

Abstract
Background
Cell-free DNA detection is becoming a surrogate assay for tumor genotyping. Biological fluids often content a very low amount of cell-free tumor DNA and assays able to detect very low allele frequency mutant with a few quantities of DNA are required. We evaluated the ability of the fully-automated molecular diagnostics platform Idylla for the detection of KRAS, NRAS and BRAF hotspot mutations in plasma from patients with metastatic colorectal cancer (mCRC).
Materials and methods
First, we evaluated the limit of detection of the system using two set of laboratory made samples that mimic mCRC patient plasma, then plasma samples from patients with mCRC were assessed using Idylla system and BEAMing digital PCR technology.
Results
Limits of detection of 0.1%, 0.4% and 0.01% for KRAS, NRAS and BRAF respectively have been reached. With our laboratory made samples, sensitivity up to 0.008% has been reached. Among 15 patients’ samples tested for KRAS mutation, 2 discrepant results were found between Idylla and BEAMing dPCR. A 100% concordance between the two assays has been found for the detection of NRAS and BRAF mutations in plasma samples.
Conclusions
The Idylla system does not reach as high sensitivity as assays like ddPCR but has an equivalent sensitivity to modified NGS technics with a lower cost and a lower time to results. These data allowed to consider the Idylla system in a routine laboratory workflow for KRAS, NRAS and BRAF mutations detection in plasma.
Introduction
Presence of cell-free nucleic acids (cfNA) in plasma has been described in 1948 by Mandel and Métais [1]. In 1977, Leon et al. described that concentration of cell free DNA (cfDNA) is higher in plasma of patients with cancer [2]. Cell-free tumor DNA (ctDNA) is mostly shed in body fluids by apoptosis, necrosis and active mechanisms. ctDNA is also found in several types of micro vesicles like exosomes and micro particles secreted by most cells including tumor cells [3]. ctDNA is commonly more fragmented than cfDNA deriving from healthy cells (160–180 bp) and fragments have a size mainly smaller than 145 nucleotides [4,5].
The research of KRAS and NRAS (RAS genes) mutations is highly important since the existence of a mutation on codons 12, 13, 59, 61, 117 or 146 is known as a resistance marker to anti-EGFR monoclonal antibodies (i.e. cetuximab and panitumumab) associated with chemotherapy in the management of patients with metastatic colorectal cancer (mCRC) [6]. Moreover, the presence of a V600E BRAF mutation is recognized as a poor prognosis factor [7], thus assessment of KRAS, NRAS and BRAF has become a standard for the management of patients with mCRC.
Formalin-fixed paraffin embedded (FFPE) tissue is recognized as the gold standard for the research of RAS and BRAF mutations. Tumor biopsy is not always possible and is an invasive procedure for patients with cancer. The patient’s follow-up and the determination of minimal residual disease also require iterative biopsies, which is not possible nor ethical using tissue. Moreover, because of the formalin fixation process, DNA extracted from FFPE tissues is sometimes too fragmented or of bad quality. The assessment of RAS and BRAF mutations using ctDNA extracted from plasma could be a fair alternative for patient quality of life improvement since a blood sample is an easier and less invasive procedure than a tissue biopsy.
ctDNA detected in plasma has been described as representative of tumor heterogeneity and several studies showed a good concordance with tissue samples. In the study conducted by Thierry et al., samples from 140 patients with mCRC have been analyzed and a concordance of 72% was showed between KRAS exon 2 status found in plasma and FFPE tissue [8,9]. In the RASANC prospective study, RAS status was determined using next-generation sequencing (NGS) on 412 paired plasma and tumor samples. An excellent concordance (kappa coefficient 0.71 [95% CI: 0.64–0.77] and accuracy 85.2% [95% CI: 81.4–88.5]) were found between plasma and tissue [10]. These different studies allowed considering the use of liquid biopsy but with a necessity of tumor tissue testing in case of negative results in plasma.
The Idylla platform is a CE-IVD fully-integrated system based on real-time polymerase chain reaction (PCR). This system has already been validated for the determination of RAS and BRAF mutations using FFPE tissues [11–15] and for the BRAF hotspot mutation detection in plasma samples [16–19].
ctDNA can represent between 0.01% and 90% of the cfDNA extracted from plasma, thus a very sensitive assay is needed for a reliable detection of low amount of ctDNA and/or low variant allele frequency [20]. In this study, we evaluated the ability and the limit of detection (LOD) of the Idylla system for the detection of KRAS, NRAS and BRAF mutations in plasma using laboratory-made samples (DNA from cell-line and from commercial controls) that mimic patients and samples from patients with mCRC.
Materials and methods
DNA from characterized cell lines
Details of ATCC cell lines used to obtain DNA with characterized mutations. Cell line details are described in Table 1 and culture conditions in S1 Table.
Table 1. Details of cell-lines used to create our plasma samples.
| Cell-line | ATCC® reference | Gene | Exon | Reference Sequence | Coding DNA Sequence mutation Amino Acid mutation |
Mutation |
|---|---|---|---|---|---|---|
| ML-2 | CVCL_1418TM | KRAS | 4 | NM_033360.2 | c.436G>A p.(Ala146Thr) |
Heterozygous |
| SW620 | CCL-227TM | KRAS | 2 | NM_033360.2 | c.35G>T p.(Gly12Val) |
Homozygous |
| HT29 | HTB-38TM | BRAF | 15 | NM_004333.4 | c.1799T>A p.(Val600Glu) |
Heterozygous |
| MZ2 | CVCL-1435TM | NRAS | 3 | NM_002525.4 | c.181C>A p.(Gln61Lys) |
Heterozygous |
DNA extracted from characterized cell lines (Table 1) was measured using the Qubit® dsDNA HS assay kit (which allows detection of concentration between 0.01 and 100 ng/μL) and the Qubit® 3.0 Fluorometer (ThermoFisher Scientific Inc, Massachusetts, USA). Extracted DNA was then diluted in TE buffer at 4ng/μL. Concentrations of the diluted samples were finally assessed using Qubit® prior to fragmentation.
DNA was then fragmented using Covaris® M220 Focused-Ultrasonicator (Covaris, Massachusetts, USA) duration 150 seconds, peak incident power 75.0 Watt, duty factor 26.0% and burst 150. Fragments sizes were then determined using Fragment analyzer® (Advanced Analytical, Ankeny, USA) and DNF-477 High Sensitivity Small Fragment Analysis Kit (1 bp– 1 500 bp). According to Thierry et al, which report that ctDNA is more fragmented than cfDNA deriving from healthy cells, with a size mainly smaller than 145 bp, targeted DNA fragments size was 145 bp [3]. Fig 1 shows fragmentation profile for ML-2 cell line; similar data were obtained for the 3 other cell lines.
Fig 1. DNA size profiles obtained using fragment analyzer®.
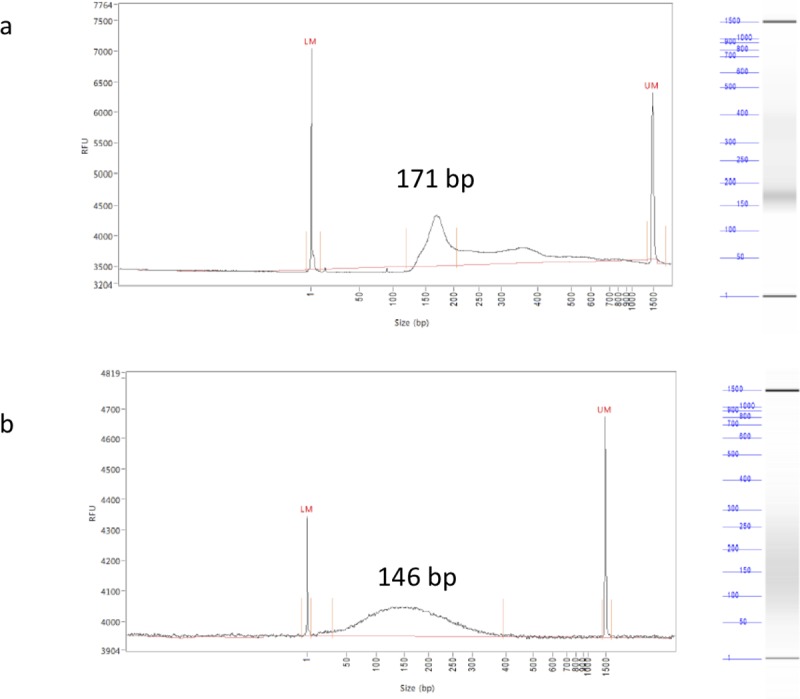
a. DNA fragments profile of DNA extracted from plasma of patient with metastatic colorectal cancer b. DNA fragments profile of DNA extracted from ML-2 cell line after fragmentation with Covaris®.
Commercial plasma (Dutscher S4180-500, Brumath, France) obtained from healthy donor was defrosted and centrifuged 10 minutes at 20°C– 6,000G.
This commercial plasma doesn’t contain ctDNA but naturally contains cfDNA whose concentration was determined using Qubit® prior addition of ctDNA solution (Table 2).
Table 2. Limit of detection of one KRAS exon 2 and one KRAS exon 4 mutations by ctKRAS mutation assay and detection of one BRAF codon 600 mutation and one NRAS exon 3 mutation by ctNRAS-BRAF mutation assay on samples mimicking plasma.
| Gene mutation | cfDNA concentration* | ctDNA concentration† | Mutated ctDNA concentration‡ | cfDNA total concentration§ | Ratio mutated copies / wild-type copies | Cq wild-type (control)|| | Cq mutated || | Mutation interpretation |
|---|---|---|---|---|---|---|---|---|
| ng/mL | ng/mL | ng/mL | ng/mL | % | ||||
| copies /mL | copies /mL | copies / mL | copies /mL | |||||
|
KRAS p.(Ala146Thr) |
460.0 | 9.00 x 10−2 | 4.50 x 10−2 | 460.09 | 1/10 000 | 27.7 | 39.2 | Detected |
| 138 000 | 28 | 14 | 138 028 | 0.010% | ||||
| 460.0 | 7.70 x 10−2 | 3.85 x 10−2 | 460.08 | 1/12 000 | 29.0 | 39.6 | Detected | |
| 138 000 | 23 | 11.5 | 138 23 | 0.009% | ||||
| 460.0 | 7.40 x 10−2 | 3.70 x 10−2 | 460.07 | 1/12 500 | 29.8 | - | Not detected | |
| 138 000 | 22 | 11 | 138 022 | 0.008% | ||||
|
KRAS p.(Gly12Val) |
460.0 | 4.00 x 10−2 | 4.00 x 10−2 | 460.04 | 1/11 500 | 28.1 | 38.3 | Detected |
| 138 000 | 12 | 12 | 138 012 | 0.009% | ||||
| 460.0 | 3.50 x 10−2 | 3.50 x 10−2 | 460.035 | 1/13 000 | 27.9 | 32.8 | Detected | |
| 138 000 | 10.5 | 10.5 | 138 010 | 0.008% | ||||
|
BRAF p.Val600Glu |
460.0 | 14.00 x 10−2 | 7.00 x 10−2 | 460.014 | 1/6 500 | 37.8 | 53.4 | Detected |
| 138 000 | 42 | 21 | 138 42 | 0.015% | ||||
| 460.0 | 10.50 x 10−2 | 5.25 x 10−2 | 460.0525 | 1/8 750 | 38.1 | 55.3 | Detected | |
| 138 000 | 32 | 16 | 138 032 | 0.011% | ||||
| 460.0 | 10.30 x 10−2 | 5.15 x 10−2 | 460.010 | 1/9 000 | 37.7 | - | Not detected | |
| 138 000 | 30 | 15 | 138 030 | 0.011% | ||||
|
NRAS p.Gln61Lys |
460.0 | 14.00 x 10−2 | 7.00 x 10−2 | 460.014 | 1/6 500 | 37.8 | 55 | Detected |
| 138 000 | 42 | 21 | 138 042 | 0.015% | ||||
| 460.0 | 13.00 x 10−2 | 6.50 x 10−2 | 460.013 | 1/7 000 | 37.7 | - | Not detected | |
| 138 000 | 40 | 20 | 138 040 | 0.014% | ||||
| 460.0 | 12.66 x 10−2 | 6.33 x 10−2 | 460.012 | 1/7 250 | 37.6 | - | Not detected | |
| 138 000 | 38 | 19 | 138 038 | 0.014% |
* cfDNA concentration: concentration of cfDNA in commercial plasma
† ctDNA concentration: concentration of ctDNA added in commercial plasma
‡ mutated ctDNA concentration = ctDNA concentration for homozygous mutation and ctDNA concentration/2 for heterozygous mutation
§ cf DNA total concentration = cfDNA concentration + ctDNA concentration. cfDNA total number of copies = cfDNA number of copies + ctDNA number of copies
|| cycle quantification.
To obtain several plasma samples with different ctDNA concentration, different volumes of fragmented DNA (300 copies/ng) solution were spiked in 1 mL of commercial plasma. A concentration range was obtained for each cell-line (Table 2, S2 and S3 Tables).
ctDNA from commercial panel control
To complete the panel of tested mutations, we used AcroMetrix Idylla ctRAS verification Panel (ThermoFisher Scientific Inc). This commercial panel of controls is composed of 9 vials containing one mutation each. Each vial contains 167 ng of fragmented DNA (160 +/- 24 bp) in normal human EDTA plasma (total volume of 1.1 mL). DNA is 5% (+/-0.50%) mutated for p.(Gly12Asp), p.(Gly13Asp), p.(Gly12Ser), p.(Gly12Val) KRAS mutations and p.(Val600Glu) BRAF mutation and 10% (+/-1.5%) mutated for p.(Gly12Asp), p.(Gly12Val), p.(Gln61Arg) and p.(Gln61Lys) NRAS mutations.
To assess the limits of detection of the Idylla system, these controls were diluted in different volumes of commercial plasma (Dutscher) to obtain a concentrations range of mutated allele fractions (Table 3).
Table 3. Limit of detection of four KRAS exon 2 mutations by ctKRAS mutation assay and limit of detection of one BRAF codon 600 mutation, two NRAS exon 2 mutations and two NRAS exon 3 mutations by ctNRAS-BRAF mutation assay on a commercial controls.
| Volume of control (μL) in 1 mL of commercial plasma |
Number of Mutated copies in the sample | Ratio mutated copies / wild-type copies % |
Cq wild-type (control) | Cq mutated | Mutation interpretation | |
|---|---|---|---|---|---|---|
|
KRAS p.(Gly12Asp) |
60.60 | 138 | 0.1% | 24.7 | 30.61 | Detected |
| 30.30 | 69 | 0.05% | 25.5 | 33.36 | Detected | |
| 12.12 | 28 | 0.02% | 26.3 | - | Not detected | |
|
KRAS p.(Gly12Ser) |
60.60 | 138 | 0.1% | 25.3 | 32.6 | Detected |
| 30.30 | 69 | 0.05% | 26.0 | 34.1 | Detected | |
| 15.15 | 35 | 0.025% | 26.5 | - | Not detected | |
|
KRAS p.(Gly12Val) |
15.15 | 35 | 0.025% | 26.4 | 36.5 | Detected |
| 9.09 | 21 | 0.015% | 27.0 | 36.97 | Detected | |
| 6.06 | 14 | 0.01% | 27.2 | - | Not detected | |
|
KRAS p.(Gly13Asp) |
90.90 | 197 | 0.15% | 25.2 | 30.3 | Detected |
| 60.60 | 138 | 0.1% | 24.9 | 30.8 | Detected | |
| 45.45 | 104 | 0.075% | 25.6 | - | Not detected | |
|
BRAF p.(Val600Glu) |
6.06 | 14 | 0.01% | 37.8 | 55.1 | Detected |
| 4.54 | 10 | 0.0075% | 37.1 | 52.7 | Detected | |
| 3.03 | 7 | 0.005% | 37.3 | - | Not detected | |
|
NRAS p.(Gly12Asp) |
151.50 | 690 | 0.5% | 34.6 | 47.0 | Detected |
| 121.20 | 552 | 0.4% | 35.0 | 50.6 | Detected | |
| 90.90 | 414 | 0.3% | 35.1 | - | Not detected | |
|
NRAS p.(Gly12Val) |
90.90 | 414 | 0.3% | 35.2 | 45.1 | Detected |
| 30.30 | 138 | 0.1% | 36.5 | 50.7 | Detected | |
| 15.15 | 69 | 0.05% | 36.7 | - | Not detected | |
|
NRAS p.(Gln61Arg) |
90.90 | 414 | 0.3% | 35.3 | 44.3 | Detected |
| 30.30 | 138 | 0.1% | 37.6 | 46.4 | Detected | |
| 15.15 | 69 | 0.05% | 37.4 | - | Not detected | |
|
NRAS p.(Gln61Lys) |
121.20 | 552 | 0.4% | 35.1 | Detected | |
| 90.90 | 414 | 0.3% | 35.8 | 48.42 | Detected | |
| 75.75 | 345 | 0.25% | 35.5 | - | Not detected |
Patient samples
Patients’ samples were selected in January 2018 from the Institut de Cancérologie de Lorraine biobank (Nancy, France) (CircuLOR-1 study (NCT02751177)). Methods were performed in accordance with the relevant guidelines and regulations and approved by the ethical and scientific board of Institut de Cancérologie de Lorraine. This study has been specifically approved by the ethical and scientific board of Institut de Cancérologie de Lorraine. All samples were anonymized prior to analysis. Plasma samples were collected from patients with a mCRC with a requested KRAS, NRAS or BRAF genotyping. cfDNA was extracted from plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany). Then, BEAMing method was performed with the OncoBEAM CRC1kit (Sysmex Inostics, Hamburg, Germany). This technique consists on a hybridization between DNA-coated beads and a sequence specific fluorescein-labeled probe which are then analyzed by flow cytométrie [21,22]. Data are processed by a software and results then reported as “no mutation detected” or “mutation detected”.
In our study, mutation detection with Idylla platform was performed on 15 samples with ctKRAS cartridge and in 3 samples with ctNRAS-BRAF cartridge.
Discrepant samples were assessed using a third assay. DNA was extracted from plasma using the QIAamp® Circulating Nucleic Acid kit (Qiagen, Hilden, Germany), libraries were then prepared with the custom STS 51-Gene kit (Sophia genetics, Saint-Sulpice, Switzerland), a capture-based target enrichment kit. Sequencing was performed on MiSeq (Illumina, San Diego, USA) and results were treated using Sophia DDM® software (Sophia genetics).
Idylla platform
Idylla platform (Biocartis, Mechelen, Belgium) is a fully cartridge-based automated platform and uses microfluidics processing with all reagents on-board.
ctKRAS cartridge can detect 21 mutations on codons 12, 13, 59, 61, 117 et 146 of KRAS gene in plasma samples. ctNRAS-BRAF cartridge can detect 18 mutations in codons 12, 13, 59, 61, 117, 146 of NRAS gene, 5 mutations in codon 600 of the BRAF gene (S4 Table).
One milliliter of plasma and 20 μL of proteinase K were added in the cartridge. Proteinase K was added proteins hydrolyzation, including fibrin which could potentially interfere with DNA analysis. The cartridge was then sealed and inserted in the instrument. Inside the cartridge, nucleic acids are amplified using real-time PCR coupled with a fluorophore-based detection system. Idylla console autoanalyses the PCR curve to determine the presence or absence of a mutation and the results are presented as either “no mutation detected” or “mutation detected” [14].
Cycle quantification (Cq) of the control and of the mutated sample are available on the result sheet.
Results
Laboratory made samples that mimic plasma
A range of dilutions between 2.30 x 10−2 ng/mL (7 copies/mL) and 2.30 x 10−1 ng/mL (69 copies/mL) (13 points) and between 3.50 x 10−2 ng/mL (10.5 copies/mL) and 4.00 x 10−2 ng/mL (12 copies/mL) (3 points) were prepared for p.(Ala146Thr) and p.(Glu12Val) KRAS mutations respectively.). The range obtained was between 3.50 x 10−2 ng/mL (10.5 copies/mL) and 7.00 x 10−2 ng/mL (21 copies/mL) for p.(Val600Glu) BRAF mutation (7 points) and between 1.67 x 10−2 ng/mL (5 copies/mL) and 7.00 x 10−2 ng/mL (21 copies/mL) for p.(Gln61Lys) NRAS mutation (7 points). All concentrations are described in the S2 and S3 Tables.
Sensitivity were 10.5 copies/mL for p.(Glu12Val) KRAS mutation, 11.5 copies/mL for p.(Ala146Thr) KRAS mutation, 16 copies/mL for p.(Val600Glu) BRAF mutation and 21 copies/mL for p.(Gln61Lys) NRAS mutation
Considering wild type DNA present in our samples, sensitivity of ctKRAS mutation assay was 1 mutated copy/12 000 wild-type copies for KRAS mutation (Table 2). Sensitivity of ctNRAS-BRAF mutation assay was 1 mutated copy/8750 wild-type copies and 1 mutated copy/6500 wild-type copies for BRAF and NRAS mutations respectively (Table 2).
No mutation has been detected in negative controls (plasma from healthy donor used for samples preparation).
Commercial panel control
Different volumes of control have been used to obtain allele frequencies mutant between 0.01% (14 copies /mL) and 0.1% (138 copies/mL) for KRAS p.(Gly12Asp) (4 points), 0.015% (21 copies/mL) and 0.15% (197 copies/mL) for KRAS p.(Gly13Asp) (3 points), 0.01% (14 copies/mL) and 0.1% (138 copies/mL) for KRAS p.(Gly12Val) (5 points) and 0.025% (14 copies/mL) and 0.1% (138 copies/mL) for KRAS p.(Gly12Ser) (6 points). Different volumes of NRAS mutated controls have been used to obtain allele frequencies mutant between 0.1% (138 copies/mL) and 0.5% (690 copies/mL) for NRAS p.(Gly12Asp) (5 points), 0.05% (69 copies/mL) and 0.5% (690 copies/mL) for NRAS p.(Gly12Val) mutations (4 points), 0.05% (69 copies/mL) and 0.3% (414 copies/mL) for NRAS p.(Gln61Arg) (3 points) and between 0.1% (138 copies/mL) and 0.4% (552 copies/mL) for NRAS p.(Gln61Lys) mutation (5 points). All concentrations are described in S5 Table. BRAF p.(Val600Glu) mutation has been tested between 0.005% (7 copies/mL) and 0.01% (14 copies/mL) (3 points). All concentrations are described in S6 Table.
LOD were 21 copies/mL for KRAS p.(Gly12Val), 69 copies/mL for KRAS p.(Gly12Asp) and KRAS p.(Gly12Ser) mutations and 138 copies for KRAS p.(Gly13Asp) mutations (Table 3).
LOD was 10 copies/mL for p.(Val600Glu) BRAF mutation.
LOD were 138 copies/mL for NRAS p.(Gly12Val) mutation, 414 copies/mL for NRAS p.(Gln61Lys) mutation and 552 copies/mL for NRAS p.(Gly12Asp) and p.(Gln61Arg) mutations (Table 3).
Patient’s samples
KRAS mutations have been detected in 12/15 samples with ctKRAS cartridges and 14/15 samples with BEAMing dPCR (Tables 4 and 5). One sample was found wild-type for KRAS, NRAS and BRAF with both techniques. Idylla system and BEAMing dPCR both found 2 NRAS mutations (Tables 4 and 5). One BRAF mutation was detected by both Idylla system and BEAMing dPCR (Tables 4 and 5).
Table 4. Results obtained with BEAMing and Idylla assays with ΔCq data for mutated samples.
| Patient | BEAMing | Idylla | ΔCq mutated Idylla |
|---|---|---|---|
| 1 | KRAS exon 2 codon 12 | KRAS p.(Gly12Asp) | 6,2 |
| 2 | KRAS exon 2 codon 12 | KRAS p.(Gly12Asp) | 5,71 |
| 3 | KRAS exon 2 codon 13 | KRAS p.(Gly13Asp) | 4,52 |
| 4 | KRAS exon 2 codon 13 | KRAS p.(Gly13Asp) | 6,23 |
| 5 | KRAS exon 3 codon 61 | KRAS p.(Gln61His) | 5,77 |
| 6 | KRAS exon 3 codon 61 | KRAS p.(Gln61Ar) ou p.(Gln61Leu) | 4,74 |
| 7 | KRAS exon 4 codon 146 | No mutation detected | / |
| 8 | KRAS exon 4 codon 146 | No mutation detected | / |
| 9 | KRAS exon 2 codon 12 | KRAS p.(Gly12Asp) | 5,21 |
| 10 | KRAS exon 4 codon 146 | KRAS p.(Ala146Pro) ou p.(Ala146Thr) ou p.(Ala146val) | 8,84 |
| 11 | KRAS exon 2 codon 12 | KRAS p.(Gly12Asp) | 8,95 |
| 12 | KRAS exon 2 codon 12 | KRAS p.(Gly12Val) | 2,85 |
| 13 | KRAS exon 2 codon 13 | KRAS p.(Gly13Asp) | 4,39 |
| 14 | KRAS exon 2 codon 13 | KRAS p.(Gly13Asp) | 4,34 |
| 15 | Wild-type for KRAS | No mutation detected | / |
| 16 | NRAS exon 3 codon 61 | NRAS p.(Gln61Arg) or p.(Gln61Lys) |
12,67 |
| 17 | NRAS exon 2 codon 13 | NRAS p.(Gly13Arg) or p.(Gly13Val) | 11,65 |
| 18 | BRAF codon 600 | BRAF p.(Val600Glu) | 12,19 |
Table 5. Concordance between BEAMing dPCR and Idylla system for KRAS, NRAS and BRAF mutation detection in plasma of mCRC patients.
| KRAS (n = 15) | |||||||
| BEAMing dPCR | |||||||
| p.(Gly12Asp) | p.(Gly12Val) | p.(Gly13Asp) | p.(Gln61His) | p.(Gln61Arg) or p.(Gln61Leu) | p.(Ala146Pro) or p.(Ala146Thr) or p.(Ala146Val) | WT | |
| ctKRAS Idylla cartridge | |||||||
| p.(Gly12Asp) | 4 | ||||||
| p.(Gly12Val) | 1 | ||||||
| p.(Gly13Asp) | 4 | ||||||
| p.(Gln61His) | 1 | ||||||
| p.(Gln61Arg) or p.(Gln61Leu) | 1 | ||||||
| p.(Ala146Pro) or p.(Ala146Thr) or p.(Ala146Val) | 1 | ||||||
| WT | 2 | 1 | |||||
| Total | 4 | 1 | 4 | 1 | 1 | 3 | 1 |
| NRAS (n = 2) | |||||||
| BEAMing dPCR | |||||||
| p.(Gln61Arg) or p.(Gln61Lys) |
p.(Gly13Arg) or p.(Gly13Val) | WT | |||||
| ctNRAS-BRAF Idylla cartridge | |||||||
| p.(Gln61Arg) or p.(Gln61Lys) |
1 | 0 | |||||
| p.(Gly13Arg) or p.(Gly13Val) | 0 | 1 | |||||
| WT | 0 | 0 | 0 | ||||
| Total | 1 | 1 | 0 | ||||
| BRAF (n = 1) | |||||||
| BEAMing dPCR | |||||||
| p.(Val600Glu) or p.(Val600Asp) |
WT | ||||||
| ctNRAS-BRAF Idylla cartridge | |||||||
| p.(Val600Glu) or p.(Val600Asp) |
1 | 0 | |||||
| WT | 0 | 0 | |||||
| Total | 1 | 0 | |||||
Two discrepant samples were found. Both samples were found carrying a codon 146 mutation of KRAS gene detected with BEAMing and not detected with Idylla. Among these 2 samples, only one was found to carry a codon 146 mutation of KRAS and the other one was found to carry no mutation using NGS.
Discussion
In this study, two different set of mutated DNA (from cell lines and from commercial controls) have been used to obtain samples that mimic blood samples from patients with mCRC. We have obtained samples with different mutant allele frequencies for 5 different KRAS mutations, 1 BRAF mutation and 4 different NRAS mutations. Then the use of Idylla system has been evaluated on 18 plasma samples from patients with mCRC. According to the manufacturer technical sheet the LODs of the Idylla system depend on the mutation and are between 0.2% and 2.8% for KRAS mutations, between 0.2% and 0.5% for BRAF mutations and between 0.6% and 2.5% for NRAS mutations (with a background of 50 000 WT copies). Our panel has been developed to cover several mutations with different LOD. We finally obtained LOD of 0.1%, 0.4% and 0.01% for KRAS, NRAS and BRAF respectively. To select mutations, we focused on exon 2 codons 12 and 13 mutations which represent the majority of KRAS mutations in mCRC [23,24]. To assess the ability of Idylla system to detect less prevalent hotspot mutations, we also added a KRAS exon 4 mutation p.(Ala146Thr). Because they also are the most described mutations in mCRC, BRAF codon 600 and NRAS exon 2 codon 12 and exon 3 codon 61 were also chosen for this evaluation [7,23]. It is important to notice that the Cq are very high for samples with very low VAF which was expectable. The manufacturer of the Idylla system warns that there is a risk of false negative result with their system for Cq higher than 25.5 for KRAS and 35.5 for NRAS/BRAF. Since the aim of this study was to estimate the LOD of the system, we made the choice to retain the samples with Cq higher than the maximum Cq recommended by the manufacturer. In clinical settings, it seems important to us that the samples have to be analyzed with another assay when Cq are higher than the maximum recommended Cq.
Samples used to mimic mCRC patient’s plasma samples have several limitations since they have been elaborated in ideal conditions. In our study, we spiked fragmented DNA in plasma samples, thus 100% of them were containing cfDNA which is not always the case in the “real-life” because of the intermittent shedding of DNA by the tumor. It has been shown that blood samples from patients with cancer may sometimes not contain ctDNA [25]. Tumor cells and ctDNA have also some cancer-associated molecular characteristics such as single-nucleotide mutation or a specific methylation profile that we were not able to reproduce in vitro [26,27]. For ctDNA use in clinical applications, adding the detection of these epigenomics or genomics features may ensure the presence of ctDNA in the sample.
We used DNA of good quality extracted from cell-lines and mechanical fragmentation to prepare 145bp DNA fragments. Quality of plasma and DNA concentrations were also well controlled. We note in this way discrepancies when using a commercial panel of controls for LOD determination. For example, NRAS p.(Gln61Lys) mutation was detected in our laboratory made samples with 21 mutated copies/mL of plasma whereas 414 copies/mL of commercial control DNA were necessary for mutation detection. Perhaps freezing / thawing cycles of commercial controls alter DNA quality. Moreover, we have to considered declared concentrations and pipetting uncertainty. These two set of samples highlight that mutation detection depends on several preanalytical factors. To understand these discrepancies it would have been necessary to measure DNA control fragment size or concentration. For a real-life application of ctDNA detection in patients with cancer, we have to consider that various pre-analytical factors can influence samples quality. Sample handling as centrifugation procedures or storage conditions can drastically influence samples quality. Thus, a standardized pre-analytical protocol is required [28]. cfDNA is mainly present on structures like nucleosomes which protect them from nuclease degradation [3]. To prevent contamination of ctDNA by DNA shed by white blood cells, several specific tubes as Streck BCT cfDNA blood collection tubes are available and allow samples handling up to 5 days [29,30].
To translate our laboratory study into clinical practice, we tested 18 plasma samples from patients with mCRC with various mutations identified using BEAMing dPCR. Among the 18 plasma samples tested, 2 discrepant results were found. One KRAS exon 4, codon 146 mutation was detected with BEAMing and not by Idylla system and NGS. At this point, it is not possible to know whether this codon 146 mutation is a BEAMing dPCR false positive or a both Idylla and NGS false negative. A most sensitive assay as ddPCR would be relevant but the technology was not available in our laboratory. One KRAS exon 4, codon 146 mutation was detected with both BEAMing dPCR and NGS and not with ctKRAS cartridge. This sample has been identified to have a low cfDNA concentration, thus using only 1 mL of plasma may not be sufficient for a detection using the Idylla system. Three milliliters of plasma were used for ctDNA extraction for BEAMing dPCR assay and then more DNA was used for amplification and detection with BEAMing, which probably allowed the detection of the mutation for this sample with low allele frequency. Moreover, the main peak size obtained with plasma from patients with a mCRC differ from the one obtained by sonication (Fig 1). Indeed, it has been reported in the literature that ctDNA fragment size depends on tumor type [31,32]. This heterogeneity between cancer types explains the difference between targeted size (based on a mean peak size observed in several cancer type) and observed size (based on our pool of mCRC patients).
Interest for cfDNA application in cancer management has grown these last years. Several studies have demonstrated a high concordance between mutational profiles of candidate genes in matched tumor and plasma DNA samples from mCRC patients. ctDNA should be an easy and reliable tool for targeted therapy introduction [8–10]. RAS mutations have been assessed using BEAMing in 146 plasma samples from patients with mCRC in the study published by Grasselli et al.; 48% of their samples were found with a mutant allele fraction from 0.01% to 1% [33]. These results show that a sensitive method is required for ctDNA detection for clinical applications. Based on our results, this LOD is reached for BRAF mutations with the Idylla system. For KRAS and NRAS mutations, Idylla system should miss mutations with mutant allele fraction around 0.01% contrary to most sensitive techniques as BEAMing dPCR which reach a sensitivity if 0.01% and digital droplet PCR (ddPCR) which reach a sensitivity of 0.001% [21,34]. Regular amplicon-based NGS assays allow the detection of variants with an allele frequency greater than 2% [35]. Recently, new bioinformatics and library preparation approaches have been described to improve NGS performance for low-allele frequency mutation detection in ctDNA [36–38]. Moreover, NGS methods have highlighted in some cases, non-hotspot mutations with a potential clinical relevance [39–42]. These non-hotspot mutations are not detected by PCR based methods like Idylla because they only focus on hotspot mutations (S4 Table). The main aim of this study was to demonstrate the aptitude of Idylla system to detect hotspot mutations with low allele frequencies. Only few plasma samples from patients with mCRC were available for comparison, which is an actual limit of our study. To assess Idylla system relevance in clinical context, more qualified patient’s samples should be analyzed. Finally, the Idylla system requires a total of 5 minutes hands-on time and 130 minutes to complete an analysis. Assays like droplet-based PCR and NGS require DNA extraction, samples preparation and results interpretation for a total workflow of 1 day for droplet-based PCR, 3 days for BEAMing dPCR and 4 days for NGS.
In conclusion, the Idylla system allows the detection of ctDNA mutations in plasma with limits of detection of 0.1% with a short turnover time. This system can easily be integrated in a routine workflow for plasma analysis and should be considered as well as more sensitive or more explorative assays like ddPCR or NGS respectively. Considering the LOD of the assay and the paucity of ctDNA in some plasma samples, it seems to be still necessary to assess mutational status on tumor tissue when no mutation is detected in plasma.
Supporting information
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
All reagents have been provided free of charge by Biocartis, manufacturer of the Idylla system.
References
- 1.Mandel P, Mettais P. Les acides nucléiques du plasma sanguin chez l'homme. C R Acad Sci Paris 1948;142: 241–243. [PubMed] [Google Scholar]
- 2.Leon SA, Shapiro B, Sklaroff DM, Yaros MJ. Free DNA in the serum of cancer patients and the effect of therapy. Cancer Res 1977;37: 646–650. [PubMed] [Google Scholar]
- 3.Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev 2016;35: 347–76. 10.1007/s10555-016-9629-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mouliere F, Rosenfeld N. Circulating tumor-derived DNA is shorter than somatic DNA in plasma. Proc Natl Acad Sci U S A 2015;112: 3178–3179. 10.1073/pnas.1501321112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mouliere F, Robert B, Arnau Peyrotte E, Del Rio M, Ychou M, Molina F, et al. High fragmentation characterizes tumour-derived circulating DNA. PLoS One 2011;6: e23418 10.1371/journal.pone.0023418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol 2016;27: 1386–1422. 10.1093/annonc/mdw235 [DOI] [PubMed] [Google Scholar]
- 7.De Roock W, De Vriendt V, Normanno N, Ciardiello F, Tejpar S. KRAS, BRAF, PIK3CA, and PTEN mutations: implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol 2011;12: 594–603. 10.1016/S1470-2045(10)70209-6 [DOI] [PubMed] [Google Scholar]
- 8.Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med 2014;20: 430–5. 10.1038/nm.3511 [DOI] [PubMed] [Google Scholar]
- 9.Thierry AR, El Messaoudi S, Mollevi C, Raoul JL, Guimbaud R, Pezet D, et al. Clinical utility of circulating DNA analysis for rapid detection of actionable mutations to select metastatic colorectal patients for anti-EGFR treatment. Ann Oncol 2017;28: 2149–2159. 10.1093/annonc/mdx330 [DOI] [PubMed] [Google Scholar]
- 10.Bachet JB, Bouché O, Taieb J, Dubreuil O, Garcia ML, Meurisse A, et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: the AGEO RASANC prospective multicenter study. Ann Oncol. 2018;29: 1211–1219. 10.1093/annonc/mdy061 [DOI] [PubMed] [Google Scholar]
- 11.Janku F, Claes B, Huang HJ, Falchook GS, Devogelaere B, Kockx M, et al. BRAF mutation testing with a rapid, fully integrated molecular diagnostics system. Oncotarget. 2015;6: 26886–94. 10.18632/oncotarget.4723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harlé A, Salleron J, Franczak C, Dubois C, Filhine-Tressarieu P, Leroux A, et al. Detection of BRAF mutation using a Fully Automated Platform and Comparison with HighResolution Melting, Real-Time Allele Specific Amplification, Immunohistochemistry and NextGeneration Sequencing Assays, for patients with Metastatic Melanoma. PLoS One 2016;11: e0153576 10.1371/journal.pone.0153576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solassol J, Vendrell J, Märkl B, Haas C, Bellosillo B, Montagut C, et al. Multi-center evaluation of the fully automated PCR-based Idylla KRAS mutation assay for rapid KRAS mutation status determination on formalin-fixed paraffin-embedded tissue of human colorectal cancer. PLoS One 2016;11: e0163444 10.1371/journal.pone.0163444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colling R, Wang LM, Soilleux E. Automated PCR detection of BRAF mutations in colorectal adenocarcinoma: a diagnostic test accuracy study. J Clin Pathol 2016;69: 398–402. 10.1136/jclinpath-2015-203345 [DOI] [PubMed] [Google Scholar]
- 15.Johnston L, Power M, Sloan P, Long A, Silmon A, Chaffey B, et al. Clinical performance evaluation of the Idylla NRAS-BRAF mutation test on retrospectively collected formalin-fixed paraffin-embedded colorectal cancer tissue. J Clin Pathol 2018;71: 336–343. 10.1136/jclinpath-2017-204629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janku F, Huang HJ, Claes B, Falchook GS, Fu S, Hong D, et al. BRAF mutation testing in cell-free DNA from the plasma of patients with advanced cancers using a rapid, automated molecular diagnostics system. Mol Cancer Ther 2016;15: 1397–404. 10.1158/1535-7163.MCT-15-0712 [DOI] [PubMed] [Google Scholar]
- 17.Schreuer M, Jansen Y, Planken S, Chevolet I, Seremet T, Kruse V, et al. Combination of debrafenib plus trametinib for BRAF and MEK inhibitor pretreated patients with advanced BRAFV600-mutant melanoma:an open-label, single arm, dual-centre, phase 2 clinicaltrial. Lancet Oncol 2017;18: 464–472. 10.1016/S1470-2045(17)30171-7 [DOI] [PubMed] [Google Scholar]
- 18.Colling R, Wang LM, Soilleux E. Validating a fully automated real-time PCR-based system for use in the molecular diagnostic analysis of colorectal carcinoma: a comparison with NGS and IHC. J Clin Pathol 2017;70: 610–614. 10.1136/jclinpath-2017-204356 [DOI] [PubMed] [Google Scholar]
- 19.Seremet T, Planken S, Schreuer M, Jansen Y, Delaunoy M, El Housni H, et al. Illustrative cases for monitoring by quantitative analysis of BRAF/NRAS ctDNA mutations in liquid biopsies of metastatic melanoma patients who gained clinical benefits from anti-PD1 antibody therapy. Melanoma Res 2018;28: 65–70. 10.1097/CMR.0000000000000415 [DOI] [PubMed] [Google Scholar]
- 20.Francis G and Stein S. Circulating Cell-Free Tumour DNA in the Management of Cancer. Int J Mol Sci 2015;16: 14122–14142. 10.3390/ijms160614122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, Diehl F, Dressman D, Vogelstein B, Kinzler KW. BEAMing up for detection and quantification of rare sequence variants. Nat Methods 2006;3: 95–97. 10.1038/nmeth850 [DOI] [PubMed] [Google Scholar]
- 22.Diehl F, Li M, He Y, Kinzler KW, Vogelstein B, Dressman D, et al. BEAMing: single-molecule PCR on microparticles in water-in-oil emulsions. Nat methods 2006;3(7): 551–9. 10.1038/nmeth898 [DOI] [PubMed] [Google Scholar]
- 23.Zhang J, Zheng J, Yang Y, Lu J, Gao J, Lu T, et al. Molecular spectrum of KRAS, NRAS, BRAF and PIK3CA mutations in Chinese colorectal cancerpatients: analysis of 1,110 cases. Sci Rep 2015;5: 18678 10.1038/srep18678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamzehzadeh L, Khadangi F, Ghayoor Karimiani E, Pasdar A, Kerachian MA, et al. Common KRAS and NRAS gene mutations in sporadic colorectal cancer in Northeastern Iranian patients. Curr Probl Cancer 2018. pii: S0147-0272(18)30038-2. [DOI] [PubMed] [Google Scholar]
- 25.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med 2008;14: 985–990. 10.1038/nm.1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng F, Su L, Qian C. Circulating tumor DNA: a promising biomarker in the liquid biopsy of cancer. Oncotarget 2016;7: 48832–48841. 10.18632/oncotarget.9453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lissa D, Robles AI. Methylation analyses in liquid biopsy. Transl Lung Cancer Res 2016;5: 492–504. 10.21037/tlcr.2016.10.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.El Messaoudi S, Rolet F, Mouliere F, Thierry AR. Circulating cell free DNA: Preanalytical considerations. Clin Chim Acta 2013;424: 222–230 10.1016/j.cca.2013.05.022 [DOI] [PubMed] [Google Scholar]
- 29.Medina Diaz I, Nocon A, Mehnert DH, Fredebohm J, Diehl F, Holtrup F. Performance of Streck cfDNA Blood Collection Tubes for Liquid Biopsy Testing. PLoS One 2016;11: e0166354 10.1371/journal.pone.0166354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henao Diaz E, Yachnin J, Grönberg H, Lindberget J. The In Vitro Stability of Circulating Tumour DNA. PLoS One 2016;11: e0168153 10.1371/journal.pone.0168153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA Comprises an In Vivo Nucleosome Footprint that Informs Its Tissues-Of-Origin. Cell 2016;164:57–68. 10.1016/j.cell.2015.11.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mouliere F, Chandrananda D, Piskorz AM, Moore EK, Morris J, Ahlborn LB, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 2018;10(466). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grasselli J, Elez E, Caratù G, Matito J, Santos C, Macarulla T, et al. Concordance of blood- and tumor-based detection of RAS mutation to guide anti-EGFR therapy in metastatic colorectal cancer. Ann Oncol 2017;28: 1294–1301. 10.1093/annonc/mdx112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem 2011;83: 8604–8610. 10.1021/ac202028g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DW, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med 2012;4: 136ra168. [DOI] [PubMed] [Google Scholar]
- 36.Newman AM, Bratman SV, To J, Wynne JF, Eclov NC, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med 2014;20: 548–554 10.1038/nm.3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nature biotechnology 2016;34: 547–555 10.1038/nbt.3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pécuchet N, Rozenholc Y, Zonta E, Pietraz D, Didelot A, Combe P, et al. Analysis of base-position error rate of next-generation sequencing to detect tumor mutations in circulating DNA. Clin Chem 2016;62: 1492–1503. 10.1373/clinchem.2016.258236 [DOI] [PubMed] [Google Scholar]
- 39.Harlé A, Filhine-Tresarrieu P, Husson M, Boidot R, Rouyer M, Dubois C, et al. Rare RAS Mutations in Metastatic Colorectal Cancer Detected During Routine RAS Genotyping Using Next Generation Sequencing. Target Oncol 2016;11: 363–70. 10.1007/s11523-015-0404-7 [DOI] [PubMed] [Google Scholar]
- 40.Franczak C, Kandathil SM, Gilson P, Husson M, Rouyer M, Demange J, et al. Uncommon mutational profiles of metastatic colorectal cancer detected during routine genotyping using next generation sequencing. Sci Rep 2019;9: 7083 10.1038/s41598-019-43646-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones JC, Renfro LA, Al-Shamsi HO, Schrock AB, Rankin A, Zhang BY, et al. Non-V600 BRAF Mutations Define a Clinically Distinct Molecular Subtype of Metastatic Colorectal Cancer. J Clin Oncol 2017;35: 2624–2630. 10.1200/JCO.2016.71.4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shinozaki E, Yoshino T, Yamazaki K, Muro K, Yamaguchi K, Nishina T, et al. Clinical significance of BRAF non-V600E mutations on the therapeutic effects of anti-EGFR monoclonal antibody treatment in patients with pretreated metastatic colorectal cancer: the Biomarker Research for anti-EGFR monoclonal Antibodies by Comprehensive Cancer genomics (BREAC) study. Br J Cancer 2017;117: 1450–1458. 10.1038/bjc.2017.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.