang D-M, Takasaki Y, Hashimoto H, Harley JB, Guthridge JM, Grossman JM, Cantor RM, Song YW, Bae S-C, Chen S, Hahn BH, Lau YL, and Tsao BP. 2010. Sex-specific association of X-linked Toll-like receptor 7 (TLR7) with male systemic lupus erythematosus. PNAS 107: 15838–15843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Liu M, Guo Q, Wu C, Sterlin D, Goswami S, Zhang Y, Li T, Bao C, Shen N, Fu Q, and Zhang X. 2019. Type I interferons promote the survival and proinflammatory properties of transitional B cells in systemic lupus erythematosus patients. Cellular & Molecular Immunology 16: 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, Schieferl S, Hom J, Jenks S, Feldman RJ, Mehr R, Wei C, Lee FE-H, Cheung WC, Rosenberg AF, and Sanz I. 2015. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nature Immunology 16: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wu X, Ye Y, Niu J, Li Y, Li X, You X, Chen H, Zhao L, Zeng X, Zhang F, Tang F, He W, Cao X, Zhang X, and Lipsky PE. 2014. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Science Translational Medicine 6: 246ra99–246ra99. [DOI] [PubMed] [Google Scholar]
- 109.Chang N-H, Li TT, Kim JJ, Landolt-Marticorena C, Fortin PR, Gladman DD, Urowitz MB, and Wither JE. 2015. Interferon-α induces altered transitional B cell signaling and function in Systemic Lupus Erythematosus. Journal of Autoimmunity 58: 100–110. [DOI] [PubMed] [Google Scholar]
- 110.Palanichamy A, Bauer JW, Yalavarthi S, Meednu N, Barnard J, Owen T, Cistrone C, Bird A, Rabinovich A, Nevarez S, Knight JS, Dedrick R, Rosenberg A, Wei C, Rangel-Moreno J, Liesveld J, Sanz I, Baechler E, Kaplan MJ, and Anolik JH. 2014. Neutrophil-Mediated IFN Activation in the Bone Marrow Alters B Cell Development in Human and Murine Systemic Lupus Erythematosus. The Journal of Immunology 192: 906–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heremans J, Garcia-Perez JE, Turro E, Schlenner SM, Casteels I, Collin R, de Zegher F, Greene D, Humblet-Baron S, Lesage S, Matthys P, Penkett CJ, Put K, Stirrups K, Thys C, Van Geet C, Van Nieuwenhove E, Wouters C, Meyts I, Freson K, and Liston A. 2018. Abnormal differentiation of B cells and megakaryocytes in patients with Roifman syndrome. Journal of Allergy and Clinical Immunology 142: 630–646. [DOI] [PubMed] [Google Scholar]
- 112.Suryani S, and Tangye SG. 2010. Therapeutic implications of advances in our understanding of transitional B-cell development in humans. Expert Review of Clinical Immunology 6: 765–775. [DOI] [PubMed] [Google Scholar]
- 113.Goyal R, Bulua AC, Nikolov NP, Schwartzberg PL, and Siegel RM. 2009. Rheumatologic and autoimmune manifestations of primary immunodeficiency disorders. Current Opinion in Rheumatology 21: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kolhatkar NS, Brahmandam A, Thouvenel CD, Becker-Herman S, Jacobs HM, Schwartz MA, Allenspach EJ, Khim S, Panigrahi AK, Prak ETL, Thrasher AJ, Notarangelo LD, Candotti F, Torgerson TR, Sanz I, and Rawlings DJ. 2015. Altered BCR and TLR signals promote enhanced positive selection of autoreactive transitional B cells in Wiskott-Aldrich syndrome. Journal of Experimental Medicine 212: 1663–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lund FE 2008. Cytokine-producing B lymphocytes—key regulators of immunity. Current Opinion in Immunology 20: 332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, and Mauri C. 2010. CD19+CD24hiCD38hi B Cells Exhibit Regulatory Capacity in Healthy Individuals but Are Functionally Impaired in Systemic Lupus Erythematosus Patients. Immunity 32: 129–140. [DOI] [PubMed] [Google Scholar]
- 117.Hamilton JA, Wu Q, Yang P, Luo B, Liu S, Li J, Mattheyses AL, Sanz I, Chatham WW, Hsu H-C, and Mountz JD. 2018. Cutting Edge: Intracellular IFN-β and Distinct Type I IFN Expression Patterns in Circulating Systemic Lupus Erythematosus B Cells. The Journal of Immunology 201: 2203–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hamilton JA, Hsu H-C, and Mountz JD. 2018. Role of Production of Type I Interferons by B Cells in the Mechanisms and Pathogenesis of Systemic Lupus Erythematosus. Discovery Medicine 25: 21–29. [PubMed] [Google Scholar]
- 119.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, Smyth MJ, Mackay CR, and Mackay F. 2007. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. Journal of Experimental Medicine 204: 1959–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cerutti A, Puga I, and Cols M. 2011. Innate control of B cell responses. Trends in Immunology 32: 202–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.He B, Santamaria R, Xu W, Cols M, Chen K, Puga I, Shan M, Xiong H, Bussel JB, Chiu A, Puel A, Reichenbach J, Marodi L, Döffinger R, Vasconcelos J, Issekutz A, Krause J, Davies G, Li X, Grimbacher B, Plebani A, Meffre E, Picard C, Cunningham-Rundles C, Casanova J-L, and Cerutti A. 2010. The transmembrane activator TACI triggers immunoglobulin class switching by activating B cells through the adaptor MyD88. Nature Immunology 11: 836–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Figgett WA, Deliyanti D, Fairfax KA, Quah PS, Wilkinson-Berka JL, and Mackay F. 2015. Deleting the BAFF receptor TACI protects against systemic lupus erythematosus without extensive reduction of B cell numbers. Journal of Autoimmunity 61: 9–16. [DOI] [PubMed] [Google Scholar]
- 123.Du SW, Jacobs HM, Arkatkar T, Rawlings DJ, and Jackson SW. 2018. Integrated B Cell, Toll-like, and BAFF Receptor Signals Promote Autoantibody Production by Transitional B Cells. The Journal of Immunology 201: 3258–3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, and Grewal IS. 2003. Loss of TACI Causes Fatal Lymphoproliferation and Autoimmunity, Establishing TACI as an Inhibitory BLyS Receptor. Immunity 18: 279–288. [DOI] [PubMed] [Google Scholar]
- 125.Yan M, Wang H, Chan B, Roose-Girma M, Erickson S, Baker T, Tumas D, Grewal IS, and Dixit VM. 2001. Activation and accumulation of B cells in TACI-deficient mice. Nature Immunology 2: 638. [DOI] [PubMed] [Google Scholar]
- 126.Steri M, Orrù V, Idda ML, Pitzalis M, Pala M, Zara I, Sidore C, Faà V, Floris M, Deiana M, Asunis I, Porcu E, Mulas A, Piras MG, Lobina M, Lai S, Marongiu M, Serra V, Marongiu M, Sole G, Busonero F, Maschio A, Cusano R, Cuccuru G, Deidda F, Poddie F, Farina G, Dei M, Virdis F, Olla S, Satta MA, Pani M, Delitala A, Cocco E, Frau J, Coghe G, Lorefice L, Fenu G, Ferrigno P, Ban M, Barizzone N, Leone M, Guerini FR, Piga M, Firinu D, Kockum I, Bomfim IL, Olsson T, Alfredsson L, Suarez A, Carreira PE, Castillo-Palma MJ, Marcus JH, Congia M, Angius A, Melis M, Gonzalez A, Riquelme MEA, da Silva BM, Marchini M, Danieli MG, Del Giacco S, Mathieu A, Pani A, Montgomery SB, Rosati G, Hillert J, Sawcer S, D’Alfonso S, Todd JA, Novembre J, Abecasis GR, Whalen MB, Marrosu MG, Meloni A, Sanna S, Gorospe M, Schlessinger D, Fiorillo E, Zoledziewska M, and Cucca F. 2017. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N Engl J Med 376: 1615–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.MacLennan ICM, Toellner K-M, Cunningham AF, Serre K, Sze DM-Y, Zúñiga E, Cook MC, and Vinuesa CG. 2003. Extrafollicular antibody responses. Immunological Reviews 194: 8–18. [DOI] [PubMed] [Google Scholar]
- 128.Vinuesa C. G. de, O’Leary P, Sze DM-Y, Toellner K-M, and MacLennan ICM. 1999. T-independent type 2 antigens induce B cell proliferation in multiple splenic sites, but exponential growth is confined to extrafollicular foci. European Journal of Immunology 29: 1314–1323. [DOI] [PubMed] [Google Scholar]
- 129.Cattoretti G, Büttner M, Shaknovich R, Kremmer E, Alobeid B, and Niedobitek G. 2006. Nuclear and cytoplasmic AID in extrafollicular and germinal center B cells. Blood 107: 3967–3975. [DOI] [PubMed] [Google Scholar]
- 130.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, and Craft J. 2008. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. Journal of Experimental Medicine 205: 2873–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Marshall JL, Zhang Y, Pallan L, Hsu M-C, Khan M, Cunningham AF, MacLennan ICM, and Toellner K-M. 2011. Early B blasts acquire a capacity for Ig class switch recombination that is lost as they become plasmablasts. European Journal of Immunology 41: 3506–3512. [DOI] [PubMed] [Google Scholar]
- 132.Allman D, Wilmore JR, and Gaudette BT. 2019. The continuing story of T-cell independent antibodies. Immunological Reviews 288: 128–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.William J, Euler C, Christensen S, and Shlomchik MJ. 2002. Evolution of Autoantibody Responses via Somatic Hypermutation Outside of Germinal Centers. Science 297: 2066–2070. [DOI] [PubMed] [Google Scholar]
- 134.Jenks SA, Cashman KS, Woodruff MC, Lee FE-H, and Sanz I. 2019. Extrafollicular responses in humans and SLE. Immunological Reviews 288: 136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Di Niro R, Lee S-J, Vander Heiden JA, Elsner RA, Trivedi N, Bannock JM, Gupta NT, Kleinstein SH, Vigneault F, Gilbert TJ, Meffre E, McSorley SJ, and Shlomchik MJ. 2015. Salmonella Infection Drives Promiscuous B Cell Activation Followed by Extrafollicular Affinity Maturation. Immunity 43: 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, Ramiscal RR, Chan TD, Gatto D, Brink R, Yu D, Fagarasan S, Tarlinton DM, Cunningham AF, and Vinuesa CG. 2011. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. Journal of Experimental Medicine 208: 1377–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, Kenny SM, Khan M, Toellner K-M, Lane PJL, and MacLennan ICM. 2007. Salmonella Induces a Switched Antibody Response without Germinal Centers That Impedes the Extracellular Spread of Infection. The Journal of Immunology 178: 6200–6207. [DOI] [PubMed] [Google Scholar]
- 138.Gerba CP, Rose JB, and Haas CN. 1996. Sensitive populations: who is at the greatest risk? International Journal of Food Microbiology 30: 113–123. [DOI] [PubMed] [Google Scholar]
- 139.Castle SC 2000. Clinical Relevance of Age-Related Immune Dysfunction. Clin Infect Dis 31: 578–585. [DOI] [PubMed] [Google Scholar]