

Abstract
In-vitro studies with different Fanconi anemia (FA) cell lines and FANC gene silenced cell lines indicating involvement of mitochondria function in pathogenesis of FA have been reported. However, in-vivo studies have not been studied so far to understand the role of mitochondrial markers in pathogenesis of FA. We have carried out a systematic set of biomarker studies for elucidating involvement of mitochondrial dysfunction in disease pathogenesis for Indian FA patients. We report changes in the mtDNA number in 59% of FA patients studied, a high frequency of mtDNA variations (37.5% of non-synonymous variations and 62.5% synonymous variations) and downregulation of mtDNA complex-I and complex-III encoding genes of OXPHOS (p<0.05) as strong biomarkers for impairment of mitochondrial functions in FA. Deregulation of expression of mitophagy genes (ATG; p>0.05, Beclin-1; p>0.05, and MAP1-LC3, p<0.05) has also been observed, suggesting inability of FA cells to clear off impaired mitochondria. We hypothesize that accumulation of such impaired mitochondria in FA cells therefore may be the principal cause for bone marrow failure (BMF) and a plausible effect of inefficient clearance of impaired mitochondria in FA.
Introduction
Mitochondria are powerhouse of cells and generate energy in the form of ATP through oxidative phosphorylation process. Mitochondria are also source of reactive oxygen species (ROS) production. Thus mitochondrial dysfunction is detrimental to the organism [1]. Previous studies have established link between mitochondrial functions and oxygen metabolism in FA. There is also an evidence of involvement of FA proteins in mitochondrial dysfunctions. Mukhopadhyay et al., 2006 found that the FANCG protein is localized in mitochondria and interacts with the mitochondrial peroxidase peroxiredoxin3 (PRDX3). In turn, cells from FA-A and FA-C subtypes also had PRDX3 cleavage and decreased peroxidase activity. These findings further supported the idea of mitochondrial involvement in the pathogenesis of FA [2]. Kumari et al., 2013 have demonstrated decrease of mitochondrial membrane potential, low ATP production, impaired oxygen consumption rate and pathological changes in the morphology of FA mitochondria. The study also showed inactivation of the enzymes responsible for energy production, detoxification of ROS and over sensitivity to DNA cross-linkers by the overproduction of ROS [3,4].
Ravera et al., 2013 have analysed the respiratory fluxes in FANCA primary fibroblasts, lymphocytes and lymphoblasts. Their study revealed that FANCA mutants show defective respiration through Complex I, diminished ATP production and metabolic sufferance with an increased AMP/ATP ratio. Treatment with N-acetyl cysteine (NAC) restored oxygen consumption to normal level [5]. Recently it was also shown that genetic deletion of Fancc blocks the autophagic clearance of viruses (virophagy) and increases susceptibility to lethal viral encephalitis. FANCC protein interacts with Parkin, is required in vitro and in vivo for clearance of damaged mitochondria, and decreases mitochondrial ROS production and inflammasome activation, ultimately leading to phenotypes such as BMF, cancers, and aging associated with mutations in FA pathway genes [6,7].
Despite of experimental evidences on FA cell lines or FANC gene silenced cells for accumulation of impaired mitochondrial activities and deregulated mitophagy, there has been no biomarker study done to elucidate involvement of mitochondrial dysfunction in FA pathogenesis. Here, we present study of mitochondrial dysfunction and impaired mitophagy in ex-vivo studies on mononuclear cells derived from peripheral blood of FA patients and also mitochondrial DNA (mtDNA) variations to understand the genetic basis of mitochondrial DNA pathogenesis in FA.
Patients and materials and methods
Patients, specimen collection and ethical clearance
Seventy FA subjects including 42 males and 28 females with a mean age of 8 years were included in the study (March, 2012 to June, 2017). The peripheral blood samples were collected in heparin (7cc) and EDTA (4cc) vacutainers from the patients and 33 age-matched healthy controls (non-FA or individuals without history of haematological abnormalities that are found in FA) with the written consent of parents (in case of minors) and adult patients and controls. The study protocols were approved by Institutional Ethics Committee for human subjects of National Institute of Immunohaematology, Parel, Mumbai. The diagnosis for FA was confirmed with the clinical examination and methods described in our previous publications for investigations like chromosomal breakage [8], FANCD2 immunoblotting [8] and complementation group determination using direct sequencing [8] and targeted exome sequencing (TES). Molecular investigation for FANC genes for a few patients was carried at MedGenome Pvt. Ltd., Karnataka, India and with collaboration with Dr. Minoru Takata’s Lab at Radiation Biology Center, Kyoto University, Kyoto, Japan. It was done by targeted gene capture using a custom capture kit. The libraries were sequenced on Illumina sequencing platform (mean coverage >80 to 100X). The identified mutations were confirmed by direct sequencing.
qPCR based mtDNA copy number change
Genomic DNA (gDNA) was used for mtDNA copy number study. The genomic DNA was extracted from peripheral blood collected in EDTA vacutainers using QIAamp DNA Blood Midi Kit (Qiagen, cat.# 51183) according to the manufacturer's instructions. TaqMan Universal PCR mastermix (Thermo Fisher Scientific cat.# 4304437) was used with final concentration of 200nM probe and 900nM of forward and reverse primers. A series of 10 fold dilution of standard DNA was prepared for standard curve and 15ng of gDNA was added in sample wells. An initial 10 minutes denaturation at 95°C followed by 40 cycles of 95°C and combined annealing-extension at 60°C was standardized as run program on ABI StepOne machine. Formula used for copy number change = 2– (mtDNA CT–gDNA CT). A set of primers and probes was used to study mtDNA copy number change, and β2-microglobulin primers and probes for nuclear genome as described by Malik Sahani et al., 2011 [9].
OXPHOS enzymes and TFAM expression using qPCR
RNA was extracted from EDTA peripheral blood using QIAamp RNA Blood Mini Kit (Qiagen, cat.#52304) according to the manufacturer's instructions. The concentrations of RNA were determined on nanodrop spectrophotometer. 1μg of RNA was reverse transcribed to first-strand cDNA by using RevertAid H minus First Strand cDNA Synthesis Kit (Thermo Scientific, cat.#K1632). For OXPHOS enzyme expression profiling was done by Real time PCR primers designed so as to amplify transcripts encoding OXPHOS enzymes (Complex I: NADH-coenzyme Q reductase, Complex III: CoenzymeQ—Cytochrome c reductase and TFAM were used (S1 Table). Kapa SYBR FAST qPCR Master Mix (2X) Green (cat# KM4103) was used to quantify transcripts on ABI StepOne machine.
Study of mtDNA variations of OXPHOS Complex-I subunits and Complex-III encoding genes
Genomic DNA was extracted from peripheral blood stored in EDTA vacutainer using QIAamp blood midi kit (Qiagen, cat.# 51183) and was used for study of mtDNA variations. Primers for amplifying Complex-I subunits and Complex-III encoding genes were designed as given by Rieder et al., 1998 [10] and processed for direct sequencing to screen the variations as described in [8]. Mitomap database was used to compare the obtained sequence data (https://www.mitomap.org/).
Mitophagy associated gene expression using qPCR
Relative quantification of mitophagy associated genes (ATG12, BECLIN1, and MAP1-LC3) was carried out with standard melt-curve protocol. The transcripts used were same as described in OXPHOS enzyme expression using qPCR section of methodology. The primers for ATG12, BECLIN1, and MAP1-LC3 were used at 250nM, 300nM and 300nM respectively, with Kapa SYBR FAST qPCR Master Mix Green (Kapa Biosystems, cat.# KM4103) to quantify transcripts on ABI StepOne machine in 20μl reaction mixture. Primer sequences were designed as described by Cotan et al., 2011 [11].
Statistical analysis
Statistical analysis was carried out using Graph Pad InStat 2 software (Graph Pad Software Inc., La Jolla, CA, U.S.A.). Statistical significance of different experiments performed for the study was determined either using Student’s t-test or using Chi-square test. A p-value ≤0.05 was considered statistically significant.
Results
mtDNA copy number change
Patients with positive chromosomal breakage investigation were screened using FANCD2 immunoblot to locate upstream or downstream complex defect (S2–S7 Tables). Based on defect in the pathway, complementation groups were assigned to the patients by molecular investigations such as direct sequencing and TES (S2–S6 Tables). Of the 70 FA patients studied for mitochondrial DNA (mtDNA) copy number change, 29(41%) patients showed no significant change in mtDNA copy number compared to controls and 41(59%) patients showed significant change in mtDNA copy number– 11 (16%) patients with low mtDNA copy number or decrease in mtDNA copy number and 30 (43%) with high copy number or increase in copy number (Fig 1A). The mean copy number changes of mtDNA were found to be 502 (p = 0.068), 404 (p = 0.057) and 573 (0.0092) for FA-A, FA-G and FA-L group patients respectively (Table 1, Fig 1B). The mean values of high and low copy number changes among patients of different complementation groups were found to be statistically significant (p<0.05) (Table 2, Fig 1C).
Fig 1. mtDNA copy number change study.

(a) Frequency distribution of mtDNA copy number change among FA patients (%), (b) Whisker and box plot for mtDNA copy number change among different FA complementation group patients. Dashed line represents mean mtDNA copy number (Pts.: Patients), and (c) mtDNA copy number change (Low and High) among patients of different FA complementation groups (ns: not significant, *: p<0.05, **: p< 0.01, and ***: p<0.00001).
Table 1. mtDNA copy number of FA patients from different complementation groups.
| Complementation group | Mean | Std Dev | Std Err | t-value | p-value |
|---|---|---|---|---|---|
| FA-A (n = 36) | 501.9504 | 426 | 74 | 1.51833 | 0.068 |
| FA-G (n = 12) | 404.3956 | 130 | 39 | 1.64734 | 0.057 |
| FA-L (n = 8) | 573.3908 | 331 | 125 | 2.58287 | 0.0092 |
| Non-FA-A, -G, and -L (n = 14) | 443.52 | 297 | 68 | 1.64378 | 0.055 |
| Controls (n = 33) | 323 | 115 | 31 |
Table 2. mtDNA copy number change (Low and High) among patients of different FA complementation groups.
| mtDNA copy number | Mean | StdDev | Std. Err | t-value | p-value | |
|---|---|---|---|---|---|---|
| FA-A | High (n = 13) | 874.02 | 466.06 | 129.26 | 4.2894 | 0.000117 |
| Low (n = 5) | 311 | 44.64 | 16.87 | -3.9697 | 0.00041 | |
| FA-G | High (n = 7) | 475.54 | 80.27 | 30.34 | 3.11507 | 0.00285 |
| Low (n = 1) | 136 | 0 | 0 | -2.2287 | 0.02136 | |
| FA-L | High (n = 4) | 806.67 | 222.08 | 111.04 | 6.01901 | <0.00001 |
| Low (n = 2) | 233 | 0 | 0 | -1.0748 | 0.15033 | |
| Non-FA-A, FA-G, and FA-L |
High (n = 6) | 736.08 | 314.19 | 128.27 | 4.39719 | 0.000174 |
| Low (n = 3) | 354 | 59.58 | 42.13 | -1.7897 | 0.04758 |
Study of mtDNA variations for OXPHOS complex-I and complex-III encoding genes
A total of 184 (115 synonymous and 69 non-synonymous) mtDNA variations of complex I subunits and complex III encoding genes of OXPHOS were detected in our study (Fig 2). Of 184 mtDNA variations, 138 (76%) were different variants and only 46 (24%) of variations were found to be frequently occurring among the FA patients. Majority of variations were results of transition changes (177/184, 96%) in the bases of DNA and a very few were transversion changes (7/184, 4%) (Table 3). Some of the frequently occurring synonymous and non-synonymous mtDNA variations observed in the study were screened to analyse if their occurrence is statistically significant compared to controls (Tables 4 and 5).
Fig 2. Types and frequency of mtDNA variations detected in Complex-I subunits and Complex-III encoding genes of OXPHOS.
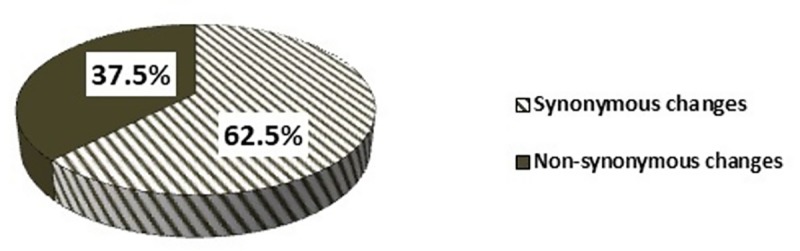
Table 3. Frequency (%) of transitions and transversions observed in different types of mtDNA variations.
| Synonymous changes (n = 115) | Non-synonymous changes (n = 69) | |
|---|---|---|
| Transitions | 112 (97.54%) | 65 (94.20%) |
| Transversions | 3 (2.46%) | 4 (5.80%) |
Table 4. mtDNA non-synonymous variations of OXPHOS complex-I subunits and complex-III encoding genes.
| Non-synonymous changes | GENES | AMINO ACID CHANGE | Transition/ Transversion | Frequency (%) | X2 score | p-value |
|---|---|---|---|---|---|---|
| 4216 T>C | ND1 | Y-H | Transition | 14 | 4.7107 | 0.029976 |
| 4225 A>G | ND1 | M-V | Transition | 3 | 4.0313 | 0.044664 |
| 4917 A>G | ND2 | N-D | Transition | 2 | 5.6738 | 0.017221 |
| 5319 A>G | ND2 | T-S | Transition | 2 | 2.1978 | 0.138208 |
| 10398 A>G | ND3 | T-A | Transition | 44 | 4.5816 | 0.032317 |
| 14323 G>A | ND6 | N-I | Transition | 2 | 60 | <0.05 |
Table 5. mtDNA synonymous variations of OXPHOS complex-I subunits and complex-III encoding genes.
| Synonymous changes | Gene | Amino acid change | Transition/ Transversion | Frequency (%) | X2 score | p-value |
|---|---|---|---|---|---|---|
| 4769 A>G | ND2 | M-M | Transition | 9 | 131.2331 | <0.05 |
| 10400 C>T | ND3 | T-T | Transition | 43 | 15.7051 | 0.000074 |
| 10873 T>C | ND4 | P-P | Transition | 25 | 52.0252 | <0.05 |
| 11083 A>G | ND4 | M-M | Transition | 2 | 52.0252 | <0.05 |
| 11467 A>G | ND4 | L-L | Transition | 5 | 1.8018 | 0.179495 |
| 11812 A>G | ND4 | L-L | Transition | 2 | 5.6738 | 0.017221 |
| 12007 G>A | ND5 | W-W | Transition | 7 | 0.5786 | 0.446865 |
| 12372 G>A | ND5 | L-L | Transition | 2 | 5.6738 | 0.017221 |
| 12705 C>T | ND5 | I-I | Transition | 7 | 108.4122 | <0.05 |
| 14905 G>A | CYTB | M-M | Transition | 2 | 5.6738 | 0.017221 |
| 15043 G>A | CYTB | G-G | Transition | 16 | 82.0513 | <0.05 |
| 15301 G>A | CYTB | L-L | Transition | 32 | 46.7532 | <0.05 |
| 11251 A>G | CYTB | L-L | Transition | 2 | 5.6738 | 0.017221 |
Mitophagy associated gene expression profiling
A significant (p = 0.02019) fold change (2.9 fold change) was observed in MAP1-LC3 gene expression of FA patients compared to controls. However, there was no significant difference in gene expression fold change (p>0.05) of Beclin1 and ATG12 genes of FA patients compared to controls (S8 Table and Fig 3A). Expression of ATG12 and Beclin1 genes among different FA complementation groups showed no significant (p>0.05) expression fold change (Fig 3B and S8 Table). Analysis of mitophagy expression according to distribution of patients among different complementation groups revealed a significant expression fold changes for MAP1-LC3 gene in FA-L group patients (3.6 fold change, p<0.05) compared to FA-A (~2.6 fold change, p = 0.082) and FA-G group patients (3.3 fold change, p = 0.074) (Fig 3B and S9 Table).
Fig 3. Study of mitophagy gene expression profiling.

(a) Gene expression for ATG12, Beclin1 and MAP1-LC3 genes (Refer S8 Table for p-values), (b) Expression fold change for mitophagy genes (ATG12, BECLIN1 and MAP1-LC3) among FA patients of different complementation group (Refer S9 Table for p-values).
OXPHOS enzyme encoding genes and TFAM gene expression
Fig 4 and S10 Table are showing the expression of OXPHOS complex-I subunits (ND1, ND2, ND3, ND4, ND4L, ND5, and ND6) and Complex-III (CYTB) encoding genes. The study revealed significant downregulation in expression compared to controls (ND1, p = 0.0132; ND2, p = 0.0072; ND3, p = 0.02268; ND4, p = 0.01765; ND4L, p = 0.00914; ND5, p = 0.04515; ND6, p = 0.02335; CYTB, p = 0.01538) (Fig 4A and S10 Table). However, no specific trend was observed in expression change of genes encoding OXPHOS complex-I subunits and complex-III encoding genes of FA patients among different complementation groups (Fig 4B and S11 Table). Expression study for TFAM gene in FA patients showed ~7.66 fold increase in levels compared to controls (S1 Fig). Complementation group-wise cross-sectional analysis showed significant upregulation in expression of TFAM gene in FA-A, FA-G and FA-L complementation groups (S1 Fig).
Fig 4. Expression profiling study for OXPHOS complex-I subunits and complex-III encoding genes.

(a) Gene expression profiling for OXPHOS complex-I subunits and complex-III encoding genes in FA patients (Refer S10 Table for p-values), (b) OXPHOS complex-I subunits and complex-III encoding gene expression in FA patients of different complementation groups (Refer S11 Table for p-values).
Discussion and conclusions
FA is a rare genetic disorder and presented with spectrum of clinical features [12]. Molecular studies have identified 22 genes associated with FA phenotype [13]. The bone marrow failure is one of the major clinical presentations in FA. However there have been several experimental proofs with no consensus result for one molecular mechanism cause underlying the BMF. Mitochondrial DNA (mtDNA) is known to be constantly challenged with ROS generated during electron transport reactions in oxidative phosphorylation event. Additionally, mtDNA lack histones and DNA damage repair system for mtDNA are not as much evolved as nuclear DNA. mtDNA copy number change has been known to be affected in many mitochondrial dysfunction syndromes and cancer conditions [14–17]. In our study, the mtDNA copy number change was observed in 59% of the FA patients where majority of them showed high copy number changes than low copy number change which suggests mitochondrial dysfunction in FA. We also carried out expression study for TFAM gene (Transcription factor A for mitochondria), a key regulator that drives mitochondrial DNA replication and transcription), to understand mitochondrial biogenesis controlling factor has any role to play [18]. We found that expression of TFAM gene was significantly upregulated in FA patients (S1 Fig). This implies that high copy number changes could be the plausible effect of compensatory mechanism of cells to cope up with energy requirement [9]. Correlation of mtDNA copy number changes with FA complementation groups suggests more significant change in copy number in FA-L group patients than FA-A and FA-G group patients. Further evaluation of same sample size of each complementation groups can lead us to understand relevance of variations in the significance of mtDNA copy number change among FA patients from different complementation group.
Mitochondrial dysfunction syndrome studies have been carried out using study of mtDNA variations especially OXPHOS reaction complex-I (NADH dehydrogenase) and complex-III (Cytochrome b) which pump electrons across inner mitochondrial membrane and generate proton gradient, the potential gradient generated is then used by ATPase-6 complex to generate energy in the form of ATP. mtDNA variations of genes encoding OXPHOS complexes have been studied and reported to form molecular pathology underlying the mitochondrial dysfunction [19, 20]. In our study, 62.5% of synonymous and 37.5% of non-synonymous changes have been observed in Complex-I subunits (ND1-ND6) and complex-III (CYTB) encoding genes. These are studied for molecular changes in FA patients and majority of the variations were result of transition changes in the nucleoside triphosphates (purine to purine or pyrimidine to pyrimidine). mtDNA variations T4216C and 10398A allele in FA patients suggest their association with mitochondrial dysfunction, as observed in other pathological diseases [21, 22]. Various mitochondrial dysfunction diseases have been shown to have impaired regulation in expression of mitochondrial Complex-I and Complex-III of OXPHOS genes [23, 24]. Gene expression study of OXPHOS complex-I and complex-III by real-time PCR suggests significant downregulation of complex-I subunits (ND1 to ND6) encoding genes and complex-III encoding genes. Various biochemical studies have been carried out for mitochondrial OXPHOS complex-I activity and have shown that the activity of complex-I is hampered in FA-A group patients [5]. Thus mtDNA copy number changes together with variations detected in mtDNA and qPCR study OXPHOS complex-I subunits and complex-III encoding genes should be considered as mitochondrial dysfunction biomarkers to track deterioration of mitochondrial associated phenotypes in FA patients.
Mitophagy is specific clearance of impaired mitochondria when cells are facing crisis. Cells prefer to clear off impaired mitochondria rather than being submissive to the cellular crisis and undergoing apoptotic cell deaths. Inefficient clearance of mitophagy events and accumulation of impaired mitochondria have been studied in FA cell lines [25].We have observed no significant changes in the expression of ATG12 and BECLIN1 genes. However, expression of MAP1-LC3 gene was found to be upregulated by ~3-fold in FA patients compared to age matched controls. The upregulated MAP1-LC3 (a useful autophagosomal marker) indicates initiation of autophagy of impaired mitochondria but inadequate expression of ATG12 protein (produces vesicle extension and completion in phagophore formation during mitophagy) and BECLIN1 (plays a significant role in cellular homeostasis and cross-regulation between apoptosis and autophagy) suggest incomplete clearance of dysfunctional mitochondria [26]. This could be a reason for accumulation of impaired mitochondria in FA cells. No specific trend was observed in expression of ATG12, Beclin1 and MAP1-LC3 genes in patients from different complementation group, suggesting inefficient mitophagy to be a generalized event rather than be associated with specific FA complementation group.
Changes in the mtDNA number (in 59% of FA patients), a high frequency of mtDNA variations (37.5% of non-synonymous variations and 62.5% synonymous variations) and downregulation of mtDNA complex-I and complex-III encoding genes of OXPHOS (p<0.05) are strong biomarkers for impairment of mitochondrial functions in FA. Deregulation of mitophagy genes (MAP1-LC3, p<0.05) suggests inability of FA cells to clear off impaired mitochondria. Accumulation of such impaired mitochondria in FA cells therefore may be the principal cause for BMF and a plausible effect of inefficient clearance of impaired mitochondria. In-vitro study of FA cell lines with mitophagy related gene silencing would strengthen the basis of this hypothesis. Together these results shed light on involvement of impaired mitochondria and deregulated mitophagy in FA pathogenesis. This signifies the need of inclusion of mitochondrial nutrients in the management strategies for FA patients.
Supporting information
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(A) Fold change for TFAM gene expression FA patients, (B) Complementation group-wise comparison of TFAM gene expression for FA patients.
(TIF)
Acknowledgments
Authors would like to thank patients and their families for participating in the study. We express our sincere gratitude to paediatricians and hemato-oncologists for referring patients to our centre. We thank Ms. Purvi Panchal and Mr. Somprakash Dhangar for carrying out qPCR experiment for TFAM gene. We would also like to extend our sincere gratitude to Dr. Minoru Takata and Dr. Minako Mori for collaborating with us to investigate FANC gene mutations in a few patients (especially for mutations of FANCB and FANCD2, and a few from FANCL).
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
Funded by Senior Research Fellowship (45/5/2014-HUM-BMS) was awarded to Ms. Avani Solanki from Indian Council of Medical Research (ICMR). The project was funded by intramural grant from National Institute of Immunohaematology (ICMR), Mumbai, India.
References
- 1.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mukhopadhyay SS, Leung KS, Hicks MJ, Hastings PJ, Youssoufi H, Plon SE. JCB: ARTICLE. 2006;175(2):225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumari U, Jun WY, Bay BH, Lyakhovich A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi Anemia cells. Oncogene. 2013;(October 2012):1–8. [DOI] [PubMed] [Google Scholar]
- 4.Lyakhovich A. Damaged mitochondria and overproduction of ROS in Fanconi anemia cells. Rare Dis. 2013;1(1):e24048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ravera S, Vaccaro D, Cuccarolo P, Columbaro M, Capanni C, Bartolucci M, et al. Biochimie Mitochondrial respiratory chain Complex I defects in Fanconi anemia complementation group A. Biochimie. 2013;95(10):1828–37. 10.1016/j.biochi.2013.06.006 [DOI] [PubMed] [Google Scholar]
- 6.Sumpter R, Levine B. Emerging functions of the Fanconi anemia pathway at a glance. 2017;2657–62. 10.1242/jcs.204909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sumpter R, Sirasanagandla S, Fernández ÁF, Wei Y, Dong X, Franco L, et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell. 2016;165(4):867–81. 10.1016/j.cell.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Solanki A, Mohanty P, Shukla P, Rao A, Ghosh K, Vundinti BR. FANCA gene mutations with 8 novel molecular changes in Indian Fanconi anemia patients. PLoS One. 2016;11(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malik AN, Shahni R, Rodriguez-de-Ledesma A, Laftah A, Cunningham P. Mitochondrial DNA as a non-invasive biomarker: Accurate quantification using real time quantitative PCR without co-amplification of pseudogenes and dilution bias. Biochem Biophys Res Commun. 2011;412(1):1–7. 10.1016/j.bbrc.2011.06.067 [DOI] [PubMed] [Google Scholar]
- 10.Rieder MJ, Taylor SL, Tobe VO, Nickerson DA. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: Analysis of the human mitochondrial genome. Nucleic Acids Res. 1998;26(4):967–73. 10.1093/nar/26.4.967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cotan D, Cordero MD, Garrido-Maraver J, Oropesa-Avila M, Rodriguez-Hernandez A, Gomez Izquierdo L, et al. Secondary coenzyme Q10 deficiency triggers mitochondria degradation by mitophagy in MELAS fibroblasts. FASEB J. 2011;25(8):2669–87. 10.1096/fj.10-165340 [DOI] [PubMed] [Google Scholar]
- 12.Faivre L, Guardiola P, Lewis C, Dokal I, Ebell W, Zatterale A, et al. Association of complementation group and mutation type with clinical outcome in Fanconi anemia. Blood. 2000;96(13):4064–70. [PubMed] [Google Scholar]
- 13.Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2017;31(3):93–9. 10.1016/j.blre.2016.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mozhey OI, Zatolokin PA, Vasilenko MA, Litvinova LS, Kirienkova E V., Mazunin IO. Evaluating the number of mitochondrial DNA copies in leukocytes and adipocytes from metabolic syndrome patients: Pilot study. Mol Biol. 2014;48(4):590–3. [PubMed] [Google Scholar]
- 15.Song J, Oh JY, Sung YA, Pak YK, Park KS, Lee HK. Peripheral blood mitochondrial DNA content is related to insulin sensitivity in offspring of type 2 diabetic patients. Diabetes Care. 2001;24(5):865–9. 10.2337/diacare.24.5.865 [DOI] [PubMed] [Google Scholar]
- 16.Liu S-F, Kuo H-C, Tseng C-W, Huang H-T, Chen Y-C, Tseng C-C, et al. Leukocyte Mitochondrial DNA Copy Number Is Associated with Chronic Obstructive Pulmonary Disease. PLoS One. 2015;10(9):e0138716 10.1371/journal.pone.0138716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thyagarajan B, Wang R, Nelson H, Barcelo H, Koh WP, Yuan JM. Mitochondrial DNA Copy Number Is Associated with Breast Cancer Risk. PLoS One. 2013;8(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Virbasius J V., Scarpulla RC. Activation of the human mitochondrial transcription factor A gene by nuclear respiratory factors: A potential regulatory link between nuclear and mitochondrial gene expression in organelle biogenesis. Proc Natl Acad Sci U S A. 1994;91(4):1309–13. 10.1073/pnas.91.4.1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanniarajan A, Govindaraj P, Carlus SJ, Aruna M, Aruna P, Kumar A, et al. Mitochondrial DNA variations associated with recurrent pregnancy loss among Indian women. Mitochondrion. 2011;11(3):450–6. 10.1016/j.mito.2011.01.002 [DOI] [PubMed] [Google Scholar]
- 20.Kabekkodu SP, Bhat S, Mascarenhas R, Mallya S, Bhat M, Pandey D, et al. Mitochondrial DNA variation analysis in cervical cancer. Mitochondrion. 2014;16:73–82. 10.1016/j.mito.2013.07.001 [DOI] [PubMed] [Google Scholar]
- 21.Kazuno AA, Munakata K, Nagai T, Shimozono S, Tanaka M, Yoneda M, et al. Identification of mitochondrial DNA polymorphisms that alter mitochondrial matrix pH and intracellular calcium dynamics. PLoS Genet. 2006;2(8):1167–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linnartz B, Anglmayer R, Zanssen S. Comprehensive Scanning of Somatic Mitochondrial DNA Alterations in Acute Leukemia Developing from Myelodysplastic Syndromes. Cancer Res. 2004;64(6):1966–71. 10.1158/0008-5472.can-03-2956 [DOI] [PubMed] [Google Scholar]
- 23.Reinecke F, Smeitink JAM, van der Westhuizen FH. OXPHOS gene expression and control in mitochondrial disorders. Biochim Biophys Acta—Mol Basis Dis. 2009;1792(12):1113–21. [DOI] [PubMed] [Google Scholar]
- 24.Salehi MH, Kamalidehghan B, Houshmand M, Yong Meng G, Sadeghizadeh M, Aryani O, et al. Gene expression profiling of mitochondrial oxidative phosphorylation (OXPHOS) complex I in Friedreich ataxia (FRDA) patients. PLoS One. 2014;9(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shyamsunder P, Esner M, Barvalia M, Wu YJ, Loja T, Boon HB, et al. Impaired mitophagy in Fanconi anemia is dependent on mitochondrial fission. Oncotarget. 2016;7(36):58065–74. 10.18632/oncotarget.11161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys. 2007;462(2):245–53. 10.1016/j.abb.2007.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(DOCX)
(A) Fold change for TFAM gene expression FA patients, (B) Complementation group-wise comparison of TFAM gene expression for FA patients.
(TIF)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.