

Microglia respond to white matter injury with different populations that prevent macrophage dispersion into spared tissue.
Abstract
Microglia and infiltrating macrophages are thought to orchestrate the central nervous system (CNS) response to injury; however, the similarities between these cells make it challenging to distinguish their relative contributions. We genetically labeled microglia and CNS-associated macrophages to distinguish them from infiltrating macrophages. Using single-cell RNA sequencing, we describe multiple microglia activation states, one of which was enriched for interferon associated signaling. Although blood-derived macrophages acutely infiltrated the demyelinated lesion, microglia progressively monopolized the lesion environment where they surrounded infiltrating macrophages. In the microglia-devoid sciatic nerve, the infiltrating macrophage response was sustained. In the CNS, the preferential proliferation of microglia and sparse microglia death contributed to microglia dominating the lesion. Microglia ablation reversed the spatial restriction of macrophages with the demyelinated spinal cord, highlighting an unrealized macrophages-microglia interaction. The restriction of peripheral inflammation by microglia may be a previously unidentified mechanism by which the CNS maintains its “immune privileged” status.
INTRODUCTION
Injury and diseases of the central nervous system (CNS) are ubiquitously associated with microglia and infiltrating macrophage activation. Despite their pervasive representation in CNS disorders, it is still unclear whether these cells are responding to or aggravating CNS insults and whether they are serving similar or different roles. Microglia and infiltrating macrophages are required during spontaneous remyelination (1) but are also detrimental by mediating autoimmune injury (2).
Further complicating the role of microglia and infiltrating macrophages are the remarkable similarities between these two distinct cell populations in diseased states (3, 4). In the uninjured state, microglia express not only classical macrophage markers such as CD45 but also several apparent microglia-specific genes, such as Tmem119, P2RY12, and Fcrls (3, 4). However, these apparent microglia-specific genes, and others, appear to decrease following disease (5). CNS-infiltrating macrophages adopt a microglia-like phenotype after exposure to the CNS of microglia-deficient mice (6). The highly plastic nature of microglia and infiltrating macrophages makes it challenging to specifically track and manipulate activated microglia and understand their specific roles with any certainty. Recently, the inducible Cre recombinase under the CX3CR1 promoter (2, 7) was demonstrated to permanently label and fate-map highly plastic microglia during their activation despite their dynamic changes in gene expression. This approach relies on the fact that bone marrow–derived macrophages (BMDM) do not retain reporter protein expression because of their ongoing replacement. In contrast, microglia turnover relies on self-renewal resulting in the long-term presence of reporter protein expression (8).
Using this targeted fate mapping strategy, we have characterized the activation profile of microglia. We demonstrate, on a single-cell level, distinct subpopulations of activated microglia, including a defined subtype with increased interferon (IFN) signaling, following lysophosphatidylcholine (LPC)–induced demyelination of the spinal cord. We found that CNS-infiltrating macrophages were far less prominent than microglia in the lesion suggesting that, until now, activated microglia have been largely misidentified as CNS-infiltrating macrophages in this demyelinating model. The extent of this misidentification across other diseases and injury models is unclear. Microglia also appear to regulate macrophage numbers within the demyelinating lesion environment as evidenced by an increase in CNS-infiltrating macrophage numbers following microglia ablation.
RESULTS
Fate mapping as a tool to specifically label microglia
Given the difficulty in distinguishing microglia from infiltrating macrophages, we used the CX3CR1creER line crossed with the Rosa26tdTom (tdTom) reporter line. Postnatal administration of tamoxifen enabled permanent labeling of microglia with tdTomato. This approach also labels meningeal and perivascular macrophages in the CNS (9), referred to here as CNS-associated macrophages (CAMs). Although postnatal tamoxifen injection in CX3CR1creER; Rosa26tdTom mice also induced tdTomato expression in leukocytes and monocytes, by 4 weeks, only rare tdTom+ leukocytes or monocytes could be detected (Fig. 1, A to D). To investigate whether leukocyte expression of tdTom changes after injury, we induced focal demyelination in the spinal cord by injecting LPC, which induces a primary myelinopathy resulting in demyelination and relative sparing of axons (10) that remyelinate 14 to 21 days after injection (11). Expression of tdTom remained low in the blood 7 days after LPC-induced injury (1.54% of CD45+ cells expressed tdTom; Fig. 1E). Microglia/CAMs (tdTom+IBA1+) were, therefore, marked while minimally labeling sources of CNS-infiltrating macrophages during CNS injury. We found a small percentage of monocytes/macrophages in the spleen and lymph node that express tdTom (3 to 5%; fig. S1), but these cells were not recruited into the blood in appreciable numbers following LPC-induced demyelination (Fig. 1E).
Fig. 1. Fate mapping as a tool to specifically label microglia/CAMs.
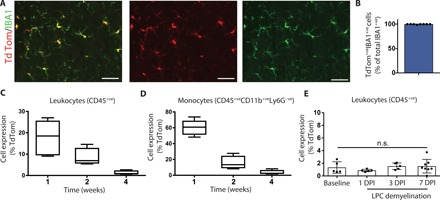
(A and B) Representative immunohistochemical images of the uninjured spinal cord demonstrated very high recombination efficiency at 4 weeks after tamoxifen (tdTom reporter, red; IBA1, green) (A). This observation was also reflected in quantification (B). (C and D) Flow quantification of reporter expression in the blood of uninjured mice demonstrated a progressive reduction in tdTom+ leukocytes (C) or monocytes (D). (E) Flow quantification of the blood of injured mice demonstrated no difference in reporter expression in CD45+ leukocytes. n = 4 (B), n = 3 to 4 (C to E). Error bars indicate ± SEM. DPI, days post-LPC injection. Scale bars, 25 μm.
Common markers to distinguish microglia from CNS-infiltrating macrophages are less sensitive after microglia activation
Using genetic fate mapping with CX3CR1creER; Rosa26tdTom mice, we measured two common distinguishing markers: CD45 that is high in leukocytes and infiltrating macrophages, compared to microglia (12); and CX3CR1, which is enriched in microglia (13). Following injury, we found that Lyve1+ meningeal or perivascular macrophages did not penetrate deep into the parenchyma (fig. S2), suggesting that these cells either minimally contribute following LPC-mediated demyelination or that they down-regulate Lyve1. To assess CD45 changes in microglia after injury, we measured CD45 expression using immunohistochemistry (fig. S3, A to D) and flow cytometry at 7 days after LPC (figs. S3, E to M, and S4). Although the average maximum intensity of CD45 protein per cell was higher in CNS-infiltrating macrophages, both CNS-infiltrating macrophages (CD45+tdTom−) and activated microglia (CD45+tdTom+) expressed high CD45 levels (fig. S3, A to D), making it difficult to differentiate these different cell types based on immunohistochemical detection of CD45 expression alone. Flow cytometry revealed a trend toward greater CD45 intensity in tdTom+ cells in the injured compared to uninjured spinal cord tissue (fig. S3, K to M). In addition, CX3CR1 levels were down-regulated in activated microglia (figs. S3, H and I, and S5), with no difference in CD11b or Ly6C mean intensity (fig. S3J). The CX3CR1creER; Rosa26tdTom mice also expressed enhanced yellow fluorescent protein (EYFP) under the CX3CR1 promoter. As expected, we find that EYFP+ cells and tdTom+ fate-mapped cells overlap (fig. S5). We also find that after injury, tdTom expression remains high, and EYFP expression is reduced in some activated microglia, suggesting that microglia express lower levels of CX3CR1 when they become activated. These results highlight that classic microglia markers (CX3CR1+CD45low) become less sensitive for activated microglia, emphasizing the usefulness of fate mapping strategies to measure and monitor microglia/CAMs in injury conditions.
Single-cell transcriptional profiling of microglia activation after LPC demyelination
Microglia activation is often considered as any morphological or biochemical change from the naïve state, but activation may be multifaceted or tailored to each specific disease or injury state. With the capacity to distinguish microglia/CAMs from CNS-infiltrating macrophages, we sought to more accurately understand the process of microglia/CAM activation by performing single-cell transcriptomic analysis on microdissected naïve and demyelinating sections of spinal ventral horn (Fig. 2). We fluorescence-activated cell sorted (FACS)–purified tdTom+ microglia/CAM from 5 days after lesion induction—to understand the early lesion response—or from naïve adult CX3CR1creER; Rosa26tdTom mice and prepared single-cell RNA-sequencing (RNA-seq) libraries for each sample in parallel using 10× Chromium technology and v2 Chemistry (Fig. 2A). Sequencing metrics for each library, quality control measures, and downstream analysis are described in the Supplementary Materials (fig. S6).
Fig. 2. Single-cell transcriptome-wide profiling of adult microglia/CAMs.
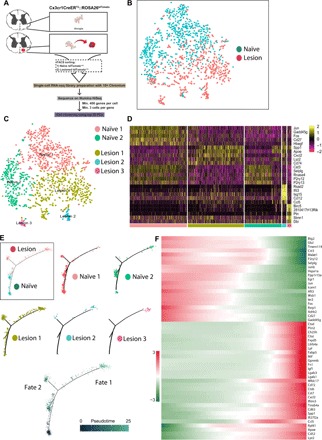
(A) FACS purification and single-cell sequencing of microglia/CAMs from adult CX3CR1creER; Rosa26tdTom mice. (B) Unsupervised graph-based clustering of single-cell RNA-seq dataset projected onto a tSNE plot. Most tdTom+ microglia/CAMs from lesioned sample aggregated distinctly away from tdTom+ microglia/CAMs from naïve sample (B, box) and formed distinct clusters (lesions 1, 2, and 3) compared to naïve (naïve 1 and 2) (C). (D) Heat map of top 10 marker genes (determined by likelihood-ratio test) for each of the five clusters. (E) Pseudotime two-dimensional minimal spanning tree clustering to understand potential intermediary microglial/CAMs states. Each dot represents single cells ordered in pseudotime color-coded either by sample (box) or by Seurat-state (naïve 1 or 2, lesion 1, 2, or 3) or pseudotime state (blue). The line connecting the dots outlines a path of transcriptional relatedness, which represents fate trajectory. Uninjured naïve branch was set as the root state to specify the start point of the trajectory (dark blue). There was a bifurcation into two subpopulations (cell fates 1 and 2) emanating after injury (light blue). (F) Top 50 pseudotime-dependent genes calculated from the full trajectory seen in (E) (q value, <1 × 10−27). Legend represents arbitrary units based on the order of single cells (the genes representing cells in the most extreme states are darker in color and are assigned a value of ±3).
First, we examined the microglia/CAM response by conducting unsupervised graph-based clustering [using the top 20 principal components (PCs)] and projected them onto a t-distributed stochastic neighbor embedding (tSNE) plot. Our analysis identified five unique cell clusters (Fig. 2C). Of these clusters, two contained cells primarily from the naïve sample (naïve 1 and 2), and three clusters were largely composed of cells from the injury sample (lesions 1, 2, and 3). We found that most microglia/CAM from the demyelinated injury sample clustered distinctly apart from the naïve cells (Fig. 2C). One cluster (lesion 1), however, was composed of cells from both the naïve and injury sample. Using markers established by Jordão and colleagues (14), we find that resident perivascular macrophages and meningeal macrophages make up only a small percentage of cells in our analysis and are predominantly found in the lesion 1 cluster (fig. S7). The lesion 1 cluster was enriched with Apoe, a gene that is commonly elevated within microglia across disease conditions (15) (Fig. 2D). We also confirmed that Apoe was up-regulated at the lesion site using in situ hybridization and immunohistochemistry (fig. S8). The lesion 1 cluster was also characterized by an increase in Spp1, or osteopontin, which is an extracellular matrix (ECM) protein that can promote microglia survival (16), and the chemokine Cxcl2. Cells in the lesion 1 cluster cells also expressed the lysosomal Lyz2, and Cd74, which play a role in the intracellular sorting of major histocompatibility complex II (17).
Lesion 2 cluster was enriched in genes associated with IFN signaling such as Ifit3b, Ifit3, Irf7, Usp18, Ifitm3, and the IFN response gene Cxcl10. IFN is classically related with antiviral activity (18) and is also associated with a small subset of microglia in the aged brain (19). Cluster 2 cells also expressed the chemokines monocyte chemotactic protein 5 (or Ccl12) and RANTES (or Ccl5) (Fig. 2D). The preferential increase of IFN elements into one subset suggests that there are different states of microglia activation following LPC demyelination, and one of these states is enriched for IFN signaling. Cluster 3 expressed genes associated with cycling microglia, including Birc5 and Stmn1 (20), suggesting that they are a proliferating pool of microglia. None of the activated clusters were preferentially enriched in M1-like proinflammatory and M2-like regulatory markers (21, 22), but instead each contained a mixture of these genes (fig. S9). Notably absent, we find no expression of iNOS (Nos2) in microglia after LPC demyelination. We do find enrichment for disease-associated microglia (DAM) markers such as Apoe, Cst7, and Spp1 that were recently characterized from Alzheimer’s disease tissue and animal models (5). Our identified injury-associated microglia are therefore likely related to these recently identified DAMs.
To further distinguish the injury-activated microglia from their naïve counterparts and to understand potential intermediary cell states, we used Monocle’s pseudotime ordering algorithm [R package (23)]. This machine learning–based approach compares all single-cell transcriptome profiles to each other in a multidimensional space and compresses the output to a two-dimensional minimal spanning tree. Each dot represents single microglia ordered in “pseudotime,” and the line connecting the dots outlines a path of transcriptional relatedness, which can represent a fate trajectory. Application of Monocle’s algorithm yielded a clear trajectory, where two distinct subtypes of injury-associated cells were apparent after injury (cell fates 1 and 2; Fig. 2E). This separate analysis therefore reaffirmed that microglia activation occurred in several states. Seurat-based algorithm clustering found that cells in lesion 1 could be further differentiated with Monocle-based analysis to reveal that a large proportion of these cells had different fates, equally distributed across fates 1 and 2. Likewise, lesion 2 clustered cells were found in fates 1 and 2. However, cells in the lesion 3 cluster were entirely destined to fate 2. The top 50 pseudotime-dependent genes (q value, <1 × 10−27) were plotted to examine expression changes along this trajectory. Cells from the naïve sample were largely at or close to pseudotime 0 (darker blue), and cells from the injured sample, especially cells from the lesion 3 cluster, were largely found further along the trajectory, toward pseudotime 25 (lighter blue) (Fig. 2, E and F). Notably, naïve microglia were enriched with classically defined homeostatic microglia markers such as P2ry12 and Tmem119 (Fig. 2, D and F) suggesting that we had successfully enriched for microglia (3, 4, 14, 19, 20). Injury-associated microglia decreased expression of several “homeostatic” genes such as Siglech, Sall1, and Tmem119 (Fig. 2E). Consistent with increased CD45 protein expression in activated microglia, we also find, by single-cell sequencing, increased expression of Ptprc—the gene for CD45—in activated microglial clusters (fig. S6).
Since injury-activated microglia acquire two distinct post-injury phenotypes, we performed branched expression analysis modeling to examine expression changes underlying transcriptional divergence between the two branches distinguishing fate 1 from fate 2 (false discovery rate, 1%). This analysis revealed that genes such as Cd69, Ifitm3/1, Cd9, and Cd72 exhibited branch-dependent enrichment toward one lineage (cell fate 1), whereas genes such as Il1b, Cxcl2, Ccl7, Tgif1, and Tlr2 exhibited enrichment toward the other (cell fate 2) (fig. S10A). We also performed pathway and gene set overdispersion analysis (PAGODA) to identify common gene sets across cells and decipher gene ontology (GO) annotations for these gene sets. This algorithm performs weighted PC analysis and highlights the gene sets for which variance explained by the first PC exceeds genome-wide background expectation. The rows of the dendrogram reflect the top five significant aspects of heterogeneity (P < 0.05) on the basis of gene sets defined by GO annotations. We noted defined subsets of cells clustering together that had gene sets consistent with IFN activation (aspect 2) or mitosis (aspect 3) (fig. S10, B and C). Together, using multiple unbiased analytical approaches, our single-cell RNA-seq dataset strongly suggests a distinct microglial subtype within the activated microglia population that are enriched in IFN signaling.
To explore microglia regulation, we also performed single-cell regulatory network inference and clustering (SCENIC) analysis, which revealed distinct gene regulatory networks (GRN) in uninjured versus injury-associated cells (Fig. 3) (24). A GRN is defined as a set of activated transcription factors (TFs) and cofactors that govern downstream gene expression to enact distinct cell states. In 1284 microglia that passed quality control filters, we identified 245 regulons that were predicted to be active, of which, 21 were mostly specific to injury-activated microglia (Fig. 3). For example, we found that the type I IFN response genes Irf7 and Irf9 were enriched in lesion-activated microglia and the hypoxia sensor Hif1a. To confirm gene expression of TFs predicted to be active within microglia subsets [as their raw unique molecular identifier (UMI) counts were sparse], we imputed the raw gene-barcode matrix using Markov affinity–based graph imputation of cells (MAGIC) to restore expression of transcripts that may have suffered from systematic undersampling of single transcriptomes (25). This analysis predicted expression (albeit at low levels for TFs such as Zfp110) for GRNs predicted by SCENIC. Notably, more regulons were active in naïve microglia (224 regulons) than in lesion-activated microglia (21 regulons), suggesting that homeostasis required a more complex transcriptional regulation than microglia activation (Fig. 3D). Together, we find that microglia activation involves a state change that is associated with changes in GRN.
Fig. 3. Single-cell GRN profiling of adult microglia/resident macrophage and activated macrophages.
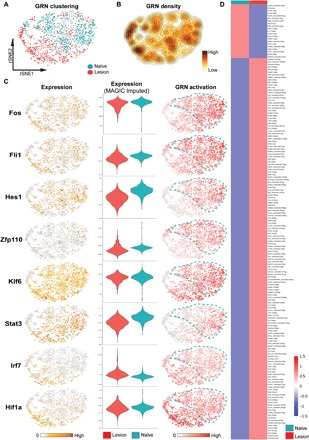
(A) Unsupervised graph-based clustering of single-cell RNA-seq dataset projected onto a tSNE plot comparing microglia isolated from naïve and LPC-lesioned mice (B) GRN density projected onto the tSNE plot. (C) Raw gene expression (left), MAGIC-imputed gene expression (center), and SCENIC analysis (right) demonstrated that injury-associated microglia/resident macrophages were distinct from uninjured conditions. (D) It was revealed that there were 245 regulons, of which several were unique, largely to the uninjured state.
Microglia progressively outnumber macrophages after CNS demyelination
To understand the contribution of microglia after white matter injury, we injected CX3CR1creER; Rosa26tdTom mice with tamoxifen postnatally, and 4 to 6 weeks later when tdTom expression was virtually undetectable in monocytes, we demyelinated the spinal cord by injecting the lipid disrupting agent LPC (10). We compared activated microglia and CNS-infiltrating macrophage dynamics at 3, 5, and 7 days after demyelination (Fig. 4A). Initially, at 3 days after LPC, microglia (CD45+ve; tdTom+ve) and CNS-infiltrating macrophages (CD45+ve; tdTom-ve) increased in density within the demyelinated lesion compared to the uninjured spinal cord (Fig. 4, B to D). Because of the predominance of cells expressing high levels of CD45 within the LPC lesion—previously assumed to be CNS-infiltrating macrophages—we were not expecting to find that by 7 days after demyelination, most of the IBA1+ cells were microglia (~85%) (Fig. 4, H and J). Seven days after LPC corresponds to a time point of active oligodendrocyte differentiation (11), suggesting that the expansion of microglia occurs in conjunction with oligodendrogenesis. As we were not expecting the reduction of CNS-infiltrating macrophages following LPC, we wondered whether elements of the CNS were intrinsically antagonistic toward CNS-infiltrating macrophages. To address this question, we investigated macrophage accumulation in the white matter of the most comparable, non-CNS environment: the peripheral nervous system (PNS). The PNS is an appropriate comparison, as the vast majority (~98%) of resident PNS macrophages, such as microglia, are labeled with tdTom up to 4 to 6 weeks after tamoxifen administration in naïve CX3CR1creER; Rosa26tdTom mice (fig. S11). Like in the CNS, LPC injection into the PNS has been demonstrated to cause defined demyelination (fig. S12) (26). However, unlike the CNS, resident PNS macrophages do not expand after LPC-induced demyelination (Fig. 4, E to G, I, and K). Instead, infiltrating macrophages accumulate, and their numbers increase for the first week following PNS demyelination. The halted accumulation of infiltrating macrophages in the CNS and their expansion in the PNS may suggest that the CNS environment limits macrophage accumulation.
Fig. 4. Microglia progressively outnumber macrophages after CNS demyelination.
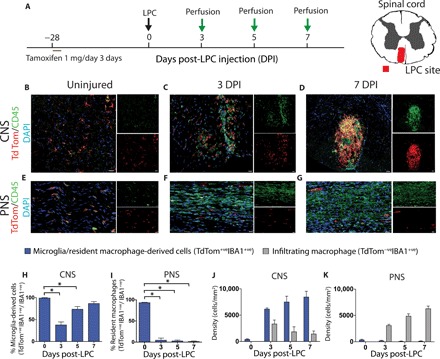
(A) Schematic depicting experimental plan. (B to G) Representative immunohistochemical images of the lesioned spinal cord (CNS; B to D) and sciatic nerve (PNS; E to G) demonstrating widespread reporter expression (red) in the CNS versus the PNS. CD45 (green); 4′,6-diamidino-2-phenylindole (DAPI) (blue). This observation was also reflected in quantification (CNS: H and J; PNS: I and K). n = 4 (0 DPI) or n = 5 to 9 (H to K); analysis of variance (ANOVA) with Tukey’s multiple comparison test (P < 0.05; H and I), two-way ANOVA with Sidak’s multiple comparison test (P < 0.05; J and K). Error bars indicate ± SEM. Scale bars, 20 μm.
Preferential microglia proliferation and limited death account for microglia expansion after CNS demyelinating injury
In this regard, we postulated that the CNS environment may limit the accumulation of CNS-infiltrating macrophages by at least two mechanisms: the selective elimination of infiltrating macrophages or the preferential expansion of activated microglia. We first explored proliferation dynamics of activated microglia and CNS-infiltrating macrophages (Fig. 5). In the CNS, microglia proliferation (% labeled with the proliferative marker Ki67) was ~14× higher than the proliferation of CNS-infiltrating macrophages (Fig. 5, A and C). Only 2.1% of CNS-infiltrating macrophages expressed Ki67, suggesting a virtual absence of proliferation by infiltrating macrophages in the CNS. By comparison, resident macrophages in the PNS expressed Ki67 at roughly the same proportion as infiltrating macrophages (Fig. 5, B and D), suggesting that the PNS environment does not induce preferential proliferation to either macrophage population. The CNS environment is therefore preferentially promoting the proliferation of microglia, which is remarkably distinct from the proliferation of macrophages that occur in the PNS after demyelination. We also examined programmed cell death dynamics in the CNS and PNS. We measured cleaved capase-3 (CC3)—a key effector of apoptosis—and, consistent with infiltrating macrophages in the CNS encountering a hostile CNS environment, we found more apoptosis of infiltrating macrophages in the CNS as compared to the PNS (Fig. 5, E to H).
Fig. 5. Preferential microglia proliferation and infiltrating macrophage apoptosis in the lesioned CNS.
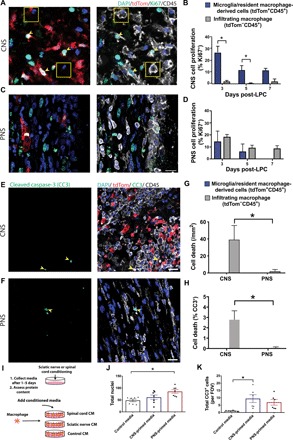
(A and C) Representative immunohistochemical images of the lesioned spinal cord (A) and sciatic nerve (C) demonstrating more proliferative (Ki67+, green) reporter-positive cells (yellow, arrowheads) compared to reporter-negative cells (white, boxes) in the CNS and no difference between cell types in the PNS. This observation was also reflected in quantification (*P < 0.02) (B and D). (E and F) Representative immunohistochemical images of the lesioned spinal cord (E) and sciatic nerve (F) at 3 days after LPC demonstrating more apoptotic (CC3+ green) infiltrating macrophage (CD45+ tdTom−, yellow, arrowheads) in the CNS versus the PNS. This observation was also reflected in quantification (*P < 0.03; G and H). (I to K) Using conditioned media (CM) from spinal cord (CNS) and sciatic nerve (PNS) on BMDM, we demonstrated a reduction in cell numbers (J) and an increase in apoptosis (CC3+ cells; K) in CNS-primed media conditions compared to PNS-primed media. n = 6 to 7, *P < 0.02. (B), n = 3 to 4 (D); two-way ANOVA with Sidak’s multiple comparison test; n = 4 (PNS) and 9 (CNS) two-way ANOVA with Sidak’s multiple comparison test (G and H); n = 6 (two independent experiments) ANOVA with Tukey’s multiple comparison test (I to K); Error bars indicate ± SEM. Scale bar, 20 μm. FOV, field of view.
Secreted factors from the CNS or PNS may account for the observed findings. We tested this by using conditioned media from spinal cords (CNS) and sciatic nerves (PNS) (Fig. 5, I to K) on BMDM, a common model for assessing macrophages, or microglia. CNS-primed media induced heightened apoptosis of BMDM compared to PNS-primed media, while PNS conditions resulted in an increase in BMDM numbers. Together, the environment of the CNS preferentially promotes proliferation of microglia and cell death of infiltrating macrophages.
Microglia exclude macrophages following LPC demyelination of the CNS
The lack of macrophage accumulation in the CNS and not the PNS led us to hypothesize that microglia actively suppress CNS-infiltrating macrophage numbers after CNS demyelination. We first examined the spatial relationship between activated microglia and CNS-infiltrating macrophages following LPC injury (Fig. 6, A to C). When microglia and CNS-infiltrating macrophages began accumulating within the LPC lesion (3 days after LPC), both cell types were dispersed throughout the lesion. However, thereafter, microglia outnumbered CNS-infiltrating macrophages and often appeared in the periphery of the lesion site with CNS-infiltrating macrophages located at the center of the site.
Fig. 6. Microglia limit toxicity and expansion into the spared white matter of CNS-infiltrating macrophages following LPC demyelination.
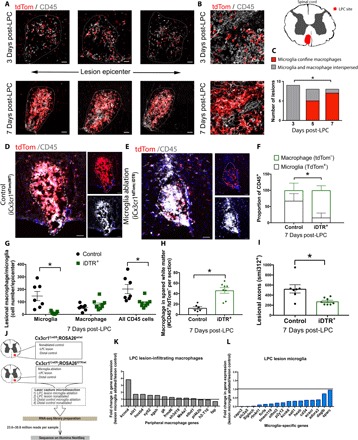
Representative immunohistochemical images of the lesioned spinal cord at 3 and 7 days after LPC (A) demonstrating confinement of monocyte-derived cells (CD45+ tdTom−, white) by microglia (red) apparent by 7 days after LPC (high resolution) (B). This observation was also reflected in quantification as the proportion of lesions with the confinement phenotype (C). Representative immunohistochemical images of the lesioned spinal cord in nonablated iCX3CR1tdTom;WT (D) and DT-ablated iCX3CR1tdTom;iDTR (E) mice with quantification of microglia ablation showing no significant compensation from infiltrating macrophages (F and G). Microglia/CAM ablation demonstrated an increase in CNS-infiltrating macrophages (CD45+ tdTom−) into the spared white matter (H) and a reduction in lesional axons (I). (J) Schematic depicting RNA-seq experimental plan. RNA-seq analysis of laser-captured microdissected lesions revealed that 10 of 13 infiltrating macrophage-associated genes were elevated after microglia ablation in the LPC lesion (K). In contrast, microglia-specific genes were reduced on average of 70% following microglia ablation (L). n = 8 to 9 (C), χ2 test; n = 7 to 8 (J), n = 3 to 4 samples each with two to five pooled animals, Mann-Whitney test; error bars indicate ± SEM. White dashed line is lesion edge. Scale bar, 20 μm. *P < 0.05.
Ablation of microglia increases the accumulation of macrophages in LPC lesions
To investigate how microglia regulate CNS-infiltrating macrophages in acute lesions, we ablated microglia/CAMs. CX3CR1creER mice were crossed with a transgenic line containing an inducible diphtheria toxin (DT) receptor (DTR) (27) and the tdTomato reporter line. Upon Cre-recombinase activity, DTR is inserted into cellular membranes to cause diphtheria-resistant mouse cells to become sensitive to DT. These CX3CR1creER; Rosa26tdTom; Rosa26iDTR (referred to as iCX3CR1tdTom;iDTR) mice or their CX3CR1creER; Rosa26tdTom; Rosa26WT control (iCX3CR1tdTom;WT) were injected with tamoxifen early postnatally—as before—and 12 weeks later, they were injected with DT starting 24 hours following LPC demyelination. Daily DT treatment of iCX3CR1tdTom;iDTR mice reduced the proportion of activated microglia 7 days after LPC demyelination within the lesion epicenter (Fig. 6, D to F).
To determine whether macrophage compensates for the loss of microglia/CAMs, we measured the number of microglia (CD45+tdTom+) and macrophage (CD45+tdTom−) within the lesion epicenter. We found that although microglia numbers were reduced in iCX3CR1tdTom;iDTR mice, there was no change in the number of macrophages within the lesion epicenter (Fig. 6G). We did, however, find that macrophages penetrated into the spared white matter adjacent to the lesion in iCX3CR1tdTom;iDTR mice, while macrophages were minimal in the spared white matter of iCX3CR1tdTom;WT control mice (Fig. 6H). The dispersion of infiltrating macrophages was confirmed with a second experiment where CX3CR1creER;Rosa26iDTR mice were treated with DT or saline (fig. S13). The ablation of microglia/CAMs was more cytotoxic and was associated with axonal loss within the lesion epicenter of iCX3CR1tdTom;iDTR mice compared to control mice (Fig. 6I).
To understand how microglia/CAM loss changes the demyelinated lesion, we also performed RNA-seq on laser-captured microdissected lesions. First, we injected CX3CR1creER;Rosa26iDTR and CX3CR1creER;Rosa26wt control mice with DT daily and subsequently collected tissue from the demyelinated lesion by laser capture microdissection (LCM). Tissue complementary DNA (cDNA) libraries were developed and sequenced (Fig. 6J). On the basis of available literature from expressional data of CNS macrophages from a peripheral origin compared to microglia (6, 28), we developed a candidate list to distinguish peripheral macrophages. Of the 13 peripheral macrophage genes that we included, 10 were elevated after microglia ablation in the LPC lesion (Fig. 6K). In contrast, microglia-specific genes were reduced, on average, by 70% following microglia ablation (Fig. 6L), demonstrating that microglia were adequately ablated within the demyelinated lesion.
To understand what molecular pathways were altered in the absence of microglia, we conducted ingenuity pathway analysis, which predicts changes in known signaling pathways based on gene expression patterns. In corroboration with the single-cell RNA-seq data, we found not only reduced expression of key IFN type I signaling molecules such as Irf7, Stat1, and Stat2 (fig. S14) but also reduction in IFN type II signaling molecules. IFN type II, such as INF-γ, can often oppose IFN type I signaling (29), but here, loss of microglia is associated with reduced IFN type I and II (i.e., IFN-γ) signaling (fig. S15).
DISCUSSION
In this study, we use the CX3CR1creER system to fate-map and selectively ablate microglia/CAMs and their progeny (2, 7). Using these transgenic tools, we make three novel findings: Microglia respond robustly after white matter injury in the spinal cord in multiple distinct activation states; microglia, not infiltrating macrophages, dominate the CNS lesion in response to LPC-induced demyelination; and activated microglia limit the dispersion of CNS-infiltrating macrophages into the spared white matter.
Microglia heterogeneity following acute demyelination
The improved tracking of microglia along with high-dimensional single-cell characterization is redefining the microglia response and providing an understanding of its heterogeneous reaction to injury and disease (5, 19). For example, in the field of Alzheimer’s research, single-cell sequencing of rodent microglia has identified a unique activation state referred to as DAM (5). While the functions of DAMs are unknown, their identification provides the first step in determining their properties.
Understanding microglia reactivity, on a single-cell level, in the CNS regenerative models can provide insights into microglia functions. We investigated the role of microglia in a resolving model of white matter injury induced by the lipid disrupting agent LPC (10). We find multiple activation states of microglia, but when we compare the activation patterns of these states, we find little overlap with proinflammatory “M1” or immunoregulatory “M2” phenotypes (4, 21). Microglia in culture can be skewed toward M1 and M2 phenotypes in the presence of granulocyte-macrophage colony-stimulating factor, IFN-γ and lipopolysaccharide, or macrophage colony-stimulating factor and interleukin-4, respectively (4). Our lack of overlap with classical M1 and M2 markers could therefore suggest that the lesion microenvironment following LPC has other, more prominent regulators of microglia phenotypes. We observe overlap with genes associated with DAMs (5), which are predicted to respond to neurodegenerative molecular patterns. In addition, we also observe overlap with genes identified by Hammond and colleagues in the aged CNS. They identified defined subsets of microglia that were enriched for several IFN response genes (19) consistent with “cluster 2” genes that we identified at 5 days after LPC (Fig. 2). Recently, type I IFN was found to boost microglia repopulation after demyelination, which was required for remyelination (30).
The general applicability of these microglia signatures after LPC injury (here and (19)) and in other disease states will need to be resolved in the future by directly comparing these datasets (5, 14). However, we find different microglia phenotypes in the spinal cord 5 days after LPC as compared to 7 days after LPC in the corpus callosum (19). Whether these differences relate to time after LPC or are due to anatomical differences is unclear. The partial phenotypic overlap between the microglial response following acute demyelination with both DAMs and aging-associated microglia suggests that parallel microglia activation mechanisms exist following demyelination, neurodegeneration, and aging.
With the identification of multiple microglia activation states (5, 19), it raises the question of whether microglia respond to several different disease conditions by forming a unique activation state or a stable and common activation subset. If so, what are the consequences of such microglia diversity? Microglia have an incredible capacity to sense their extracellular environment. Hickman and colleagues determined, using RNA-seq and gene ontology, that microglia express 1299 potential sensome genes representing more than 5% of their transcriptome, thereby enabling their capacity to measure hundreds or even thousands of different molecules (31). How microglia integrate and respond to these complex networks of receptor systems is unclear. One possibility is that because of a convergence in signaling, microglia respond to their sensome by forming a finite number of distinct subsets. However, equally possible is that microglia may respond to each molecule or combination of molecules that it senses in a distinct way. It remains an important task to describe this phenotypic heterogeneity to decipher signatures driving adaptive responses to restore CNS function such as the phagocytosis of inhibitory myelin debris (32) or maladaptive responses such as mediating autoimmune injury (2).
How microglia may limit macrophage dispersion
Here, we made the unexpected observation that in response to CNS injury, there is a specific expansion of microglia but not CNS-infiltrating macrophages. Infiltrating macrophages enter the CNS through damaged or permeable blood vessels within the first few days after LPC in the CNS. After entry into the CNS, they do not proliferate, and their density is gradually reduced, in part because of cell death. The preferential loss of CNS-infiltrating macrophages does not occur in a similar demyelinating insult to the PNS, suggesting that CNS elements prevent peripheral macrophage accumulation. Aspects of these CNS versus PNS differences can be explained by the knowledge that resident macrophages of the PNS are a distinct lineage of myeloid cells from microglia and likely respond to injury differently. Here, we find that microglia become activated, then surround, and confine infiltrating macrophages. The ablation of microglia allows for the dispersion of CNS-infiltrating macrophages into the spared white matter, suggesting that activated microglia interfere with peripheral macrophage dynamics by one or several potential mechanisms including the entry, proliferation, survival, or migration of these cells. Microglia may not directly suppress or kill infiltrating macrophages to permit their dispersion into the spared CNS but may indirectly regulate macrophage. For example, loss of microglia after traumatic spinal cord injury alters the astrocyte response (33), which is important in confining infiltrating macrophages into the lesion epicenter (33, 34).
While the functional consequences of this is still unclear, we hypothesize that microglia restrict macrophage access into the CNS as a means to limit peripheral CNS inflammation or resolve and transition inflammation toward an immunoregulatory phenotype. Limiting macrophage expansion similarly may represent a key mechanism by which the CNS maintains its “immune privileged” status. Whether limiting peripheral inflammation is beneficial in the CNS is unclear and may be context dependent. For example, infiltrating macrophages were recently found to suppress the inflammatory activity of microglia via prostaglandin E2 (PGE2) signaling, which was beneficial following spinal cord injury (35).
Microglia may limit the expansion of CNS-infiltrating macrophages by a number of distinct mechanisms, including the secretion of molecules that either kill, stop proliferation, or prevent infiltration of infiltrating macrophages. From our single-cell sequencing data, we found the increased expression of several molecules that prevent macrophage buildup in other organs including IGF1 (36), LGALS3 (37), and CCL5 (38). With regards to macrophage migration, we found that following microglia ablation, the vast majority of chemokines are down-regulated, suggesting that there is no major increase in chemokines (and most cytokines) in the absence of microglia.
Another factor that affects macrophage migration that is regulated by microglia is the ECM. We find that microglia may protect the ECM of the CNS by expressing the protease inhibitor Timp2, which inhibits several matrix metallopeptidases (MMPs) and ADAM12 peptidase (39). Following inflammation, immune cells secrete proteinases such as those from the MMP family, in part, to cross the basement layer of blood vessels into the CNS parenchyma (40). By expressing and secreting Timp2, activated microglia may prevent the breakdown of basement membrane or other ECM components that restrict the entry of movement of macrophages into and around the CNS. Fibronectin is one such ECM protein enriched within the basement membrane (41). We find that fibronectin is increased in activated microglia (Fig. 2), and it could serve to inhibit macrophage migration. However, certain ECM molecules targeted by Timp2 such as chondroitin sulfate proteoglycans can also promote macrophage migration (42).
Microglia may also limit the expansion of CNS-infiltrating macrophages into the CNS by protecting and repairing the blood-brain barrier (BBB). Microglia rapidly envelope damaged blood vessels, and their ablation or impairing their migration can impede the repair of the BBB (43). In the PNS, the mechanisms that regulate tethering, recruitment, and extravasation are likely distinct, which may account for the larger accumulation of infiltrating macrophages into the PNS after LPC demyelination. If microglia repair the BBB, then microglia ablation may delay BBB repair and allow greater entry of blood leukocytes. Future work will investigate the mechanisms by which activated microglia limit CNS-infiltrating macrophage expansion.
Conclusion
We use a targeted model of microglia fate mapping to characterize the activation state of microglia in acute demyelination and demonstrate that microglia preferentially accrue and monopolize the CNS lesion site, which is in stark contrast to the ongoing accumulation of peripheral macrophages following PNS injury. Microglia within the lesion site respond heterogeneously, forming multiple activation states. These activated microglia in the CNS expand and confine CNS-infiltrating macrophages to minimize their penetration into the spared white matter. Deleting microglia promotes the axonal loss that occurs following LPC demyelination, suggesting that microglia are neuroprotective. These observations may be of particular relevance for understanding the beneficial aspects of neuroinflammation as a means to find directed therapies for white matter diseases.
MATERIALS AND METHODS
Animal information
We initially obtained CX3CR1creER (JAX 021160), Ai9 (Rosa26tdTom; JAX 007905), and Rosa26iDTR (JAX 007900) mice from Jackson mice. Male and female CXCR1CreER; Rosa26tdTom mice were used for LPC in the spinal cord (total n = 24) or sciatic nerve (total n = 11). We investigated CX3CR1creER; Rosa26tdTom; Rosa26iDTR (iCXCR1tdTom;iDTR) mice (n = 9) and CX3CR1creER; Rosa26tdTom; Rosa26WT (iCXCR1tdTom;WT) control mice (n = 7) for histological assessment of cellular ablation. In a second study, we investigated (total n = 27) CXCR1creER; Rosa26iDTR/wt mice to measure histological change after microglia ablation with DT treatment or saline controls. We investigated (n = 16 grouped into four samples for sequencing) CXCR1creER; Rosa26iDTR/wt or (n = 7 grouped into three samples for sequencing) CXCR1creER; Rosa26wt/wt. All animal experiments were conducted in accordance with procedures that were reviewed and approved by animal subcommittees at the University of Calgary.
Animal surgery
Spinal cord injections
We described LPC injections more thoroughly elsewhere (10). Briefly, LPC (0.5-μl volume at 1%; L1381, Sigma-Aldrich) was injected into the T3-T4 ventral spinal cord white matter with a Hamilton 34-gauge needle using ketamine (200 mg/kg) and xylazine (10 mg/kg) to anesthetize mice. The spinal cord was injected lateral to the midline at 5° to a depth of 1.3 mm. Following needle withdrawal, the overlying musculature and skin were closed with sutures. Postoperative buprenorphine (0.05 mg/kg) was administered as an analgesic. After their recovery, mice were returned to their cages until appropriate time of euthanasia. Mice were euthanized via transcardial perfusion. Tissue was collected and fixed overnight with 4% paraformaldehyde (PFA) before cryoprotecting in a 30% sucrose solution, freezing, and cryosectioning onto slides.
Nerve surgeries
All surgeries were carried out under isoflurane (2 to 5%) inhalation anaesthetic. Subcutaneous injections of buprenorphine (0.05 mg/kg) were used to treat postoperative pain on the day of surgery and the following day. Before surgery, the mouse hindlimb area was shaved and disinfected using 70% ethanol. The sciatic nerve was exposed at the sciatic notch as viewed with a dissection microscope (M651, Leica) and injected with 1% LPC (2-μl volume) using a 30-gauge Hamilton syringe.
Animal treatments
We injected CXCR1CreER; Rosa26tdTom, CXCR1CreER; Rosa26iDTR; iCXCR1tdTom;WT, or iCXCR1tdTom;iDTR mice with 1 mg of tamoxifen (20 mg/ml; T5648, Sigma) dissolved in corn oil (C8267) intraperitoneally starting from P12 to P16 for two to three consecutive days. Preliminary evidence suggests that a single injection of tamoxifen was associated with >95% recombination. We injected CXCR1CreER; Rosa26iDTR/wt or CXCR1CreER; Rosa26wt/wt mice (for gene expression with LCM) or iCXCR1tdTom;iDTR and iCXCR1tdTom;WT control mice (for histological assessments) with 1 μg of DT intraperitoneally daily starting 24 hours after LPC surgery and administered daily until euthanasia. In a second study, we administered 1 μg of DT intraperitoneally or phosphate-buffered saline (PBS) control into CXCR1CreER; Rosa26iDTR mice for 3 days before surgery and then at 2, 4, and 6 days after LPC injection.
Tissue and immunohistochemistry
Animals were perfused with PBS and then 4% PFA, before incubating tissue overnight in 4% PFA and cryopreserved in 30% sucrose. For immunohistochemistry, tissue was rehydrated in PBS. Slides were incubated for 30 to 60 min with blocking solution composed of PBS with 10% horse serum, 1% bovine serum albumin, 0.1% cold fish gelatin, 0.1% Triton X-100, and 0.05% Tween 20. Slides were then incubated overnight in a PBS antibody solution containing 1% bovine serum albumin, 0.1% cold fish gelatin, and 0.5% Triton X-100 with the following primary antibodies: polyclonal rabbit anti-Iba1 (1:1000; Wako, no. 019-19741), polyclonal rat anti-CD45 (1:100; BD Pharmingen, no. 550539), anti-CC3 (1:200; Cell Signaling Technology, no. 9661S), rabbit polyclonal anti-Ki67 (1:1000; Abcam, no. AB15581), rat anti-F4/80 (1:200; Invitrogen, no. MF48000), goat anti-ApoE (1:400; Millipore, AB947), rabbit anti-Lyve1 (1:500; Abcam, ab14917), chicken anti-GFP (1:2000; Abcam, ab13970), and monoclonal rabbit Tmem119 (1:400; Abcam, no. AB209064). Afterward, slides were washed in PBS and incubated for 1 to 2 hours with donkey anti-rabbit, mouse, rat F(ab)2 fragment secondary antibodies congregated to Alexa Fluor 488, 594, or 647. Certain sections were also stained with 4′,6-diamidino-2-phenylindole. Representative images were collected with either a Nikon A1 confocal microscope or an Olympus FluoView FV10i confocal microscope. In all cases, bleed-through was negligible.
LCM and RNA extraction
Animals were perfused with RNA-free PBS, dissected, and flash-frozen in optimal cutting temperature compound. Tissue was collected on slides and stored at −80°C. Before laser capture, tissue was incubated in 70% ethanol and stained in 1% Cresyl violet and then washed in 70% and 100% ethanol. Lesion and distal tissue were visualized and microdissected with a Zeiss PALM MicroBeam. LPC lesions were captured on the basis of their reduced Cresyl violet staining and increased hypercellularity in the lesion and the meninges. By contrast, distal sections were collected if there were no cellular infiltrates present in the meninges. For each sample, roughly 90 to 120 sections were collected from two to five animals. For distal and lesion groups, there were four samples (CXCR1CreER; Rosa26iDTR/wt) and three samples (CXCR1CreER; Rosa26wt/wt). RNA was isolated using an RNeasy micro kit (74004, QIAGEN) according to the manufacturer’s instructions. All subsequent steps were conducted by the University of Calgary Genomics core. RNA integrity was measured with a TapeStation assay, and all values had an RNA integrity number greater than 6.8. All samples were reverse-transcribed, and multiple displacement amplification was conducted by the University Core DNA services (University of Calgary) as described previously (44). Briefly, RNA libraries were prepared for sequencing using standard Illumina protocols. Illumina-compatible libraries were then prepared as per the REPLI-g single-cell RNA library prep kit protocol. A single pool containing equal amounts of each library was subjected to on-board cluster formation and sequencing on an Illumina NextSeq 500 sequencer with a high-output v2 75-cycle sequencing kit (FC-404-2005) as per the standard Illumina protocols. The number of assigned reads for each sample was between 23.6 million and 30.8 million.
After sequencing, FASTQ files were mapped to the mm10 (Mus musculus) reference genome, using BWA-MEM Galaxy Version 0.7.17.1 (45). All mapping was completed using the “single-end” option, with all other parameters set to default. The resulting sequence alignment map (SAM) files were internally fed through the bundled “SAMtools” and “Bamtools” of the Galaxy Project (https://usegalaxy.org/) (46) to generate BAM files of mapped reads for each sample. All BAM files were imported into an R3 environment and analyzed for differential expression using the package ‘DESeq2’ (47). Each comparison was made using default parameters and was exported to different comma-separated value files. Differentially expressed genes were determined using the cutoff of adjusted P < 0.1 after Benjamini-Hochberg correction of multiple testings. Differentially expressed genes from each comparison were uploaded into the ingenuity pathway environment and analyzed using the “Core Analysis” tool under false discovery rate threshold and default parameters (48).
Flow cytometry
CXCR1CreER; Rosa26tdTom mice were injected with a terminal dose of ketamine and xylazine. Mice were flushed with PBS, their spinal cords were removed, and the LPC lesion sites were microdissected 7 days after LPC. Tissue was diced and digested in a solution containing papain (1.54 μg/ml; Worthington, Lakewood, NJ), l-cysteine (360 μg/ml; Sigma-Aldrich) and DNAseI (703 μg/ml; Roche) for 25 min at 37°C. Tissue was then dissociated by trituration in 20% fetal bovine serum (FBS) and filtered through a 70-μm filter before staining. For blood flow cytometry, blood was collected through cardiac puncture and placed in a tube with heparin before diluting sample with Hank’s buffered salt solution (HBSS). Cells were exposed to Fc block (1:100, αCD16/32; BD Pharmingen) for 30 min at 4°C, and the following antibodies were used to stain tissue at 1:50 for 45 min at 4°C: CD45-PerCP (BD Pharmingen; clone 30-F11), CD11b-FITC (BD Pharmingen; clone M1/70), Ly6C-V450 (BD Horizon; clone Al-21), and Ly6G-APC-Cy7 (BD Pharmingen; clone 1A8). For blood staining, red blood cells were subsequently lysed by addition of BD FACS Lysing Solution (BD Biosciences) while rocking for 12 min at room temperature. Cells were washed and fixed in 1% buffered formalin for flow cytometry analysis (conducted by University of Calgary Flow Cytometry Core Facility). Data were analyzed using FlowJo 10.0.8 (FlowJo).
Cell culture
Mice were sterilized and cleared of hair and skin. Femurs were obtained, and then cells from bone marrow were collected by flushing five times with ice-cold calcium and magnesium-free PBS. Samples were centrifuged for 5 min at 200g, supernatant was discarded and then resuspended in media and plated at 500,000 cells/ml. Media included recombinant M-CSF (100 ng/ml; R&D Systems), 1% penicillin-streptomycin (Gibco), 2% minimum essential medium (MEM) nonessential amino acid solution (Gibco), and 10% FBS (Sigma-Aldrich) in high-glucose Dulbecco’s modified Eagle’s medium (DMEM). The day after plating, once cells had settled, media was changed. Once cells had reached 80% confluence (~5 days after plating), half of the media was changed, and then at days 7 to 10, cells were replated onto 96-well plates at 50,000 cells/ml. Cells were treated with media preconditioned with sciatic nerve tissue or spinal cord tissue for 3 to 5 days. To prepare media for conditioned media experiments, equal wet weights of healthy sciatic nerves or white matter from thoracic spinal cords were used. Briefly, tissue was dissected from C57BL/6 mice (n = 8 to 10), then maintained in DMEM and F12 (3:1), B27 supplement (1%), penicillin (100 μg/ml)–streptomycin (100 U/ml), and 10% FBS for up to 5 days before filtering (0.2 μm), and snap-frozen at −80°C. Depleted media alone controls were treated identically except no explants were added. Three days post-CM treatment, macrophages were washed with sterile PBS and then fixed in 4% PFA for ~10 min. For immunocytochemistry, cells were permeabilized using 0.5% triton X-100 and 5% BSA for 1 to 2 hours at room temperature. Primary antibodies were incubated overnight (1:200) at room temperature. Unbound primary antibodies were washed, and cells were subsequently incubated with Alexa-conjugated secondary antibodies (1:200, Invitrogen) at room temperature for 1 to 2 hours and washed, and nuclei were counterstained with Hoechst (1:1000; Sigma-Aldrich) and imaged using ImageXpress (Molecular Devices). The sum of cells in 12 images (20×) per well was obtained and then normalized to obtain a percentage value for each well. This value was subsequently averaged across three replicates per condition. Experiments were replicated on three independent days.
Single-cell sequencing analysis
FACS, single-cell library preparation, sequencing, and processing
The ventral horns of CXCR1CreER; Rosa26tdTom mice (n = 2 naïve and n = 9 LPC) were microdissected and enzymatically dissociated with Collagenase-4 (2 mg/ml; Worthington) at 37°C for 40 min with trituration every 15 to 20 min. Cell suspensions were titurated in DMEM with 10% FBS and then filtered through a 70-μm nylon cell strainer to obtain a single-cell suspension. Cell debris was removed with debris removal solution according to the manufacturer’s instructions (Miltenyi Biotec). Cells were stained with NucBlue and eBioscience Fixable Viability Dye eFluor 780 (Thermo Fisher Scientific) for 20 min and washed in HBSS. Liberated cells were resuspended in 1% BSA in HBSS before FACS purification. For FACS purification/sorting, singlet cells were captured using side scatter and forward scatter before selecting for nucleated cells that were viable. Wild-type cells were used as a negative control to determine gates for detection of tdTomato-fluorescence.
We collected one population of CXCR1CreER; Rosa26tdTom cells from naïve mice (3200 cells) and one population from lesioned mice (3209 cells) and prepared single-cell sequencing libraries for each sample in parallel. All cells were processed according to 10× Genomics Chromium Single Cell 3′ Reagent Guidelines (v2 Chemistry; https://support.10xgenomics.com/single-cell-gene-expression). Briefly, cells were partitioned into nanoliter-scale gel bead–in-emulsions (GEMs) using 10× GemCode Technology. Primers containing (i) an Illumina R1 sequence, (ii) a 16–base pair (bp) 10× barcode, (iii) a 10-bp UMI, and (iv) a poly-dT primer sequence were incubated with partitioned cells resulting in barcoded, full-length cDNA from poly-adenylated mRNA. Silane magnetic beads were used to remove leftover biochemical reagents/primers, and then cDNA was amplified by polymerase chain reaction (PCR). Enzymatic fragmentation and size selection were used to optimize cDNA amplicon size before library construction. R1 (read 1 primer sequence) were added during GEM incubation, whereas P5, P7, a sample index (i7), and R2 (read 2 primer sequence) were added during library construction. Quality control and quantification were performed using a Kapa Library Quantification quantitative PCR kit (Kapa Biosystems). Sequencing was performed using Illumina HiSeq 4000 and standard 10× Genomics Chromium Single Cell 3′ Reagent v2 Chemistry sequencing protocols. This resulted in an average read depth of 121,000 reads per cell.
Reads were processed using the 10× Genomics Cell Ranger Single Cell 2.0.0 pipeline with default and recommended parameters, as previously described (49). FASTQs generated from Illumina sequencing output were aligned to the mouse GRCm38.p5 (primary assembly) reference genome using the STAR algorithm. Next, Gene-Barcode matrices were generated for each sample by counting UMIs and filtering noncell-associated barcodes. Last, samples were aggregated, with intermediary depth normalization, to generate a gene-barcode matrix containing 1285 barcoded cells and gene expression counts. This output was then imported into the Seurat (v1.4.0.15) R toolkit for quality control and downstream analysis of our single-cell RNA-seq experiment (50) (fig. S6; GEO: GSE115803).
We sequenced each sample to an average depth of 76 million reads, and all samples were subsequently normalized to a total of 96,570 reads per cell and aggregated to a total of 1285 barcoded cells. We excluded low-quality cells with <400 genes detected and sparse genes, those identified in less than three cells. Notably, we found sparse labeling of the dendritic cell, monocyte-derived cell, or border-associated macrophage markers Siglec 1, CD38, and CD43 (51) (only 19 cells), suggesting that we sequenced a relatively pure population of activated microglia. All functions were run with default parameters, unless specified otherwise. Gene expression was log-normalized to a scale factor of 10,000. UMI counts, gene counts, and percentage of mitochondrial genes detected in single-cell data were plotted to determine any outliers of cells (there were none). Significant principle components were then determined using elbow plot (Seurat v1.4.0.15). The “elbow” in this plot reflects a transition from informative PCs to PCs that offer comparatively little information. On this basis, PCs 1 to 20 were used for downstream analysis. Samples were projected onto tSNE plots and color codes based on sample ID or unbiased clustering (at resolution = 0.3). Expression of selected microglia marker genes, as well as a monocyte gene, was used to confirm cell identities. The top marker genes for each cell cluster were identified using the likelihood ratio test for single-cell gene expression, and the top 10 markers for each cluster (based on average difference compared to remaining cells) were selected and plotted on a heatmap.
Monocle’s (2.4.0) pseudotime ordering algorithm was used (23). All genes in the matrix were used as input genes for pseudotime ordering, and DDRTree dimensionality reduction was performed using default parameters. The orderCells function was run with the uninjured branch set as the root state to specify the start point of the trajectory. In this way, we could track the trajectory of microglia activation from the naïve microglia starting point. The top 50 pseudotime-dependent genes (q value, <1 × 10−27) were plotted. For PAGODA analysis, we implemented in the SCDE package (52) by importing a log-normalized Seurat data frame.
We applied SCENIC (v 1.0.1) pipeline with mouse mm9 genome for cis-regulatory analysis directly onto our normalized Seurat matrix (24). Briefly, this pipeline contains three sequential steps, each using a specific R/bioconductor package: (i) GENIE3 (v 1.4.0) identifies coexpression between TFs and its downstream targets. (ii) RcisTarget (v 1.2.0) performs TF-motif enrichment analysis to infer target genes of TFs (termed “regulons”) identified by GENIE3. (iii) AUCell (v 1.4.0) scores the activity of each regulon for every cell in the matrix. Last, the AUCell score distribution for a particular regulon is used to binarize regulon activity as “active” or “inactive.” We set our analysis at 50 PCs and 50 perplexity. To confirm gene expression of predicted TFs, we used the library.size.normalize() function to normalized by library size and then performed transcriptome-wide imputation using magic() function from Rmagic package.
In situ hybridization
For in situ hybridization, we used an RNAscope multiplex assay (Advanced Cell Diagnostics). The RNAscope probes targeted base pairs ranging from 83 to 1245 (NM_009696.3, catalog no. 313271-C3). The probe used as a negative control targets the nonspecific bacterial dihydrodipicolinate reductase (dapB) gene, which is not expressed in mammalian tissues, and was negative. The double Z target probe system of RNAscope technology allows each probe to reliably detect RNA at single-molecule resolution while preventing amplification of nonspecific signals. The probe is magnified by branching amplification and then developed using an alkaline phosphatase reaction with the Fast Red substrate. Briefly, sections were processed following the standard RNAscope singleplex protocol. Sections were first permeabilized with a broad-spectrum protease for 30 min. Samples were then hybridized with target-specific probes for 2 hours at 40°C, washed, then hybridized with amplifiers, and visualized using the Fast Red substrate via alkaline phosphatase reaction. Immediately after, slides were imaged as above.
Statistics
Statistical significance (*P < 0.05) was determined using one or two-way analysis of variance (ANOVA) with post hoc testing (details in figure legends), unless otherwise noted (Prism 6.0, GraphPad Software). All data were presented as means ± SEM.
Supplementary Material
Acknowledgments
We thank J. Wang for technical support, as well as the Hotchkiss Brain Institute AMP core facility, the flow cytometry core, and the UCDNA sequencing and genetic analysis laboratory at the University of Calgary. Funding: This study was supported by operating grants from Canadian Institutes for Health Research (CIHR), MS Society of Canada, Alberta Heritage Foundation For Medical Research, University of Alberta start-up funds for J.R.P. and a CRIO Team grant from Alberta Innovates Health Solutions (AIHS). J.R.P. was supported by fellowships from CIHR, T. Chen Fong, AIHS, and the Donna Joan Oxford award from the MS Society of Canada. J.R.P. is also supported by funds from the University of Alberta. J.A.S. was supported by an Alberta Children’s Hospital Research Institute postdoctoral fellowship. K.S.R. was supported by a Vanier Canada Graduate Scholarship and a studentship from the University of Calgary Cummings School of Medicine. N.J.M. was supported by the MS Society of Canada and a studentship by the University of Calgary. S.S. was supported by CIHR graduate and summer studentships. K.T. and T.N.F. were supported by the MS Society of Canada. A.V.C. was supported by fellowships from AIHS. J.B. is supported by the Calgary Firefighters Burn Treatment Society Chair in Skin Regeneration and Wound Healing. P.K.S. and V.W.Y. are supported by Canada Research Chair (Tier 1) awards. Author contributions: J.R.P. and J.A.S. initiated the project. J.R.P., J.A.S., E.Z., K.T., S.S., C.S.B., M.H., J.Y., K.L., and S.J. performed immunohistochemistry, staining, cell culture, and tissue quantification. J.R.P., J.A.S., K.S.R., and Y.D. conducted and analyzed flow cytometry experiments. J.R.P., N.J.M., K.S.R., and C.S.B. conducted spinal cord surgeries and tissue processing. J.A.S. conducted sciatic nerve surgeries. J.R.P., J.A.S., and C.S. conducted flow cytometry for single-cell sequencing. J.A.S. conducted single-cell sequencing analysis. J.R.P. conducted LCM, and T.N.F. conducted sequencing LCM bioinformatics. A.V.C. and V.H. aided in animal experiments. A.J. helped with single-cell RNA sequencing analysis and created the website. J.R.P., J.A.S., and V.W.Y. co-wrote the manuscript. K.S.R., N.J.M., S.S., S.J., R.M., B.J.K., P.K.S., and J.B. reviewed the manuscript. J.R.P., R.M., P.K.S., J.B., and V.W.Y. supervised the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. For single-cell RNA-seq, we have the following GEO submission: Transcriptional profiling of 5 days post-LPC lesion after microglia ablation is available at www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121484. For our laser capture RNA-seq experiment, we have the following GEO submission: Single-cell transcriptomics of Cx3cr1 fate-mapped cells following lysolecithin-induced demyelination in the spinal cord. GEO: GSE115803 (link: www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115803). These data can be viewed graphically at the following website: www.biernaskielab.ca/Injury_Microglia/
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/3/eaay6324/DC1
Fig. S1. Minimal expression of Cx3Cr1creER-TdTom in monocytes or macrophages in the spleen and lymph node.
Fig. S2. Lack of meningeal or perivascular macrophage marker Lyve1.
Fig. S3. Common markers to distinguish microglia and macrophages are less sensitive after microglia activation.
Fig. S4. Gating strategy for flow cytometry.
Fig. S5. Microglia express CX3CR1 and tdTomato.
Fig. S6. RNA-seq experiment metrics and quality control.
Fig. S7. Sparse CAM present following single-cell RNA sequencing of fate mapped cells (tdTom+).
Fig. S8. Activated microglia express ApoE.
Fig. S9. M1 and M2 genes in scRNA sequencing dataset.
Fig. S10. Distinct activated microglia subphenotypes.
Fig. S11. Fate mapping as a tool to specifically label resident macrophage in sciatic nerve.
Fig. S12. CNS and PNS LPC injections.
Fig. S13. Infiltrating macrophages expand in CNS when microglia/CAMs are ablated following LPC demyelination.
Fig. S14. Cytosolic pattern recognition receptors reduced in the absence of microglia.
Fig. S15. IFN type I and type II reduced in the absence of microglia.
REFERENCES AND NOTES
- 1.Miron V. E., Boyd A., Zhao J.-W., Yuen T. J., Ruckh J. M., Shadrach J. L., van Wijngaarden P., Wagers A. J., Williams A., Franklin R. J. M., ffrench-Constant C., M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 16, 1211–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldmann T., Wieghofer P., Müller P. F., Wolf Y., Varol D., Yona S., Brendecke S. M., Kierdorf K., Staszewski O., Datta M., Luedde T., Heikenwalder M., Jung S., Prinz M., A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 16, 1618–1626 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Bennett M. L., Bennett F. C., Liddelow S. A., Ajami B., Zamanian J. L., Fernhoff N. B., Mulinyawe S. B., Bohlen C. J., Adil A., Tucker A., Weissman I. L., Chang E. F., Li G., Grant G. A., Hayden Gephart M. G., Barres B. A., New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. U.S.A. 113, E1738–E1746 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butovsky O., Jedrychowski M. P., Moore C. S., Cialic R., Lanser A. J., Gabriely G., Koeglsperger T., Dake B., Wu P. M., Doykan C. E., Fanek Z., Liu L. P., Chen Z., Rothstein J. D., Ransohoff R. M., Gygi S. P., Antel J. P., Weiner H. L., Identification of a unique TGF-β–dependent molecular and functional signature in microglia. Nat. Neurosci. 17, 131–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T. K., David E., Baruch K., Lara-Astaiso D., Toth B., Itzkovitz S., Colonna M., Schwartz M., Amit I., A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Bennett F. C., Bennett M. L., Yaqoob F., Mulinyawe S. B., Grant G. A., Gephart M. H., Plowey E. D., Barres B. A., A combination of ontogeny and CNS environment establishes microglial identity. Neuron 98, e1178, 1170–1183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parkhurst C. N., Yang G., Ninan I., Savas J. N., Yates J. R. III, Lafaille J. J., Hempstead B. L., Littman D. R., Gan W. B., Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ajami B., Bennett J., Krieger C., Tetzlaff W., Rossi F. M. V., Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538–1543 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Goldmann T., Wieghofer P., Jordão M. J. C., Prutek F., Hagemeyer N., Frenzel K., Amann L., Staszewski O., Kierdorf K., Krueger M., Locatelli G., Hochgerner H., Zeiser R., Epelman S., Geissmann F., Priller J., Rossi F. M. V., Bechmann I., Kerschensteiner M., Linnarsson S., Jung S., Prinz M., Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797–805 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plemel J. R., Michaels N. J., Weishaupt N., Caprariello A. V., Keough M. B., Rogers J. A., Yukseloglu A., Lim J., Patel V. V., Rawji K. S., Jensen S. K., Teo W., Heyne B., Whitehead S. N., Stys P. K., Yong V. W., Mechanisms of lysophosphatidylcholine-induced demyelination: A primary lipid disrupting myelinopathy. Glia 66, 327–347 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Duncan G. J., Plemel J. R., Assinck P., Manesh S. B., Muir F. G. W., Hirata R., Berson M., Liu J., Wegner M., Emery B., Moore G. R. W., Tetzlaff W., Myelin regulatory factor drives remyelination in multiple sclerosis. Acta Neuropathol. 134, 403–422 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Stirling D. P., Yong V. W., Dynamics of the inflammatory response after murine spinal cord injury revealed by flow cytometry. J. Neurosci. Res. 86, 1944–1958 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Paschalis E. I., Lei F., Zhou C., Kapoulea V., Thanos A., Dana R., Vavvas D. G., Chodosh J., Dohlman C. H., The role of microglia and peripheral monocytes in retinal damage after corneal chemical injury. Am. J. Pathol. 188, 1580–1596 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jordão M. J. C., Sankowski M., Brendecke S. M., Sagar, Locatelli G., Tai Y.-H., Tay T. L., Schramm E., Armbruster S., Hagemeyer N., Groß O., Mai D., Çiçek Ö., Falk T., Kerschensteiner M., Grün D., Prinz M., Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363, eaat7554 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Krasemann S., Madore C., Cialic R., Baufeld C., Calcagno N., El Fatimy R., Beckers L., O'Loughlin E., Xu Y., Fanek Z., Greco D. J., Smith S. T., Tweet G., Humulock Z., Zrzavy T., Conde-Sanroman P., Gacias M., Weng Z., Chen H., Tjon E., Mazaheri F., Hartmann K., Madi A., Ulrich J. D., Glatzel M., Worthmann A., Heeren J., Budnik B., Lemere C., Ikezu T., Heppner F. L., Litvak V., Holtzman D. M., Lassmann H., Weiner H. L., Ochando J., Haass C., Butovsky O., The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, e569, 566–581.e9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu H., Liu X., Zhong Y., The effect of osteopontin on microglia. Biomed. Res. Int. 2017, 1879437 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koch N., Moldenhauer G., Hofmann W. J., Möller P., Rapid intracellular pathway gives rise to cell surface expression of the MHC class II-associated invariant chain (CD74). J. Immunol. 147, 2643–2651 (1991). [PubMed] [Google Scholar]
- 18.González-Navajas J. M., Lee J., David M., Raz E., Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 12, 125–135 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hammond T. R., Dufort C., Dissing-Olesen L., Giera S., Young A., Wysoker A., Walker A. J., Gergits F., Segel M., Nemesh J., Marsh S. E., Saunders A., Macosko E., Ginhoux F., Chen J., Franklin R. J. M., Piao X., McCarroll S. A., Stevens B., Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity 50, 253–271.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li Q., Cheng Z., Zhou L., Darmanis S., Neff N. F., Okamoto J., Gulati G., Bennett M. L., Sun L. O., Clarke L. E., Marschallinger J., Yu G., Quake S. R., Wyss-Coray T., Barres B. A., Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron 101, 207–223.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.David S., Kroner A., Repertoire of microglial and macrophage responses after spinal cord injury. Nat. Rev. Neurosci. 12, 388–399 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Jha A. K., Huang S. C. C., Sergushichev A., Lampropoulou V., Ivanova Y., Loginicheva E., Chmielewski K., Stewart K. M., Ashall J., Everts B., Pearce E. J., Driggers E. M., Artyomov M. N., Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N. J., Livak K. J., Mikkelsen T. S., Rinn J. L., The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aibar S., González-Blas C. B., Moerman T., Huynh-Thu V. A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., van den Oord J., Atak Z. K., Wouters J., Aerts S., SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Dijk D., Sharma R., Nainys J., Yim K., Kathail P., Carr A. J., Burdziak C., Moon K. R., Chaffer C. L., Pattabiraman D., Bierie B., Mazutis L., Wolf G., Krishnaswamy S., Pe’er D., Recovering gene interactions from single-cell data using data diffusion. Cell 174, 716–729.e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghabriel M. N., Allt G., Schmidt-Lanterman incisures. II. A light and electron microscope study of remyelinating peripheral nerve fibres. Acta Neuropathol. 52, 97–104 (1980). [DOI] [PubMed] [Google Scholar]
- 27.Buch T., Heppner F. L., Tertilt C., Heinen T. J. A. J., Kremer M., Wunderlich F. T., Jung S., Waisman A., A Cre-inducible diphtheria toxin receptor mediates cell lineage ablation after toxin administration. Nat. Methods 2, 419–426 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Cronk J. C., Filiano A. J., Louveau A., Marin I., Marsh R., Ji E., Goldman D. H., Smirnov I., Geraci N., Acton S., Overall C. C., Kipnis J., Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 215, 1627–1647 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baruch K., Deczkowska A., David E., Castellano J. M., Miller O., Kertser A., Berkutzki T., Barnett-Itzhaki Z., Bezalel D., Wyss-Coray T., Amit I., Schwartz M., Aging-induced type I interferon response at the choroid plexus negatively affects brain function. Science 346, 89–93 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lloyd A. F., Davies C. L., Holloway R. K., Labrak Y., Ireland G., Carradori D., Dillenburg A., Borger E., Soong D., Richardson J. C., Kuhlmann T., Williams A., Pollard J. W., des Rieux A., Priller J., Miron V. E., Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 22, 1046–1052 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hickman S. E., Kingery N. D., Ohsumi T. K., Borowsky M. L., Wang L.-c., Means T. K., El Khoury J., The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Natrajan M. S., Komori M., Kosa P., Johnson K. R., Wu T., Franklin R. J. M., Bielekova B., Pioglitazone regulates myelin phagocytosis and multiple sclerosis monocytes. Ann. Clin. Transl. Neurol. 2, 1071–1084 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellver-Landete V., Bretheau F., Mailhot B., Vallières N., Lessard M., Janelle M. E., Vernoux N., Tremblay M. È., Fuehrmann T., Shoichet M. S., Lacroix S., Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 10, 518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wanner I. B., Anderson M. A., Song B., Levine J., Fernandez A., Gray-Thompson Z., Ao Y., Sofroniew M. V., Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J. Neurosci. 33, 12870–12886 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greenhalgh A. D., Zarruk J. G., Healy L. M., Baskar Jesudasan S. J., Jhelum P., Salmon C. K., Formanek A., Russo M. V., Antel J. P., McGavern D. B., McColl B. W., David S., Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLOS Biol. 16, e2005264 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spadaro O., Camell C. D., Bosurgi L., Nguyen K. Y., Youm Y.-H., Rothlin C. V., Dixit V. D., IGF1 shapes macrophage activation in response to immunometabolic challenge. Cell Rep. 19, 225–234 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pejnovic N. N., Pantic J. M., Jovanovic I. P., Radosavljevic G. D., Milovanovic M. Z., Nikolic I. G., Zdravkovic N. S., Djukic A. L., Arsenijevic N. N., Lukic M. L., Galectin-3 deficiency accelerates high-fat diet-induced obesity and amplifies inflammation in adipose tissue and pancreatic islets. Diabetes 62, 1932–1944 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rudemiller N. P., Patel M. B., Zhang J. D., Jeffs A. D., Karlovich N. S., Griffiths R., Kan M. J., Buckley A. F., Gunn M. D., Crowley S. D., C-C motif chemokine 5 attenuates angiotensin II-dependent kidney injury by limiting renal macrophage infiltration. Am. J. Pathol. 186, 2846–2856 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rawlings N. D., Barrett A. J., Thomas P. D., Huang X., Bateman A., Finn R. D., The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46, D624–D632 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stuve O., Chabot S., Jung S. S., Williams G., Yong V. W., Chemokine-enhanced migration of human peripheral blood mononuclear cells is antagonized by interferon beta-1b through an effect on matrix metalloproteinase-9. J. Neuroimmunol. 80, 38–46 (1997). [DOI] [PubMed] [Google Scholar]
- 41.Stüve O., Dooley N. P., Uhm J. H., Antel J. P., Francis G. S., Yong V. W., Williams G., Interferon β-1b decreases the migration of T lymphocytes in vitro: Effects on matrix metalloproteinase-9. Ann. Neurol. 40, 853–863 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Stephenson E. L., Mishra M. K., Moussienko D., Laflamme N., Rivest S., Ling C.-C., Yong V. W., Chondroitin sulfate proteoglycans as novel drivers of leucocyte infiltration in multiple sclerosis. Brain 141, 1094–1110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taylor S., Mehina E., White E., Reeson P., Yongblah K., Doyle K. P., Brown C. E., Suppressing Interferon-γ stimulates microglial responses and repair of microbleeds in the diabetic brain. J. Neurosci. 38, 8707–8722 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stratton J. A., Holmes A., Rosin N. L., Sinha S., Vohra M., Burma N. E., Trang T., Midha R., Biernaskie J., Macrophages regulate Schwann cell maturation after nerve injury. Cell Rep. 24, 2561–2572.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 45.Li H., Durbin R., Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Afgan E., Baker D., Batut B., van den Beek M., Bouvier D., Čech M., Chilton J., Clements D., Coraor N., Grüning B. A., Guerler A., Hillman-Jackson J., Hiltemann S., Jalili V., Rasche H., Soranzo N., Goecks J., Taylor J., Nekrutenko A., Blankenberg D., The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, W537–W544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krämer A., Green J., Pollard J. Jr., Tugendreich S., Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zheng G. X., Terry J. M., Belgrader P., Ryvkin P., Bent Z. W., Wilson R., Ziraldo S. B., Wheeler T. D., McDermott G. P., Zhu J., Gregory M. T., Shuga J., Montesclaros L., Underwood J. G., Masquelier D. A., Nishimura S. Y., Schnall-Levin M., Wyatt P. W., Hindson C. M., Bharadwaj R., Wong A., Ness K. D., Beppu L. W., Deeg H. J., McFarland C., Loeb K. R., Valente W. J., Ericson N. G., Stevens E. A., Radich J. P., Mikkelsen T. S., Hindson B. J., Bielas J. H., Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Macosko E. Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A. R., Kamitaki N., Martersteck E. M., Trombetta J. J., Weitz D. A., Sanes J. R., Shalek A. K., Regev A., McCarroll S. A., Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mrdjen D., Pavlovic A., Hartmann F. J., Schreiner B., Utz S. G., Leung B. P., Lelios I., Heppner F. L., Kipnis J., Merkler D., Greter M., Becher B., High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 48, 380–395.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 52.Fan J., Salathia N., Liu R., Kaeser G. E., Yung Y. C., Herman J. L., Kaper F., Fan J.-B., Zhang K., Chun J., Kharchenko P. V., Characterizing transcriptional heterogeneity through pathway and gene set overdispersion analysis. Nat. Methods 13, 241–244 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/3/eaay6324/DC1
Fig. S1. Minimal expression of Cx3Cr1creER-TdTom in monocytes or macrophages in the spleen and lymph node.
Fig. S2. Lack of meningeal or perivascular macrophage marker Lyve1.
Fig. S3. Common markers to distinguish microglia and macrophages are less sensitive after microglia activation.
Fig. S4. Gating strategy for flow cytometry.
Fig. S5. Microglia express CX3CR1 and tdTomato.
Fig. S6. RNA-seq experiment metrics and quality control.
Fig. S7. Sparse CAM present following single-cell RNA sequencing of fate mapped cells (tdTom+).
Fig. S8. Activated microglia express ApoE.
Fig. S9. M1 and M2 genes in scRNA sequencing dataset.
Fig. S10. Distinct activated microglia subphenotypes.
Fig. S11. Fate mapping as a tool to specifically label resident macrophage in sciatic nerve.
Fig. S12. CNS and PNS LPC injections.
Fig. S13. Infiltrating macrophages expand in CNS when microglia/CAMs are ablated following LPC demyelination.
Fig. S14. Cytosolic pattern recognition receptors reduced in the absence of microglia.
Fig. S15. IFN type I and type II reduced in the absence of microglia.