

Abstract
Background:
Mutations affecting DNA polymerases have been implicated in genomic instability and cancer development, but the mechanisms by which they may impact the immune system remain largely unexplored.
Objective:
To establish the role of POLD1, encoding the DNA polymerase δ 1 catalytic subunit, as the cause of a primary immunodeficiency in an extended kindred.
Methods:
We performed whole-exome and targeted gene sequencing, lymphocyte characterization, molecular and functional analyses of the DNA polymerase delta (Polδ) complex, and T and B cell antigen receptor repertoire analysis.
Results:
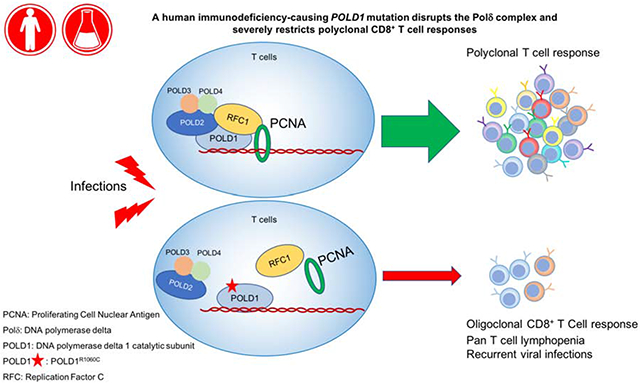
We identified a missense mutation (c. 3178C>T; p.R1060C) in POLD1 in three related subjects who presented with recurrent, especially herpetic, infections and T cell lymphopenia with impaired T cell but not B cell proliferation. The mutation destabilizes the Polδ complex, leading to ineffective recruitment of replication factor C to initiate DNA replication. Molecular Dynamics simulation revealed that the R1060C mutation disrupts the intramolecular interaction between the POLD1 CysB motif and catalytic domain, and also between POLD1 and the DNA polymerase δ subunit POLD2. The patients exhibited decreased naïve CD4 and especially CD8 T cells in favor of effector memory subpopulations. This skewing was associated with oligoclonality and restricted T cell receptor beta chain V-J pairing in CD8+ but not CD4+ T cells, suggesting that POLD1R1060C differentially impacts peripheral CD8+T cell expansion and possibly thymic selection.
Conclusion.
These results identify gene defects in POLD1 as a novel cause of T cell immunodeficiency.
Keywords: DNA Polymerase Delta, POLD1, Replication Factor C, Primary Immunodeficiency, Whole Exome Sequencing
Graphical Abstract
Capsule Summary
A Loss of function mutation in POLD1 causes impaired assembly and function of the DNA polymerase delta (Polδ) complex, resulting in a T cell immunodeficiency.
Introduction
DNA replication is a fundamental process for maintaining cellular homeostasis 1. DNA polymerase δ (Polδ), one of the three family B polymerases in eukaryotes, is essential for the leading and lagging strand synthesis 2-4. In mammals, Polymerase δ is a heterotetramer that includes four subunits: POLD1-4 5. POLD1 functions as the catalytic subunit, which is endowed with both polymerase and exonuclease activities and which plays a critical role in several synthetic and DNA-repair processes 6, 7. Total POLD1 deficiency is embryonic lethal in mice, while deficiency of POLD1 exonuclease activity in Pold1exo/exo mice (mutator mice), results in a high rate of DNA replication errors 8. POLD2, POLD3, and POLD4 are accessory subunits that interact with other nuclear proteins to regulate the activity and stability of the Polδ complex 9-11. In particular, POLD2 serves as a scaffold by interacting with POLD1 and the other Polδ subunits 12. Additionally, the Polδ complex coordinately interacts with a number of proteins that enable its function, including DNA replication factor C (RFC) and Proliferating Cell Nuclear Antigen (PCNA) 13.
Studies have shown that mutations in POLD1 in mice and humans lead to genomic instability, hypermutator phenotype and carcinogenesis 14-16. Damaging heterozygous mutations in POLD1 proof-reading (exonuclease) domain have been identified in inherited colorectal cancers 17. A POLD1 heterozygous single amino acid deletion that maps to the catalytic domain and which abrogates the DNA polymerase but not the exonuclease activity has been identified in a developmental disorder of mandibular hypoplasia, sensorineural hearing loss, progeroid features and lipodystrophy with insulin resistance 18, 19. Thus, mutations affecting different domains of POLD1 give rise to distinct disorders and phenotypes.
Here we identify a novel POLD1 mutation that affects the stability of the Polδ complex, resulting in a disorder distinct from those caused by other POLD1 mutations. The patients presented with a combined immunodeficiency disorder associated with T cell lymphopenia, CD8+ T cell oligoclonality and repertoire restriction, indicative of a particularly important role for POLD1 in CD8 T cell expansion.
Methods
Patient Studies.
All study participants were recruited after obtaining informed consent at the referring institution (Necmettin Erbakan University, Meram Medical Faculty, Konya, Turkey), and the studies were conducted at the Boston Children’s Hospital under approved protocol #04-09-113R.
Whole exome sequencing (WES).
WES was performed on genomic DNA of 2 affected siblings (P1 and P2), their parents, and their healthy sister through Axeq (Rockville, Md). The Agilent SureSelect Target enrichment kit was used for exon capture (Agilent Technologies, Santa Clara, Calif). Paired-end sequencing was performed with an Illumina HiSeq2000 instrument (Illumina, San Diego, Calif), which generated 150 base pair reads. Average coverage in WES was 53X, covering 96% of the coding regions. Analysis of WES data was performed using “Variant Explorer Pipeline” (VExP)20 to narrow down potential candidate variants. VExP is a validated comprehensive system that integrates existing methods, genetic information, and probabilistic models into an automated pipeline for the identification of disease genes. Raw data were processed, filtered and analyzed according VExP recommendations (see supplementary information). Candidate genes that passed the criteria for the 2 affected samples were further evaluated by our research team (Table E1 in the Online Repository). The identified POLD1 c. 3178C>T mutation was confirmed by Sanger sequencing by first generating a 404 base pair amplicon from genomic DNA using the following primers: forward primer 5'-AGAAGCTGGGATTGGCAGT-3' and reverse primer 5'-GAGAGGCCTTGGAGTCAGAG-3'. The amplicon was then sequenced for the presence of the mutation using the following primer: 5'-GCCTACATGAAGTCGGAGGA-3'. Sanger sequencing analysis was also used to screen other family members for the POLD1 mutation.
Antibodies and flow cytometry.
Anti-human monoclonal antibodies (mAbs) to the following antigens were used for staining: CD3 (SK7), CD4 (SK3), CD8 (SK1), CD45RA (HI100), CD45RO (UCHL1), CCR7 (150503), CD31 (L133.1), TCR A/B (WT31) and TCR G/D (11F2), CD16+56 (B73, 1MY3I), CD19 (SI25C1), IgD (IA6-2), CD27 (L128) (BD Biosciences) and the appropriate isotype controls. The monoclonal antibody against POLD1(sc-17776) was obtained from Santacruz. Antibodies against V5 was purchased from Biolegend (903801), POLD2 (HPA026745) and RFC1 (HPA046116) were purchased from Sigma-Aldrich, POLD3 (A301-244A-M) was from Bethyl Laboratories and β-actin was from Cell Signaling Technology. Whole blood was incubated with mAbs against surface markers for 30 min on ice. Intracellular staining with FOXP3 was performed using eBioscience Fixation/Permeabilization buffer according to the manufacturer’s instructions. Data were collected with a Fortessa cytometer (BD Biosciences) and analyzed with FlowJo software (TreeStar).
Cell culture and transfection.
HEK293T (American tissue culture collection), and Fibroblast cells were cultured using DMEM (Invitrogen) plus 10% FBS (Gibco), supplemented with 1% penicillin-streptomycin (Invitrogen). Peripheral blood samples (5 ml) were collected from each subject and stored in tubes containing EDTA. PBMCs were isolated by Ficoll-Paque PLUS (GE Healthcare) gradient centrifugation, and cultured in RPMI 1640 with 10% FBS and 1% pen-strep (with Non-Essential Amino Acid, Sodium Pyruvate) with anti-CD3/CD28 beads (from Invitrogen). B cell proliferation was quantified by culturing PBMCs after stimulation with anti-CD40 mAb (10 μg/mL; R&D, 6245-CL-050) plus IL-21 (30 ng/mL; Peprotech, 200-21) for 5 days. Lipofectamine 2000 (Invitrogen) was used for transient transfection of HEK293T Cells according to the manufacturer’s instructions.
BrdU cell proliferation assay.
Bromodeoxyuridine / 5-bromo-2'-deoxyuridine (BrdU) incorporation assay in patient peripheral blood mononuclear cells (PBMCs) was performed with the Phase-Flow™ FITC BrdU Kit from Biolegend (Catalog No. 370704) following the manufacturer’s instructions. The same kit included 7-aminoactinomycin D (7-AAD) for staining for total DNA.
Lentiviral transfections.
HEK293T cells plated on 100-mm dishes were transfected with the indicated lentiviral expression plasmid (14ug) together with the GAG (10ug), the REV (5ug) and the VSV (2ug). After 48h, the viral particles were collected, filtered by 0.45um membrane filter and used to infect the indicated cells in the presence of polybrene (6 ug/ml). After transfection for 48h, cells were cultured in complete RPMI 1640 medium and ready for BrdU assay.
Plasmids.
POLD1 _pLX307 and pLX307 vector were purchased from Addgene. The POLD1 C3178T mutation was introduced into the POLD1 plasmid with the Q5 Site-Directed Mutagenesis Kit (NEB) using the following primers: 5'-GCAGTGCCAGtGCTGCCAGGG-3' Forward primer and 5'GTCCAGAGGCGCGAGAAGC-3' Reverse primer. POLD1 mutants were confirmed using Sanger sequencing.
Immunoprecipitation assay and immunoblot analysis.
For immunoprecipitation assay, cells extracts were prepared by using RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate) supplemented with a complete protease inhibitor cocktail (Roche), a PhosSTOP phosphatase inhibitor cocktail (Roche). Lysates were incubated with the appropriate antibody for four hours to overnight at 4°C before adding protein A/G agarose for 2 hr. The immunoprecipitates were washed three times with the same buffer and eluted with SDS loading buffer by boiling for 5 min.
For immunoblot analysis, the samples were subjected to SDS-PAGE. The resolved proteins were then electrically transferred to a PVDF membrane (Millipore). Immunoblotting was probed with indicated antibodies. The protein bands were visualized by using a SuperSignal West Pico chemiluminescence ECL kit (Pierce). Signal intensities of immunoblot bands were quantified by Image J software.
Molecular Modeling and Simulation.
We performed molecular dynamics (MD) simulation for the WT and mutant POLD1 to relax the protein structure obtained from homolog modeling. The simulation region starts from the 795th residue. All MD simulations were performed using the AMBER 14 suite of programs 21. POLD1 protein was immersed into a simulated water box with the initial structure taken from molecular docking. The force field for protein is AMBER parm10 force field 21, and the SPC/E model was adopted for water 22. Chloride ions were added to neutralize the system whose force field is obtained from Joung et al.23 The sizes of the simulation boxes are about 81 Å *65 Å *93 Å. For the WT and mutant POLD1, we carried out NPT ensemble simulation with a 10 ns long trajectory after the initial equilibration. The Berendsen weak-coupling barostat was used to control the pressure at 1 bar 24. The temperature was controlled using Langevin dynamics at 300 K. SHAKE algorithm was used to constrain all bonds involving hydrogen atoms 25. A cutoff of 10.0 was adopted for nonbonding interactions. For the long-range electrostatic interactions, the Particle Mesh Ewald method was applied.
The overall protein secondary and tertiary structure persist during the simulation, suggesting that homolog modeling gives a good prediction of POLD1 structure. A hydrogen bond is considered formed only if the distance between the heavy atom of hydrogen bond donor and acceptor is less than 3.5 Å and the N–H–O angle is greater than 135°.
Repertoire analysis.
CD4+ T(CD3+CD4+) cells, CD8+ (CD3+CD8+) T cells and B (CD3− CD19+) cells were isolated from fresh PBMCs obtained from POLD1R1060C patients and healthy controls. Genomic DNA was isolated with QIAamp DNA Blood Mini Kit (QIAGEN) and sent to Adaptive Biotechnologies (Seattle, WA) for immune repertoire analysis. A multiplex PCR protocol was used to amplify the CDR3 of the sorted lymphocytes using a standard quantity of DNA as the template. The Illumina platform was used for sequencing of the PCR products. The sequences were aligned to a reference genome, and T cell receptor β chain (TRB) and Immunoglobulin heavy chain (IGH) variable, diversity, and joining (VDJ) gene definitions were based on the International ImMunoGeneTics system (IMGT)26. The data were filtered and clustered using the relative frequency ratio between similar clones and a modified nearest neighbor algorithm to merge closely related sequences and remove both PCR and sequencing errors, as described 27, 28. Productive unique and total sequences were analyzed for TCR and BCR V, D, and J usage with ImmunoSEQ Analyzer™ set of online tools and with R (version 3.6.1). Physicochemical properties of amino acid sequences were analyzed using the Alakazam package29.Sequencing of the TRB and IGH rearranged products was completed successfully in all samples (Table E3 in the Online Repository). The primary sequencing data are available at https://clients.adaptivebiotech.com/pub/cui-2019-jaci.
Statistical Analysis.
Aggregate results are presented as means + S.E.M.. Comparison between groups was carried out with 1-way ANOVA with Bonferroni post-test analysis, as indicated. Differences in mean values were considered significant at a P value of less than 0.05.
Results
Identification of a novel immunodeficiency associated with a novel POLD1 mutation.
P1 is a 14 years old female, born to consanguineous (first cousins) Turkish parents (Fig 1, A), who suffered from recurrent upper and lower respiratory tract infections (URTI and LRTI, respectively) starting early in life. At 3 years of age she was diagnosed with sensorineural hearing loss following evaluation for language delay. She underwent adenotonsillectomy at 5 1/2 years of age because of recurrent URTI and serous otitis media. She suffered from severe Chickenpox at 6 years of age that eventually resolved. Thereafter, she started experiencing LRTI at a frequency of 4-5 times/year, mainly in winter, requiring intravenous antibiotic therapy. Starting age 9 years, she had recurrent oral herpes infections every 1-2 months as well as several episodes of recurrent herpes zoster for which she received acyclovir therapy. Investigation into her recurrent herpetic infections revealed marked lymphopenia, for which she was referred for immunological evaluation at 12 years of age. Her immunological workup was particularly notable for profound CD3+CD4+ lymphopenia. She had mildly decreased serum IgG and low IgA and IgM antibody concentrations, while her tetanus vaccine-related antibody responses were absent despite having been fully vaccinated (Table 1). She was started on immunoglobulin replacement therapy, which resulted in the resolution of LRTI and markedly decreased herpetic infections.
Figure 1. Characterization of a POLD1 mutation in kindred with combined immunodeficiency.

A. Patient Pedigree and familial segregation of the mutant POLD1 allele. Double lines connecting parents indicate consanguinity. Probands are indicated as P1-P3. Squares, Male subjects; circles, female subjects; solid symbols, patients; half-filled symbols, heterozygous. B. Sanger sequencing fluorograms of the germline c. 3178 C>T POLD1 mutation in patients P1-P3 and the parents of P1 and P2 compared with the equivalent DNA sequences in an unaffected healthy sibling (HS). C. Schematic representation of POLD1 protein. The different domains are depicted as follows: the nuclear localization signal (NLS) in red, the exonuclease in light green, the polymerase domain in light blue and the cysteine-rich metal-binding domains (CysA/B) in orange. The identified R1060C mutation and its position within the CysB polypeptide sequence is indicated by red star. D. Immunoblot analysis of POLD1/2/3 protein expression in PBMCs (left) and primary fibroblasts (right) of P1, P2 and a healthy control subject (HC). E. Quantitation of POLD1/2/3 protein expression in primary fibroblasts of P1 and P2, normalized for β-actin expression and expressed as fold change compared to that of HC fibroblasts (n=3; open circles). Results represent means ± S.E.M.**p<0.01; ***, p<0.001; ****, p<0.0001, by one-way ANOVA with Bonferroni post-test analysis.
Table1.
Hematological and immunological parameters in POLD1R1060C patients.
| P1 | P2 | P3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Age | 12yrs | 13yrs (On IVIG) |
Normal range for age |
3yrs | 4yrs | 5yrs | Normal range for age |
29yrs | Normal range for age |
| CBC | |||||||||
| WBC (cells/μl) | 4300 | 4860 | 4500-13000 | 3260 | 3070 | 1830 | 5000-14500 | 6720 | 4000-10000 |
| Neutrophils(cells/μl) | 3100 | 3430 | 1500-7300 | 1970 | 1740 | 900 | 1500-8000 | 5370 | 1500-7300 |
| Lymphocytes(cells/μl) | 600 | 430 | 1200-5200 | 765 | 971 | 470 | 1500-7000 | 780 | 800-5500 |
| Eosinophils cells/μl) | 20 | 260 | 200-600 | 2 | 134 | 130 | 200-600 | 70 | 200-600 |
| Hemoglobin(g/dL) | 12.8 | 14.2 | 12.1-17.2 | 13.0 | 13.1 | 13 | 11.1-17.2 | 11.9 | 12.1-17.2 |
| Platelets(cells/μl) | 344000 | 348000 | 150000-400000 | 360000 | 323000 | 244000 | 150000-400000 | 152000 | 150000-400000 |
| Immunoglobulins | |||||||||
| IgA (mg/dL) | 23 | 23 | 72-177 | 26 | 28 | 46-129 | 42.1 | 65-176 | |
| IgM (mg/dL) | 49 | 55 | 63-164 | 97 | 97 | 50-146 | 126 | 86-175 | |
| IgG (mg/dL) | 723 | 911 | 822-1323 | 588 | 568 | 722-1195 | 902 | 944-1506 | |
| IgE (IU) | 17 | 19 | 18 | 19 | 68 | 18 | |||
| Specific antibody response | |||||||||
| Anti-Tetanus IgG (IU/mL) | 0.01 | 0.65 | >0.1 | 0.01 | 0.2* | >0.1 | 0.1 | >0.1 | |
| Isohemaglutinin titer (dilutions) | 1/16 | 1/16 | >1/16 | 1/32 | 1/64 | >1/16 | 1/16 | >1/16 | |
| Anti Hepatitis B antibodies (IU) | 319 | >10 | 38 | 624 | >10 | 0.06 | >10 | ||
| Lymphocyte subset analysis | |||||||||
| CD3+ cells/μl | 330 | 177 | 1000-2600 | 337 | 437 | 100 | 1400-6200 | 252 | 1000-2600 |
| CD3+CD4+ cells/μl | 102 | 66 | 530-1500 | 145 | 184 | 57 | 700-2200 | 101 | 530-1500 |
| CD3+CD8+ cells/μl | 186 | 89 | 330-1100 | 176 | 233 | 105 | 490-1300 | 135 | 330-1100 |
| CD19+ cells/μl | 120 | 151 | 110-570 | 337 | 456 | 239 | 390-1400 | 85 | 110-570 |
| CD16+CD56+ cells/μl | 12 | 46 | 70-480 | 61 | 58 | 61 | 130-720 | 402 | 70-480 |
| CD3+TCR α/β+ cells/μl | 213 | 700-2800 | 466 | 600-4300 | 600-3300 | ||||
| CD3+ TCR γ/δ+ cells/μl | 54 | 39-540 | 39 | 27-960 | 25-200 | ||||
| CD4+CD45RA+CD31+% | 13.0 | 32.9-61.5 | 48.0 | 52-67 | 5 | 9.8-43.2 | |||
| CD19+IgD+CD27−% | 62.6 | 51.3-82.5 | 61.8 | 47.3-77.0 | 64 | 48.4-79.7 | |||
| CD19+IgD−CD27+% | 7.0 | 2.06 | 8.7-25.6 | 6.1 | 1.12 | 10.9-30.4 | 1.86 | 8.3-27.8 | |
| CD19+ IgD+CD27+% | 0.53 | 4.6-18.2 | 0.49 | 5.2-20.4 | 0.47 | 7.0-23.8 | |||
| CD19+CD24+CD38+ % | 5.48 | 5.3-18.9 | 4.43 | 7.4-23.7 | 0.7 | 2.2-13.3 | |||
post-booster vaccination.
Patient P2 is the younger sister of P1 and presented at age 3 with history of fever and cough. Her weight (12,3kg; 10th-25th percentile) and height (98 cm; 50th-75th percentile) were in normal range for her age. Her laboratory results revealed marked lymphopenia, hypogammaglobulinemia and low CD3+CD4+ T cell number similar to her older sister (Table 1). She also had an unprotective tetanus antibody response on initial presentation despite her prior vaccination that normalized on booster vaccination. She is currently 5 years of age and still suffers from recurrent croup and oral herpes infections, for which she is treated symptomatically. Auditory testing revealed normal hearing.
P3 is a 29 years old paternal aunt of P1 and P2 (Fig 1, A). She had encephalitis following a primary varicella infection at 3 years of age, from which she was left with mental retardation, hearing deficit and speech delay. In the ensuing years, she has had recurrent oral herpetic infections, for which she is treated symptomatically. She also has URTI in winter, requiring oral antibiotic therapy.
Pedigree analysis suggested an autosomal recessive inheritance of the disease. To identify the underlying gene defect, we performed whole exome sequencing for the whole family. Knowing that the family is consanguineous, we executed homozygosity mapping using their WES data (see Supplementary Methods for more information). Five regions of homozygosity were identified, and only the largest one found on chromosome 19, 14Mb in size, contains mutations with a minor allele frequency <0.01 that segregate within family (Fig E1 and Table E2 in the Online Repository). Of the nine genes thus identified in this region, including six candidate genes with homozygous recessive mutations, the most promising was a homozygous C>T substitution in exon 26 of POLD1 was identified (c.3178C>T; p.R1060C; based on POLD1 isoform 1, NM_001256849.1) (Table E1 in the Online Repository). This missense homozygous mutation has not been previously reported in genomAD, ExAC, dbSNP or 1000Genome and was confirmed by Sanger sequencing (Fig 1, B). Both parents were heterozygous for the mutation. Her sister P2, who also suffered from recurrent infections, was found homozygous for the same mutation by Sanger sequencing, as was a paternal aunt P3, who suffered from an early childhood encephalitis post varicella infection. A number of extended family members had colon and rectal cancers but did not carry the mutant allele.
The R1060C substitution localizes to the CysB motif at C-terminus of POLD1, away from the exonuclease and polymerase domains (Fig 1, C). Immunoblot analysis revealed decreased POLD1 expression in P1 and P2 compared to healthy sibling in PBMCs and fibroblasts (Fig 1, D and E). Other components of the Polδ complex are also decreased, including POLD2 and POLD3. Thus, the POLD1R1060C mutation exerted a global effect on the Polδ complex.
POLD1R1060C affects the stability of polymerase δ complex.
DNA polymerases have been classified into several families based on their amino acid sequences. In polymerase family B, both Polα and Polδ are key enzymes for eukaryotic nuclear DNA replication30. The structure of the family B DNA polymerases has been conserved throughout evolution, although their primary sequence varies. The structure of DNA polymerase alpha 1, catalytic subunit (POLA1) is well studied31. To clarify the role of the R1060C missense substitution in POLD1 from the family with immunodeficiency syndrome, we generated the model of full-length POLD1 using Swiss modeling based on the crystal structure of POLA1, which has a sequence identity of 25.9%. Most of the structural elements aligned well between the POLD1 model and the POLA1 structure, with an average root-mean-square deviation (RMSD) of 0.315 Å (Fig 2, A). Based on the POLD1 model, we introduced the R1060C mutation in PyMol followed by molecular dynamics (MD) simulation for the wild-type and mutant POLD1 to relax the protein structure obtained from homolog modeling with a time scale of 10 ns. The simulation region starts from the residue 795 in consideration of domain of interests and computational cost. After we extract 10-ns snapshots of wild-type POLD1 and the R1060C mutant protein, we found a significance distance change between the CysB motif and the DNA polymerase type B family catalytic subunit (POLBc) delta domain (Fig 2, B). The CysB motif in POLD1 shows an intact interaction with the POLBc delta domain while the C1060 mutant lost the interaction. Detailed interface analysis showed the hydrogen bonds between E830 and R1060, R968 and R1060 backbone, R808 and Q1059, and the hydrophobic interaction between V832 and I1078 dominate the mutual interaction between these two domains (Fig 2, C). By calculating the distance between residue 1060 and 830 in WT and mutant POLD1, we found that the R1060-E830 interaction maintains a shorter distance than C1060-E830 after the structural relaxation (Fig E2, A in the Online Repository), indicating that this interaction pair may be critical to maintain the CysB- POLBc_delta interaction. Also, hydrogen bond tracing revealed that the interaction between R1060 and E830 is highly stable during the whole simulation trajectory (Fig E2, B in the Online Repository), which provided further evidence that R1060 is critical for the intramolecular interaction of POLD1. Because of the decrease expression of POLD2 in patient’s polymerase δ, we hypothesized that the POLD1R1060C mutation affects the stability of polymerase δ complex. Considering that the weaker intramolecular interaction in the mutant protein may further affects the recruitment of downstream POLD subunits, we built both the wild-type and mutant model of POLD1 10 ns snapshots/POLD2 complex based on the crystal structure of the POLA1/POLA2 complex (PDB ID: 5EXR). Overall, the docking model clearly indicated the cooperation between the CysB motif and the POLBc_delta domain in recruiting POLD2. This cooperative interaction is weakened by the R1060C mutant, which may disrupt POLD1:POLD2 complex formation (Fig 2, D).
Figure 2. POLD1R1060C mutation disrupts POLD1-intrinsic and POLD1-POLD2 molecular interactions.

A. The protein structure model of full-length POLD1 using Swiss modeling based on the crystal structure of POLA1. B. Distance between the CysB motif and POLBc_delta domain in WT POLD1 and the POLD1R1060C mutant protein are measured by 10-ns snapshots. C. Detailed interface analysis of POLD1 structure. The hydrogen bonds between E830 and R1060, R968 and R1060 backbone, and R808 and Q1059 were in blue and red. D. Docking model showing the cooperation between the CysB motif and the POLBc_delta domain in recruiting POLD2 and its disruption by the R1060C mutation. E. HEK293T cells were transfected with the indicated plasmids. Twenty-four hours after transfection, cell lysates were immunoprecipitated with an anti-V5 antibody and then immunoblotted with the indicated antibodies. F. Quantitation of co-immunoprecipitated POLD2 protein by Empty vector, WT POLD1 and POLD1R1060C in E, expressed as fold change compared to that of WT POLD1 (n=3; open circles). Results represent means ± S.E.M. ***, p<0.001; ****, p<0.0001, by one-way ANOVA with Bonferroni post-test analysis.
To validate the results obtained with molecular modeling, we examined the capacity of recombinant tagged WT and mutant POLD1 expressed in HEK293T cells to interact with the endogenous POLD2. Co-immunoprecipitation studies showed that the R1060C mutation interfered with the formation of the POLD complex, as indicated by the capacity of WT POLD1 but not the POLD1R1060C mutant protein to co-precipitate with POLD2 despite equal expression of the respective POLD1 proteins (Fig 2, E, F). Furthermore, we examined the capacity of POLD1R1060C mutant protein to interact with other components of the DNA replication complex. During DNA replication, Polδ associates with replication factor C (RFC), a step necessary for loading Proliferating Cell Nuclear Antigen (PCNA) onto DNA to initiate DNA replication. However, unlike WT POLD1, POLD1R1060C failed to effectively coprecipitate with RFC, indicative of poor association (Fig 2, E). Overall, these results indicate that that the POLD1R1060C mutation interfered with molecular interactions involved in Polδ assembly and the formation of the overall DNA replication complex.
POLD1R1060C results in lower cell proliferation.
To determine whether the R1060C mutation in POLD1 impacted DNA replication and cell proliferation, we analyzed the proliferative activities of control and patient T cells upon treatment of PBMC with anti-CD3+anti-CD28 mAb for 3 days followed by a 4-hour stimulation with phorbol myristate acetate (PMA) and ionomycin32. The cells were pulsed with BrdU, which labels newly synthesized DNA characteristic of the cell cycle S phase 33. Results showed that patient T cells, but not B cells, were profoundly deficient in BrdU labeling following stimulation, indicative of poor S phase DNA replication (Fig 3, A-D). To determine whether the poor proliferation of patient T cells was directly related to the POLD1 defect, we undertook rescue experiments in which patient T cells were expanded with anti-CD3+anti-CD28 mAb treatment, followed by transfection with a lentiviral vector encoding either wild-type (WT) POLD1 or the POLD1R1060C mutant. Both the WT POLD1 and the POLD1R1060C mutant were equally expressed in the transfected cells (Fig 3, E). Results showed that DNA replication was rescued in WT POLD1–transduced patient cells, with a significant increase in the frequency of cells in the S phase, whereas the POLD1R1060C failed to do so (Fig 3, F, G). Collectively, our studies indicate that the POLD1R1060C mutant severely decreased DNA replication.
Figure 3. Defective proliferation of POLD1R1060C T cells and its rescue by WT POLD1 expression.

A. Representative flow cytometric analysis of BrdU+ P1 and P2 T cells versus those of a healthy control (HC) and healthy sibling (HS). B. Frequencies of BrdU+ T cells in healthy control versus P1-P3 cell cultures (n=3; open circles). C. Representative flow cytometric analysis of BrdU+ patient (Pt) B cells versus those of a healthy control (HC). D. Frequencies of BrdU+ B cells in healthy control versus P1-P3 cell cultures (n=3; open circles). E. Immunoblot analysis of POLD1 expression in transduced cells. F. Representative dot plot analysis of T cells transfected with either Empty vector, POLD1 WT or POLD1 R1060C lentiviral plasmid. Data are representative of three experiments. G. Frequencies of BrdU+ T transfected with either Empty vector, POLD1 WT or POLD1 R1060C lentiviral plasmid in F. (n=3; open circles). Results are means ± S.E.M. ****, p<0.0001; ns, not significant, by unpaired t test in B and D; *, p<0.05; ns, not significant, by one-way ANOVA with Bonferroni post-test analysis in G.
Increased frequency of memory T cells in POLD1R1060C subjects.
In view of their recurrent infections and immune disorder phenotypes, we further examined the cohort of POLD1R1060C subjects for cell abnormalities. Consistently, all three POLD1R1060C subjects showed sharp skewing of peripheral CD4 and CD8 T cell towards a memory (CD45RA−CD45RO+) phenotype (Fig 4, A-D). The frequencies of naïve CD45RA+CCR7+ CD4+T cells and especially CD8+T cells were profoundly decreased in POLD1R1060C subjects, whereas the frequencies of CD45RA−CCR7−CD4+ and CD8+ T cells [CD45RA− effector memory (TEM)] were increased. The frequencies of CD45RA+CCR7− CD8+ [CD45RA+ effector memory (TEMRA)] T cells were similar in patient and control subjects, while the CD4+ TEMRA were marginally increased in patients. The frequencies of CD45RA−CCR7+ CD4+ [central memory (TCM)] T cells and CD8+ T cells were mildly decreased compared to healthy control group without achieving statistical significance. (Fig 4, E-H).
Figure 4. Increased frequency of memory T cells in POLD1R1060C subjects.

A. Flow cytometric analysis of circulating CD45RO−CD45RA+ and CD45RO+CD45RA− CD4+ T cells in a control and a POLD1R1060C subject. B. Frequency of activated CD45RO+CD45RA− within the peripheral blood CD4+ T cell pool of controls and POLD1R1060C subjects. C. Flow cytometric analysis of circulating CD45RO−CD45RA+ and CD45RO+CD45RA− CD8+ T cells in a control and a POLD1R1060C subject. D. Frequency of activated CD45RO+CD45RA− within the peripheral blood CD8+ T cell pool of controls and POLD1R1060C subjects. E. Flow cytometric analysis of circulating TEMRA CD45RA+CCR7− and naïve CD45RA+CCR7+ CD4+ T cells in a control and a POLD1R1060C subject. F. Frequency of circulating naïve CD45RA+CCR7+, central memory CD45RA−CCR7+, effector memory CD45RA−CCR7− and TEMRA CD45RA+CCR7− within the peripheral blood CD4+ T cell pool of controls and POLD1R1060C subjects. G. Flow cytometric analysis of circulating TEMRA CD45RA+CCR7− and naïve CD45RA+CCR7+ CD8+ T cells in a control and a POLD1R1060C subject. H. Frequency of circulating naïve CD45RA+CCR7+, central memory CD45RA−CCR7+, effector memory CD45RA−CCR7− and TEMRA CD45RA+CCR7− within the peripheral blood CD8+ T cell pool of controls and POLD1R1060C subjects. ns, not significant; *, p<0.05; ***, p<0.001 by Student’s unpaired two tailed t test.
Flow cytometric analysis of the regulatory T cells (Treg) in the peripheral blood of POLD1R1060C patients showed a mild but non-significant increase in the distribution of CD4+CD25hiCD127lo T cells (Fig E3, A and B in the Online Repository). The percentage of induced Treg was found similar in POLD1R1060C patients and controls (Fig E3, C and D in the Online Repository). The expression of several canonical Treg cell markers, including Foxp3, CTLA-4, and Helios, was normal in patients compared to controls (Fig E3, E and F in the Online Repository). Overall, these results establish a predominance of effector memory T cell subpopulations in POLD1R1060C subjects in the context of T cell lymphopenia.
Restricted T cell receptor (TCR) V-J pairing in POLD1R1060C CD8+T cells.
Given that the POLD1R1060C mutation gave rise to severe T lymphopenia with effector T cell skewing, we examined the patient CD4 and CD8 T cell populations for evidence of abnormalities in their T cell receptor repertoire. Specifically, we asked if POLD1 may play a role in the nonhomologous end joining as reflected in VDJ recombination. Accordingly, we analyzed TRB of cell-sorted CD4+ and CD8+ T cells, and IGH of CD19+ B cells for evidence of VDJ recombination defects and oligoclonality. Repertoire analysis showed that the V-J pairing patterns and productive entropy (a measure of clonal diversity) were similar between POLD1R1060C and control CD4+ T cells and B cells (Fig 5, A and B, and Fig E4, A and B in the Online Repository). In contrast, the POLD1R1060C CD8+T cell subset displayed restricted V-J gene combinations and lower productive entropy (Fig 5, A and B). Analysis of the productive frequencies of top 100 as well as the total clonotypes further revealed severe oligoclonality among patient CD8+ but not CD4+ T cells (Fig 5, C and D). Altogether, these observations suggested a selective impact of POLD1R1060C on CD8 T cell clonal expansion in the periphery and possibly an additional effect during thymic development.
Figure 5. Restricted usage of Vβ and Jβ genes in POLD1R1060C T Cells.

A. Frequencies of specific Vβ and Jβ pairing in unique TRB clonotypes of CD8+T cells and CD4+Tcells from healthy controls and patients. White represents the absence of a given Vβ and Jβ pairing. Blue reflects a low frequency while red represents a higher frequency of usage. B. Productive entropy of CD8+ and CD4+ T cells in healthy controls versus patients (n=3; open circles). C. Productive frequencies of top 100 most abundant clonotypes of CD8+ and CD4+ T cells in healthy controls versus patients. D. Tree map of clonality in CD8+ and CD4+ T cells. Each square represents a specific V-J pairing. The area of each square is proportional to the total frequency of the corresponding TRB clonotypes in each sample. ns, not significant; *, p<0.05; **, p<0.01 by Student’s unpaired two tailed t test.
To determine whether POLD1 is involved in somatic hypermutation (SHM), we analyzed SHM patterns in B cells. We found no difference between patients and controls in the IGHV-IGHJ pairing, D gene usage, productive entropy and the proportion of unique IGH rearrangements carrying at least one mutation (Fig E4, A-D in the Online Repository). The rate of SHM in the V segment was similar in both of the productive IGH rearrangements and non-productive IGH rearrangements of POLD1R1060C patients (Fig E4, E in the Online Repository). Finally, the in silico translated amino acid sequences of the IGH CDR3 region no differences in length and tyrosine contents between POLD1R1060C patients and healthy controls (Fig E4, F and G in the Online Repository), whereas the hydrophobicity analysis showed a borderline statistically significant marginal difference between POLD1R1060C patients and healthy controls (p=0.049, Fig E4, F in the Online Repository). These findings, together with the normal B cell proliferation shown earlier, confirmed that the POLD1R1060C mutation spared the B cell compartment.
Discussion
In this study, we report a homozygous missense mutation in POLD1, encoding the catalytic subunit of the ubiquitous DNA polymerase Polδ, in three patients from an extended consanguineous kindred who presented with recurrent infections with T cell lymphopenia and poor antibody responses. Functional and structural analysis indicated that the mutation impaired the interaction of POLD1 with other Polδ subunits, resulting in poor Polδ complex formation and defective DNA replication. Our results thus establish this mutation as a novel cause of primary combined immunodeficiency.
The mutant POLD1R1060C protein failed to associate with Polδ accessory subunit POLD2 or with the DNA replication complex component RFC, necessary for loading PCNA onto DNA to initiate DNA replication34. Structural modeling revealed the mutation disrupted a critical molecular interaction between POLD1 and POLD2, in agreement with the co-precipitation studies showing failure of the mutant POLD1R1060C protein to associate with POLD2. While the mutation is located at the C-terminus of POLD1 away from the catalytic and DNA exonuclease domains, a secondary effect of the mutation on the catalytic function of POLD1 cannot be excluded and requires further investigation. Analysis of patient T cells indicated the POLD1R1060C mutation impaired DNA replication and cell proliferation, an effect that was rescued by lentivirus-mediated expression of a WT but not the mutant POLD1R1060C protein. In contrast, B cell proliferation was spared.
A key finding in our studies is that the POLD1R1060C mutation not only impaired T cell proliferation but was also associated with restricted TCR V-J pairing and severe oligoclonality in CD8+ T cells. Importantly, these abnormalities appeared limited to the CD8+ T cell compartment, and did not involve the CD4+ T cells or the B cells. These findings suggest that Polδ differentially impact CD8 T cells, severely limiting their peripheral expansion and possibly affecting their thymic development. A differential impact of POLD1R1060C on the CD8+ T cell compartment would be consistent with the heightened susceptibility of the patients to herpetic and respiratory viral infections. In contrast, the patients have so far been spared infections associated with severe CD4+ T cell depletion such as Pneumocystis jiroveci. This sparing may reflect the continued presence of diverse, albeit profoundly lymphopenic, CD4 T cell populations and the absence of concurrent debilitating disease(s).
In contrast to the T cell defects noted in the patients, the B cell compartment appeared to be completely spared in terms of B cell proliferation, IgH V-J pairing, D gene usage and somatic hypermutation. Furthermore, indices such as the proportion of tyrosine residue of the IGH-CDR3 region and the IgH hydrophobicity profile, both associated with B cell self-reactivity, were either similar (the former) or marginally affected (the latter). In view of these findings, the presence of mild hypogammaglobulinemia in the patients with waning antibody responses to vaccines that can be rescued by booster immunization is best explained by suboptimal T cell help.
Previous studies have identified different heterozygous mutations affecting the catalytic and exonuclease domains of POLD1 17, 35, 36. The former gives rise to a distinct multisystem developmental characterized by subcutaneous lipodystrophy, deafness, mandibular hypoplasia, while the latter results in familial colorectal cancers. Interestingly, neither of these two types of POLD1 mutations resulted in immunodeficiency. In contrast, the kindred reported herein had very minimal manifestations aside from the immunodeficiency, notably partial sensorineural hearing loss in P1. This sharp segregation of disease phenotypes as a function of the respective domains targeted by mutations suggests the POLD1R1060C mutation, which locates away from the enzymatic domains, may allow for residual Polδ activities to rescue the multisystem and mutator phenotypes associated with the catalytic and proof-reading activities in different tissues. However, the Polδ complex may play a non-redundant role in protein-protein interactions with other components of the double stranded break repair (DSBR) relevant to VDJ recombination in CD8+T cells. The nature of such interactions involving the Polδ complex in DSBR and their role in T cell development requires further investigation.
In addition to the POLD1 mutation reported herein, a splice junction mutation in POLE2, encoding the DNA polymerase epsilon subunit 2 (POLE2) has been described in a child with dysmorphic features and combined immunodeficiency and autoimmunity, with absence of circulating B cells, T cell lymphopenia and neutropenia 37. POLE2 is an accessory subunit in the Polε complex38, and while the protein expression of mutant POLE2 was unaffected in the child, the mutation may adversely affect the Polε complex assembly and/or its interactions with other proteins relevant to DNA replication. The TRB repertoire in the POLE2 patient appeared largely normal, indicating that the restricted V-J pairing in the TRB of CD8+ T cells in the POLD1R1060C patients was specific to this gene defect. Overall, these results point to an emerging subgroup of primary immunodeficiency disorders caused by genetic lesions in different DNA polymerases. The full spectrum of these disorders and their unique and shared attributes require further investigation.
Supplementary Material
Key Messages:
A POLD1 mutation disrupted the assembly of the Polδ complex and resulted in T cell immunodeficiency.
Patient CD8+ T cells exhibited severe oligoclonality and restricted T cell receptor beta chain V-J pairing.
Acknowledgements
This work was supported by National Institutes of Health grants 5R01AI085090 and 5R01AI128976 (to T.A.C.)
Abbreviations.
- BrdU
Bromodeoxyuridine / 5-bromo-2'-deoxyuridine
- LRTI
lower respiratory tract infection
- PCNA
Proliferating Cell Nuclear Antigen
- Polδ
DNA polymerase delta
- POLA1
DNA polymerase alpha 1 catalytic subunit
- POLBc
DNA polymerase type B family catalytic domain
- POLD1
DNA polymerase delta 1 catalytic subunit
- RFC
Replication Factor C
- TCR
T cell receptor
- IGH
Immunoglobulin heavy chain
- Teff
T effector cells
- Treg
Regulatory T cells
- URTI
upper respiratory tract infection
- WES
Whole exome sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare no conflict of interest.
References
- 1.Coleman WB, Tsongalis GJ. Multiple mechanisms account for genomic instability and molecular mutation in neoplastic transformation. Clin Chem 1995; 41:644–57. [PubMed] [Google Scholar]
- 2.Chilkova O, Stenlund P, Isoz I, Stith CM, Grabowski P, Lundstrom EB, et al. The eukaryotic leading and lagging strand DNA polymerases are loaded onto primer-ends via separate mechanisms but have comparable processivity in the presence of PCNA. Nucleic Acids Res 2007; 35:6588–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greenough L, Menin JF, Desai NS, Kelman Z, Gardner AF. Characterization of family D DNA polymerase from Thermococcus sp. 9 degrees N. Extremophiles 2014; 18:653–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henneke G, Flament D, Hubscher U, Querellou J, Raffin JP. The hyperthermophilic euryarchaeota Pyrococcus abyssi likely requires the two DNA polymerases D and B for DNA replication. J Mol Biol 2005; 350:53–64. [DOI] [PubMed] [Google Scholar]
- 5.Podust VN, Chang LS, Ott R, Dianov GL, Fanning E. Reconstitution of human DNA polymerase delta using recombinant baculoviruses: the p12 subunit potentiates DNA polymerizing activity of the four-subunit enzyme. J Biol Chem 2002; 277:3894–901. [DOI] [PubMed] [Google Scholar]
- 6.Makarova KS, Krupovic M, Koonin EV. Evolution of replicative DNA polymerases in archaea and their contributions to the eukaryotic replication machinery. Front Microbiol 2014; 5:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song J, Hong P, Liu C, Zhang Y, Wang J, Wang P. Human POLD1 modulates cell cycle progression and DNA damage repair. BMC Biochem 2015; 16:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uchimura A, Hidaka Y, Hirabayashi T, Hirabayashi M, Yagi T. DNA polymerase delta is required for early mammalian embryogenesis. PLoS One 2009; 4:e4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee MY, Zhang S, Lin SH, Wang X, Darzynkiewicz Z, Zhang Z, et al. The tail that wags the dog: p12, the smallest subunit of DNA polymerase delta, is degraded by ubiquitin ligases in response to DNA damage and during cell cycle progression. Cell Cycle 2014; 13:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tumini E, Barroso S, Calero CP, Aguilera A. Roles of human POLD1 and POLD3 in genome stability. Sci Rep 2016; 6:38873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murga M, Lecona E, Kamileri I, Diaz M, Lugli N, Sotiriou SK, et al. POLD3 Is Haploinsufficient for DNA Replication in Mice. Mol Cell 2016; 63:877–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Y, Meng X, Zhang S, Lee EY, Lee MY. Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PLoS One 2012; 7:e39156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strzalka W, Ziemienowicz A. Proliferating cell nuclear antigen (PCNA): a key factor in DNA replication and cell cycle regulation. Ann Bot 2011; 107:1127–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daee DL, Mertz TM, Shcherbakova PV. A cancer-associated DNA polymerase delta variant modeled in yeast causes a catastrophic increase in genomic instability. Proc Natl Acad Sci U S A 2010; 107:157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Church DN, Briggs SE, Palles C, Domingo E, Kearsey SJ, Grimes JM, et al. DNA polymerase epsilon and delta exonuclease domain mutations in endometrial cancer. Hum Mol Genet 2013; 22:2820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu D, Frederiksen JH, Liberti SE, Lutzen A, Keijzers G, Pena-Diaz J, et al. Human DNA polymerase delta double-mutant D316A;E318A interferes with DNA mismatch repair in vitro. Nucleic Acids Res 2017; 45:9427–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet 2013; 45:136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weedon MN, Ellard S, Prindle MJ, Caswell R, Lango Allen H, Oram R, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet 2013; 45:947–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sasaki H, Yanagi K, Ugi S, Kobayashi K, Ohkubo K, Tajiri Y, et al. Definitive diagnosis of mandibular hypoplasia, deafness, progeroid features and lipodystrophy (MDPL) syndrome caused by a recurrent de novo mutation in the POLD1 gene. Endocr J 2018; 65:227–38. [DOI] [PubMed] [Google Scholar]
- 20.Schmitz-Abe K, Li Q, Rosen SM, Nori N, Madden JA, Genetti CA, et al. Unique bioinformatic approach and comprehensive reanalysis improve diagnostic yield of clinical exomes. Eur J Hum Genet 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Case DAea. AMBER 14. University of California, San Francisco: 2014. [Google Scholar]
- 22.Berendsen HJC, Grigera JR, Straatsma TP. The Missing Term in Effective Pair Potentials. J Phys Chem 1987; 91:6269–71. [Google Scholar]
- 23.Joung IS, Cheatham TE. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B 2008; 112:9020–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berendsen HJC, Postma JPM, Vangunsteren WF, Dinola A, Haak JR. Molecular-Dynamics with Coupling to an External Bath. J. Chem. Phys 1984; 81:3684–90. [Google Scholar]
- 25.Ryckaert J-P, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys 1977; 23:327–41. [Google Scholar]
- 26.Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol 2012; 882:569–604. [DOI] [PubMed] [Google Scholar]
- 27.Carlson CS, Emerson RO, Sherwood AM, Desmarais C, Chung MW, Parsons JM, et al. Using synthetic templates to design an unbiased multiplex PCR assay. Nat Commun 2013; 4:2680. [DOI] [PubMed] [Google Scholar]
- 28.Robins HS, Campregher PV, Srivastava SK, Wacher A, Turtle CJ, Kahsai O, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009; 114:4099–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 2015; 31:3356–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia-Diaz M, Bebenek K. Multiple functions of DNA polymerases. CRC Crit Rev Plant Sci 2007; 26:105–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doublie S, Zahn KE. Structural insights into eukaryotic DNA replication. Front Microbiol 2014; 5:444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li CJ. Flow Cytometry Analysis of Cell Cycle and Specific Cell Synchronization with Butyrate. Methods Mol Biol 2017; 1524:149–59. [DOI] [PubMed] [Google Scholar]
- 33.Dolbeare F, Gratzner H, Pallavicini MG, Gray JW. Flow cytometric measurement of total DNA content and incorporated bromodeoxyuridine. Proc Natl Acad Sci U S A 1983; 80:5573–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Podust VN, Tiwari N, Stephan S, Fanning E. Replication factor C disengages from proliferating cell nuclear antigen (PCNA) upon sliding clamp formation, and PCNA itself tethers DNA polymerase delta to DNA. J Biol Chem 1998; 273:31992–9. [DOI] [PubMed] [Google Scholar]
- 35.Bertocci B, De Smet A, Berek C, Weill JC, Reynaud CA. Immunoglobulin kappa light chain gene rearrangement is impaired in mice deficient for DNA polymerase mu. Immunity 2003; 19:203–11. [DOI] [PubMed] [Google Scholar]
- 36.Bertocci B, De Smet A, Weill JC, Reynaud CA. Nonoverlapping functions of DNA polymerases mu, lambda, and terminal deoxynucleotidyltransferase during immunoglobulin V(D)J recombination in vivo. Immunity 2006; 25:31–41. [DOI] [PubMed] [Google Scholar]
- 37.Lieber MR. The polymerases for V(D)J recombination. Immunity 2006; 25:7–9. [DOI] [PubMed] [Google Scholar]
- 38.Elsayed FA, Tops CMJ, Nielsen M, Ruano D, Vasen HFA, Morreau H, et al. Low frequency of POLD1 and POLE exonuclease domain variants in patients with multiple colorectal polyps. Mol Genet Genomic Med 2019; 7:e00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fiorillo C, D'Apice MR, Trucco F, Murdocca M, Spitalieri P, Assereto S, et al. Characterization of MDPL Fibroblasts Carrying the Recurrent p.Ser605del Mutation in POLD1 Gene. DNA Cell Biol 2018. [DOI] [PubMed] [Google Scholar]
- 40.Frugoni F, Dobbs K, Felgentreff K, Aldhekri H, Al Saud BK, Arnaout R, et al. A novel mutation in the POLE2 gene causing combined immunodeficiency. J Allergy Clin Immunol 2016; 137:635–8 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraszewska J, Garbacz M, Jonczyk P, Fijalkowska IJ, Jaszczur M. Defect of Dpb2p, a noncatalytic subunit of DNA polymerase varepsilon, promotes error prone replication of undamaged chromosomal DNA in Saccharomyces cerevisiae. Mutat Res 2012; 737:34–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.