

We converted a potent antagonistic sdAb JN241 to an agonistic sdAb JN241-9 to human APJ by structure-guided rational design.
Abstract
Developing antibody agonists targeting the human apelin receptor (APJ) is a promising therapeutic approach for the treatment of chronic heart failure. Here, we report the structure-guided discovery of a single-domain antibody (sdAb) agonist JN241-9, based on the cocrystal structure of APJ with an sdAb antagonist JN241, the first cocrystal structure of a class A G protein–coupled receptor (GPCR) with a functional antibody. As revealed by the structure, JN241 binds to the extracellular side of APJ, makes critical contacts with the second extracellular loop, and inserts the CDR3 into the ligand-binding pocket. We converted JN241 into a full agonist JN241-9 by inserting a tyrosine into the CDR3. Modeling and molecular dynamics simulation shed light on JN241-9–stimulated receptor activation, providing structural insights for finding agonistic antibodies against class A GPCRs.
INTRODUCTION
G protein–coupled receptors (GPCRs) represent a major family of human drug targets (1–3). Nearly one-third of currently marketed drugs, including 70 small molecules and 30 peptide ligand analogs, target GPCRs. However, there are only two GPCR monoclonal antibodies (mAbs) approved by the U.S. Food and Drug Administration, including erenumab, a calcitonin gene–related peptide receptor antagonist, for migraine prevention (4), and mogamulizumab, a CCR4 neutral binder with antibody-dependent cellular cytotoxicity in vivo, for adult T cell leukemia-lymphoma and peripheral T cell lymphoma (5). Despite the substantial potential for therapeutic opportunities within this target class, there is a scarcity of successful large-molecule drugs due to the difficulties associated with generating functional mAbs against GPCRs. The first challenge that GPCRs present for antibody discovery is that the receptors have poor thermostability and high conformational flexibility, which makes obtaining purified form of the native receptors technically very difficult. This combined with an inherently poor immunogenicity and antigenicity associated with the small extracellular loops (ECLs) of these receptors pose further challenge for producing functional mAbs. These challenges are even more significant for class A GPCRs, one of the most important GPCR families for drug discovery. Unlike class B GPCRs that have large N-terminal domains to serve as immunogens and tool antigens (6–11), most class A GPCRs have a short N terminus. Peptides derived from the short N termini or the small ECLs of class A GPCRs as immunogens and/or antigens often lead to the generation of peptide-specific mAbs that rarely recognize the native form of the receptors (12). Nevertheless, a handful antagonistic mAbs against class A GPCRs have been reported using target-overexpressing cells as immunogens in mouse immunization, followed by hybridoma or phage display screening (1, 13–17). However, to the present day, no agonistic mAbs against class A GPCRs or any GPCRs have been described in the literature.
Unlike antagonistic mAbs that can function by sterically blocking natural ligand binding, agonistic mAbs need to functionally mimic natural ligands to activate the receptors. A long CDR3 loop is presumably required to reach the deep ligand-binding pocket to activate the receptor. Single-domain antibodies (sdAbs) derived from camelid heavy chain–only antibodies often have extended CDR3 loops (18). Therefore, they may be advantageous over conventional mAbs or antibody fragments derived from the mouse or human antibody repertoires in targeting the cavities of GPCRs. Several antagonistic sdAbs targeting the extracellular side of class A GPCRs were isolated from camelid immune sdAb libraries (4, 19, 20) or scaffold-based synthetic sdAb library (21) by phage display. However, agonistic sdAbs against class A or other class GPCRs have not been reported. We decided to look for sdAb agonists for human apelin receptor (APJ), a Gαi-coupled class A GPCR. APJ and its endogenous ligands, Apelin and Apela, are widely expressed in cardiovascular system. APJ signaling mediates the fluid homeostasis and cardiovascular function (22–24). Developing long-duration biologics that agonize the APJ receptor is a potential therapeutic approach for the treatment of chronic heart failure (25, 26). To explore this therapeutic approach, we have completed a discovery project to raise anti-APJ antibodies by actively immunizing camelids with thermally and/or conformationally stabilized APJ proteins (27, 28). The sdAbs raised in camelids were then screened for potent antagonistic and agonistic sdAbs. This project yielded antagonistic sdAbs, but no sdAbs with agonist activity were identified. This finding led us to develop a novel structure-guided rational design strategy to convert an orthosteric sdAb antagonist to an sdAb with potent agonist activity.
RESULTS
Generation of a potent sdAb antagonist JN241 against human APJ
We reconstituted thermally and conformationally stabilized APJ proteins in nanodiscs and proteoliposomes. APJ nanodiscs were used as immunogens to generate immune repertoires in camels. APJ proteoliposomes were used as antigens to isolate APJ-specific sdAbs from the camel immune sdAb library by phage display. This strategy yielded 186 unique sdAbs with the CDR3 lengths ranging from 13 to 23 amino acids and the affinities ranging from single-digit nanomolar to picomolar for human APJ. Consistent with previous reports, 106 APJ-specific sdAbs were found to be potent antagonists, 80 were neutral binders, and none displayed agonist activity. APJ antagonistic sdAbs directly competed with Apelin 13 for binding to the receptor in radioligand-binding assay. We extensively characterized one of the most potent antagonistic sdAbs, JN241, which harbors the longest CDR3 (23 amino acids in length). In a Biacore assay, JN241 exhibits high affinity [Kd (dissociation constant) = 83 pM] for human APJ (fig. S1A). In a functional assay, JN241 antagonized the action of Apelin 13 with half maximal inhibitory concentration (IC50) values of 5.9 and 7.0 nM in the adenosine 3′,5′-monophosphate (cAMP) assay (Fig. 1A) and β-arrestin recruitment assay (Fig. 1B), respectively. In radioligand-binding assay, JN241 blocked the binding of 125I–Apelin 13 with Ki (inhibition constant) of 0.1 nM (Fig. 1C). Together, these data suggest that JN241 is a potent competitive antagonist of human APJ.
Fig. 1. Potent APJ competitive sdAb JN241 antagonist and its cocrystal structure with human APJ.
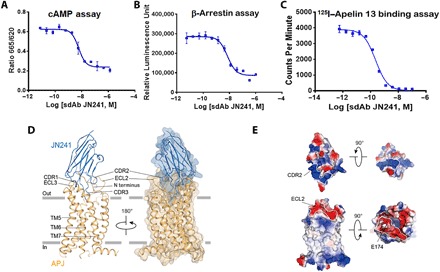
(A and B) Antagonistic activity of JN241 in cAMP (A) and β-arrestin (B) assays. (C) Competition binding of JN241 to APJ by displacement of 125I–Apelin 13 in radioligand-binding assay. (D) Side view of APJ (orange) and JN241 (blue) complex, parallel to the cell membrane. Transparent surface shows the close contact between APJ and JN241. (E) The electrostatic potential surfaces of APJ and JN241. The extensive positively charged surface of the CDR2 and negatively charged surface of the ECL2, contributing to the complex formation, are marked up by white lines. RLU, relative luminescence units; CPM, counts per minute.
Structural determination of APJ-JN241 complex
JN241 thermally stabilized the APJ receptor, allowing us to cocrystallize the complex using lipidic cubic phase (LCP) and solve the crystal structure of APJ-JN241 complex at 3.2-Å resolution by molecular replacement (figs. S1, B to D, and S2; Fig. 1D; and table S1). Overall, the receptor uses canonical seven transmembrane (TM) helix bundle structure as seen with class A GPCRs. JN241 adopts a “wine stopper” shape, with all three CDR loops making extensive contacts on the extracellular side of the receptor and with the CDR3 occupying the ligand-binding pocket. A positively charged surface of JN241, consisting of the CDR2 and CDR3, formed an electrostatic interaction with negatively charged lip peripheral to the orthosteric site (Figs. 1E and 2, A to C). In addition, networks of hydrogen bonds formed between Y29 and S30 in CDR1 with APJ TM6 and TM7 and between T52, S54, and R55 in CDR2 with E174ECL2 and N175ECL2 in APJ ECL2 (Fig. 2, B and C, and table S2). JN241 CDR3 deeply inserted into the orthosteric site and formed a contact surface area of 320 Å2, almost half of the total contact surface area, 700 Å2, of JN241 (table S2). JN241 residue E104, which contributes to the negatively charged tip of the CDR3, is well accommodated in the positively charged APJ ligand-binding pocket and forms electrostatic and hydrogen bonds with R1684.64 and Y2646.51 (Fig. 2D).
Fig. 2. The close views of APJ-JN241 complex interfaces.
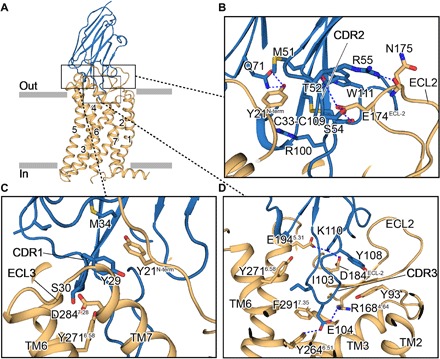
(A) Side view of APJ (orange) and JN241 (blue) complex. (B to D) Close views of the complex interfaces between APJ and JN241 CDR1 (C), CDR2 (B), and CDR3 (D). The interfaces were stabilized by hydrogen bonds (blue dashed lines) and hydrophobic interactions between several key residues (sticks) (see the main text).
Identification of critical residues for APJ-JN241 interaction
We sought to understand how each interaction contributes to JN241 binding and function. We tested the binding of JN241 to a series of APJ mutants carrying alanine substitutions in key residues identified by the structural analysis (table S2). Mutation of E174 ECL2 substantially disrupted the binding of JN241 and its inhibitory activity in ligand-induced receptor internalization (fig. S3), in agreement with the critical location of this residue in the structure and electrostatic interaction formed by this residue (Fig. 2B). We further generated and tested a panel of JN241 mutants with alanine substitutions in key residues identified by the structural analysis for binding to the APJ receptor and the residues within a distance of 5 Å from APJ residues. We found that three residues—S30, T52, and W111—in CDR1, CDR2, and CDR3, respectively, are indispensable for JN241 binding to APJ and its antagonist function. Alanine substitution of any one of these three residues abolished the binding and antagonist function of JN241. According to the complex structure, residue S30 resides in CDR1 and interacts with Y2716.58 and S2756.62 in TM6 and D2847.28 in TM7 of APJ through hydrogen bonds. Residue T52 from CDR2 interacts with E174ECL2 from APJ through hydrogen bonds. W111 from CDR3 does not have direct interaction with APJ residues, but it interacts with M51 in CDR2 through hydrogen bond and forms hydrophobic interactions with C33 in CDR1, L50 in FR2, and V98 and C109 in CDR3, which are important for stabilizing the CDR3 loop conformation (table S2). These data suggest that all three CDR loops of JN241 contribute to the binding and JN241-induced receptor antagonism through the interactions with the extracellular side of APJ and APJ ECL2 is critical for mediating JN241 binding and antagonist function. Unexpectedly, we did not find any single alanine substitution to the CDR3 loop residues in the ligand-binding pocket significantly affected JN241 binding. Thus, the interaction of JN241 with the extracellular side of the receptor largely contributes to the JN241 binding and JN241-mediated receptor antagonism.
Structure-guided rational design and conversion of JN241 into an agonist
To gain further insight into APJ-JN241 complex structure, we compared the binding mode of JN241 with that of potent peptide agonist AMG3054 (29). In contrast to AMG3054, JN241 CDR3 occupies the ligand-binding pocket with E104 interacting with R1684.64 and Y2646.51 from APJ but does not induce the hydrophobic interactions with the orthosteric subpocket (Fig. 3, A and C). A previous study showed that the APJ mutants Y35A, W85A, and R168A abolished the agonist activity of AMG3054, suggesting that the hydrophobic interactions with Y351.39 and W852.60 and a polar interaction with R1684.64 are critical for receptor activation. We reasoned that introduction of aromatic ring–containing amino acids, such as Y, F, and W, by site mutation or insertion into the tip of CDR3 could possibly establish the missing hydrophobic interactions, and if so, we may be able to convert sdAb JN241 from an antagonist into an agonist (fig. S4). To test this hypothesis, we generated and tested a panel of nine JN241 single insertion or site mutants for antagonist and agonist activities (table S3). Prominently, a single tyrosine insertion between E104 and S105 in CDR3 of JN241 resulted in a full agonist JN241-9 with median effective concentration (EC50) values of 36 and 47 nM in cAMP assay and β-arrestin recruitment assay, respectively. Insertion of a phenylalanine between E104 and S105 converted JN241 into a partial agonist JN241-7 with an EC50 of 151 nM in cAMP assay, while insertion of a bulky tryptophan residue at the same position (JN241-8) failed to convert JN241 into an agonist (Fig. 3, D to F, and table S3). Directly mutating E104 or S105 to tyrosine or phenylalanine did not convert JN241 to an agonist (table S3). The two site mutants carrying tryptophan substitutions, JN241-2 and JN241-5, also retained strong antagonist activity (table S3).
Fig. 3. Structure-guided rational design and experimental validation for the conversion of sdAb JN241 antagonist into agonist.
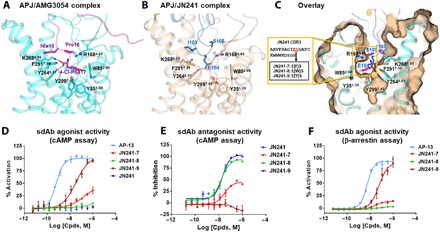
(A and B) Binding mode of peptide agonist AMG3054 in APJ-AMG3054 cocrystal structure (A) in comparison to that of JN241 CDR3 in APJ-JN241 cocrystal structure (B) at the orthostric site. AMG3054-bound APJ and AMG3054 are colored in cyan and magenta, respectively. JN241-bound APJ and JN241 are colored in light orange and sky blue, respectively. Hydrogen bonds are shown as black dashed lines. Key residues in the C-terminal AMG3054 (Nle15, Pro16, and 4-Cl-Phe17) and in JN241 CDR3 (I103, E104, and S105) contributing to the polar interactions and hydrophobic interactions in APJ orthosteric site are shown in sticks. (C) Surface overlay of the orthosteric site in APJ-JN241 and APJ-AMG3054 complex crystal structures. Three JN241 mutants, JN241-7, JN241-8, and JN241-9 with F, W, and Y insertion, respectively, between E104 and S105 in CDR3 were shown in the embedded box. (D and E) Functional test of JN241-7, JN241-8, and JN241-9 in the absence (D) or presence (E) of Apelin 13 (AP-13) by cAMP assay in comparison to parental sdAb JN241. (F) Functional test of JN241-7, JN241-8, and JN241-9 for agonistic activities by β-arrestin assay in comparison to AP-13. The function assays were repeated three times, and representative data are shown.
Molecular mechanism of the conversion from sdAb antagonist into agonist
We performed molecular modeling and simulation of wild-type (WT) APJ with antagonistic sdAb JN241 or agonistic sdAb JN241-9 to understand the molecular mechanisms of the conversion. Molecular models of APJ in complex with JN241 or JN241-9 revealed different binding modes at the orthosteric site. The WT APJ–JN241-9 complex model showed additional hydrophobic interactions introduced by the inserted tyrosine with Y351.39, W852.60, Y882.63, and Y2997.43 (Fig. 4, A and B), mimicking the hydrophobic interactions of 4-Cl-Phe17 in AMG3054 with APJ subpocket residues (29). Compared with the APJ-JN241 complex, the simulation of the APJ–JN241-9 complex led to an outward movement of TM6 as an early event of receptor activation (Fig. 4, C and D). We observed that the inserted tyrosine in JN241-9 interacts with Y2997.43 in TM7 and W852.60 and Y882.63 in upper TM2 through hydrophobic interactions and with Y2997.43 by additional hydrogen bond, causing the upward movement of Y2997.43. These interactions at the orthosteric site lastly led to the outward swing of TM6 intracellular portion (Fig. 4, E and F).
Fig. 4. Molecular modeling of WT APJ in complex with JN241 or JN241-9.
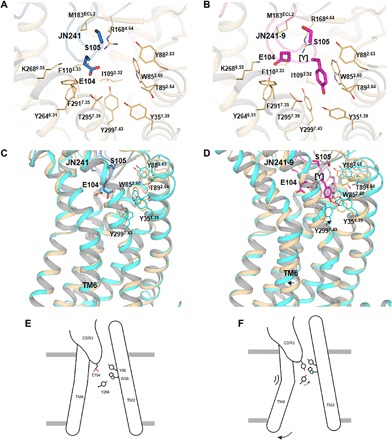
(A and B) Comparison of JN241 (A) and JN241-9 (B) in binding mode at the APJ orthosteric site: (A) model of WT APJ (in light orange) in complex with JN241 (in sky blue) that was built on the basis of APJ-JN241 cocrystal structure and (B) model of WT APJ (in light orange) in complex with JN241-9 (in magenta). (C and D) Comparison of conformational changes of WT APJ-JN241 (C) and WT APJ–JN241-9 (D) complex models before and after 240-ns MD simulation: (C) WT APJ-JN241 complex model before (in light orange and sky blue) and after (in cyan and light blue) simulation and (D) WT APJ–JN241-9 complex model before (in light orange and magenta) and after (in cyan and light pink) simulation. The proteins are shown as cartoons. Residues E104 and S105 in JN241 and residues E104, inserted tyrosine [Y], and S105 in JN241-9 are shown as sticks. APJ residues are shown as lines. The movement of Y2997.43 and TM6 of APJ is labeled with arrows. (E and F) Cartoon illustrations of JN241-mediated APJ antagonism (E) and JN241-9–mediated APJ agonism (F).
JN241-9 notably mimics peptide agonist AMG3054 in binding to APJ at the orthosteric site (29). The binding of JN241-9 or AMG3054 to APJ further resembles the binding of angiotensin II analog S1I8 to AT2R (angiotensin II receptor type II) (30) or AT1R (angiotensin II receptor type I) (31) and the binding of endothelin 1 to the endothelin receptor (32). All these peptides adopt a “C terminus down” orientation in the ligand-binding pocket, and the C-terminal residue largely determines the ligand binding and agonist activity. Deletion or substitution of the C-terminal residue with alanine leads to the loss of peptide ligand binding to the respective receptor and the loss of ligand-mediated receptor signaling or internalization (33, 34). Therefore, we further compared the residues involved in the receptor-ligand interactions at the orthosteric site among these peptide ligand GPCRs. The “Y35-W85-Y88-Y299” subpocket crucial for APJ ligand binding is conserved in AT1R and AT2R (fig. S5 and table S4) (30, 31), suggesting that the strategy for converting antagonistic sdAb JN241 into agonistic sdAb JN241-9 as described in this study may be more generally applied to designing antibody agonists to those GPCR targets that have peptidyl ligands.
DISCUSSION
We report herein the cocrystal structure of the class A GPCR APJ in complex with an orthosteric antibody antagonist and the first example of using structure-guided design to engineer for an antibody agonist. To accomplish this, we first determined that all three CDR loops of sdAb JN241 contribute to its binding and antagonist function. The mechanistic understanding of JN241 functional activity was further refined by the structural information and shown to involve engagement of APJ ECL2, which is consistent with the observations reported for other GPCRs (16, 35). In comparison, we observed that sdAbs binding to APJ N terminus alone were nonfunctional, supporting the notion that antibodies binding the N terminus alone cannot fully antagonize class A GPCRs (19, 36). Griffiths et al. (21) previously reported three CXCR4 antagonistic i-bodies that bind deep in the binding pocket of the receptor as revealed by epitope mapping. Together, class A GPCR antagonism may be mediated by an antibody binding to the ECL2, to the ligand-binding pocket, or to a complex epitope comprising the residues from the both.
We then successfully leveraged the structural information and a mutational analysis of key contact residues in the paratope region to engineer functional variants of JN241. Structural comparison between the APJ-AMG3054 complex and the APJ-JN241 complex prompts us to design a panel of insertion and site mutation variants to convert JN241 to an agonistic sdAb by mimicking the peptide agonist binding at the orthosteric site. One of these variants JN241-9 was shown to have agonist activity and characterized further.
The mechanism by which JN241-9 induces activation of APJ seems to mainly involve the CDR3 that deeply inserted into the ligand-binding pocket. The hydrophobic interactions and the hydrogen bond formation between JN241-9 CDR3 and the orthosteric subpocket appear to be critical for the agonist function. Despite the long CDR3 (23 amino acids) of JN241, insertion of a tyrosine residue is required for converting JN241 into a full agonist. For the partial agonist JN241-7, the inserted phenylalanine may form hydrophobic interactions but may not form hydrogen bond with Y2997.43 due to the lack of hydroxyl group. The bulky tryptophan residue inserted in JN241-8 may cause a steric clash in the subpocket, leading to the failure in inducing receptor conformational changes for activation. Cocrystallization of human APJ with JN241-9 or JN241-7 and resolution of these complex structures in future may further help us understand the mechanisms of antibody-mediated receptor agonism and facilitate structure-based rational design of antibody or peptide agonists.
In summary, we report a successful story of structure-guided discovery of a novel sdAb agonist against class A GPCRs. Our approach combines the power of active immunization with high-resolution structure determination and rationale design to engineer sdAbs with novel functional activities. We have successfully demonstrated this approach on the class A GPCR APJ, and it is our view that a similar approach could be applied to other GPCR targets and could drive the discovery of potent and selective GPCR antibodies with high therapeutic potential.
MATERIALS AND METHODS
Immunogen and antigen preparation
The APJ463 chimeric receptor was constructed with a truncated C terminus from position 331 to 380, a cleavable N-terminal fusion of Bril, and three site mutations—T117N, C325L, and C326M—were generated. A His10 tag was added to the C terminus for purification purposes. This design therefore leaves the ECLs and intracellular loops (ICLs) of APJ intact. APJ463 proteins were expressed in Sf9 insect cells. Membrane preparation, membrane solubilization, and purification from the insect cells were performed as reported previously (29). Protein purity and monodispersity were estimated by SDS–polyacrylamide gel electrophoresis (PAGE) and analytical size exclusion chromatography. Detergent-solubilized APJ463 proteins (micelles) were reconstituted in nanodiscs or liposomes as follows.
In nanodisc reconstitution and purification, the lipid 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine [(POPC) Avanti Polar Lipids] was dried under a nitrogen beam to form a thin layer film, which was then solubilized as a 100 mM solution in 25 mM Hepes (pH 7.4), 150 mM NaCl, and 100 mM sodium cholate. The individual components (APJ-His:MSP1D1:lipid) were mixed with an optimized molar ratio of 1:8:480 in 25 mM Hepes (pH 7.4), 500 mM NaCl, 0.05% n-dodecyl-ß-d-maltoside, and 0.005% cholesteryl hemisuccinate (CHS). The detergent was removed by incubating the mixture with SM-2 Biobeads (Bio-Rad) for at least 2 hours at 4°C. The beads were discarded, and an His affinity chromatography was performed on the supernatant by incubation with Ni-nitrilotriacetic acid resin for 1 hour at 4°C to remove empty nanodiscs. The nanodisc complex was then eluted in 25 mM Hepes (pH 7.4), 150 mM NaCl, and 300 mM imidazole, and the eluate was run on a Superdex 200 10/300 GL (GE Healthcare) using 25 mM Hepes (pH 7.4) and 150 mM NaCl as a running buffer. The fractions of the peak corresponding to the nanodisc complex were pooled. Purified nanodiscs were characterized for ligand binding. APJ-reconstituted nanodiscs were competent for Apelin 13 ligand binding.
In proteoliposome preparation, POPC (Avanti Polar Lipids) was dissolved in chloroform and dried under a stream of nitrogen, followed by incubation overnight under a vacuum. The buffer [25 mM Hepes and 150 mM NaCl (pH 7.4)] was added to the dried lipids, and the mixture was left for 1 hour at room temperature (RT). After vigorous vortexing, liposomes were obtained by extrusion through a polycarbonate filter (100 nm) using a mini-extruder device (Avanti Polar Lipids). Liposomes were first destabilized by addition of 4 mM β-DDM and incubation for 1 hour at RT. This detergent concentration is just above the Rsat value. For DDM, Rsat is 1:1 (mol/mol) and Rsol is 1:1.6 (mol/mol). The β-DDM–solubilized APJ receptor was then added at a protein-to-lipid ratio of 1: 1000 (molar ratio), and the mixture was incubated for 1 hour. The detergent was removed by incubating the mixture with SM-2 Biobeads overnight at 4°C. Proteoliposomes were recovered by ultracentrifugation (100,000g for 30 min) and resuspended in 25 mM Hepes and 150 mM NaCl (pH 7.4).
Camel immunization
A Bactrian camel (Camelus bactrianus) was immunized with APJ463 nanodiscs by subcutaneous injection at a dose of 0.1 mg per injection at five to six sites in the neck region. Institutional Animal Care and Use Committee guidelines were followed with animal subjects. Following primary injection using Freund complete adjuvant, four boosts were carried out at 3-week interval using Freund incomplete adjuvant. Five milliliters of camel blood was collected before immunization and 3 days after each injection for serum titration for APJ463 nanodiscs and membrane scaffold proteins (MSP) by indirect enzyme-linked immunosorbent assay (ELISA).
Library construction and phage display
The peripheral blood mononuclear cells (PBMCs) were prepared from the final bleed of the immunized camel and the heavy chain variable region of heavy chain only antibody (VHH) genes amplified by reverse transcription polymerase chain reaction (PCR) and nest PCR using the total RNAs extracted from the PBMCs as templates. A pair of primers that anneal to heavy-chain antibody leader sequence (pLead, 5′-GTCCTGGCTGCTCTTCTACAAGG-3′) and CH2 (pCH2, 5′-GGTACGTGCTGTTGAACTGTTCC-3′) were used for the first PCR. A second pair of primers that anneal to VHH FR1 (pFR1, 5′-CAGCCGGCCATGGCCSAKGTGCAGCTGGTGGAGTCTGG-3′) and FR4 (pFR4, 5′-ATGATGATGTGCGGCCGCTGAGGAGACRGTGACCWG-3′). The PCR products were digested with BssHII and NheI, gel-extracted, and cloned to a modified pHEN2 phagemid vector, resulting in a large immune sdAb library containing 2 × 109 individual clones. The camel immune sdAb phage library was panned against 50 nM biotinylated APJ463 liposomes in solution. Following three rounds of panning, the second and third round panned libraries were screened for APJ-specific sdAbs by phage ELISA. Positive clones for APJ463 liposomes were further tested for specificity by including irrelevant GPCR liposomes as antigen by phage ELISA.
Expression and purification of sdAbs and sdAb-Fc fusions
Isolated sdAb genes were subcloned to a T7-driven expression vector modified from pComb3 phagemid vector. Soluble sdAbs were expressed in Escherichia coli strain TG1 by induction with 0.5 mM isopropyl-β-d-thiogalactopyranoside overnight at 30°C. Overnight culture was centrifuged at 6000 rpm for 20 min, and the pellet was resuspended in 50 ml of 1× PBS supplemented with proteinase inhibitor. Polymixin B sulfate (P0972, Sigma-Aldrich) was added to the suspension (2,000,000 U/ml in H2O, 2,000,000 U/1 liter of culture pellet) to release to periplasmic sdAbs by incubation at RT for 60 min with gentle shaking. Following centrifugation at 6000g for 10 min, the sdAb-containing supernatant was transferred to a new tube and filtered with a 0.45-μm filter. His-tagged soluble sdAbs were purified by immobilized metal affinity chromatography as follows: 10 mM imidazole (final concentration) was added to the supernatant. The supernatant was then mixed with prewashed Ni-NTA resin (QIAGEN) (0.3 ml of resin/liter of culture) and incubated at RT for 30 to 60 min. The resin was washed three times with 1× PBS containing 20 mM imidazole. Bound sdAbs were eluted by 1 ml of elution buffer (1× PBS supplemented with 250 mM imidazole), and the eluates were dialyzed against 1× PBS to remove imidazole. Antibody concentration was measured by NanoDrop (E = 28; molecular weight, 15) or bicinchoninic acid (BCA) assay (Pierce BCA Protein Assay Reagent, microplate mode).
For production of sdAb-Fc fusion proteins, selected sdAb genes were subcloned to a modified pTT5 mammalian expression vector containing human Fc. Expression was performed in 293F transient expression system (Invitrogen), and sdAb-Fc fusion proteins were purified by protein A affinity purification.
Flow cytometry for epitope characterization
WT APJ and site mutant plasmids were used to transiently transfect 293FT cells. Cells were stained with JN241 and phycoerythrin conjugated to anti-His antibodies as secondary antibody. The expression level of each site mutant was determined by directly staining the cells with Alexa Fluor 488 conjugated to anti–hemagglutinin (HA) antibodies. Relative GeoMean was calculated relative to the parental cells. The ratio of relative GeoMean of JN241 staining to anti-HA staining was calculated.
Biacore binding assay for paratope characterization
In each cycle of binding test between JN241 mutants with APJ nanodisc, the NTA chips (28994951, GE Healthcare) were preconditioned with 1-min pulses of 350 mM EDTA at pH 8.3. NiCl2 (500 μM) was then injected for 90 s. His-tagged APJ nanodiscs were captured at a testing surface around 400 response unit (RU) captured level for testing. Four different doses (12.5, 25, 50, and 100 nM) of JN241 mutant antibodies were sequentially injected into both control and testing flow chambers. The binding curve (resonance unit against time) was obtained after deduction of the signaling from control surface and shown in the sensorgram. After antibody injection, 10 mM glycine-HCl (pH 1.5) and 350 mM EDTA were sequentially injected to regenerate the surface for the next round of testing.
Cell-based functional assays
Stable cell lines overexpressing human APJ (cat. no. 93-0250C2, DiscoveRx) were used in both cAMP assay and β-arrestin recruitment assay in 384-well format. Cells were maintained using the AssayComplete Cell Culture Kit from DiscoveRx (cat. no. 92-3107G). APJ cAMP assay was carried out using a Dynamic 2 cAMP kit (Cisbio), which detects the intracellular cAMP level. Briefly, 1500 cells in 10 μl of assay buffer (1× PBS, 0.1% bovine serum albumin, and 0.5 mM 3-isobutyl-1-methylxanthine) were seeded in each well and incubated with 2.5 μl per well of antibody samples at 37°C for 15 min and then stimulated with Apelin 13 at different concentrations (0 nM for agonist mode, four times EC20 for positive allosteric modulator mode, or four times EC80 for antagonist mode) in assay buffer with 10 μM Forskolin. After stimulation for 30 min, 5 μl of cAMP-d2 and 5 μl of anti–cAMP-Cryptate diluted in lysis buffer were added to the wells for cAMP detection. After incubation at RT for 1 hour, signals were read by EnVision (PerkinElmer) and calculated as a ratio of relative fluorescence units (RFU665nm)/RFU620nm. Raw data were normalized on the basis of data from positive and negative control wells. Dose-response curves were fitted using GraphPad Prism with a four-parameter Sigmoidal function. APJ β-arrestin assay was carried out using the same human APJ cell line and the PathHunter Detection Kit (cat. no. 93-0001, DiscoveRx). Briefly, 5000 cells in 20 μl of AssayComplete Cell Plating 2 Reagent (cat. no. 93-0563R2A, DiscovRx) were plated into each well and cultured for 24 hours. Antibody samples (2.5 μl) were added into the wells, and cells were incubated for 30 min at 37°C. Cells were then stimulated by the addition of 2.5 μl of 10× Apelin 13 at different concentrations (0 nM for agonist mode, 10× EC20 for positive allosteric modulator mode, and 10× EC80 for antagonist mode), followed by incubation at 37°C for 90 min. Twelve microliters of Detection Reagent Working Solution (cat. no. 92-0009, DiscoveRx) were then added to the wells, and the plate was incubated at RT for 60 min. Chemiluminescent signals were read in EnVision (PerkinElmer), and the raw data were normalized on the basis of control wells. Dose-response curves were fitted using GraphPad Prism with a four-parameter Sigmoidal function.
Radioligand-binding assay
Radioligand 125I–Apelin 13 competition binding assays were performed in a 96-well filtration plate according to the manufacturer’s protocol. WT APJ membrane was used for binding to 0.05 nM of 125I–Apelin 13 in the presence of various concentrations of samples in serial dilutions. Radioactivity was determined by TopCount. Dose curves were fit with One-site Fit Ki using GraphPad Prism software.
Receptor internalization assay
HA-tagged APJ was transiently expressed on the surface of U2OS cells that stably expresses β-arrestin–green fluorescent protein. Before the induction by agonists, the surface-existed HA-tagged APJ was labeled by fluorescence conjugated to anti-HA antibody, and the image was captured after induction for 30 min by 100 nM Apelin 13 in the presence or absence of 1 μM JN241. The representative images were shown. Nonstimulated control group was also imaged to show the initial cellular distribution of the receptor and β-arrestin.
APJ-JN241 complex formation and crystallization
Membrane preparation, membrane solubilization, and purification of engineered APJ proteins from insect cells were performed as reported (29). The purified APJ receptor was supplemented with soluble JN241 at 1:2 molar ratio and concentrated in a 100-kD molecular mass cutoff concentrator from Sartorius to 40 mg/ml. Protein purity and monodispersity were estimated by SDS-PAGE and analytical size exclusion chromatography. The concentrated APJ-JN241 complex samples were reconstituted into LCP by mixing with molten lipid in a mechanical syringe mixer. The APJ-JN241 complex–LCP mixture containing 40% (w/w) protein solution, 54% (w/w) monoolein (Sigma-Aldrich), and 6% (w/w) cholesterol (Avanti Polar Lipids) was used to make transparent LCP. Crystallization trials were performed in 96-well glass sandwich plates (Nova) with 40-nl drops and 800-nl precipitant solution mixed in each well using a Graphen crystallization robot, followed by sealing with a glass coverslip. The plates were stored and imaged in an imager (RockImager 1000, Formulatrix) at 20°C. Crystals appeared around 1 day and reached maximum size of ~100 μm within 1 week in 100 mM MES (pH 6.1), 26% poly(ethylene glycol) dimethyl ether 500, 125 mM MgCl2, and 100 mM NaCl. Crystals were harvested directly from the LCP drop with 100-μm MiTeGen micromounts and flash-frozen in liquid nitrogen.
Data collection and structure determination
Crystal diffraction data were collected at the SPring-8 beam line 41XU (Hyogo, Japan) with a Pilatus3 6M detector (x-ray wavelength, 1.0000 Å). The crystals were exposed to a 10-μm beam for 0.2 s and 0.2° oscillation per frame. To prevent radiation damage, data were collected in wedges of 10° to 30° with attenuation at 590 μm before moving onto a different site either on the same crystal or on a new crystal. The data from nine crystals of APJ-JN241 complex were integrated and scaled using XDS41. The initial molecular replacement was performed with Phaser (37) using the 2.6-Å APJ receptor structure [Protein Data Bank (PDB) ID 5VBL] with rubredoxin and sdAb (PDB ID 4NC2). Model building and refinement were carried out using the computer graphic program Coot (38) and the crystallographic refinement software package Phenix (39). Stereochemistry of the model was evaluated by MolProbity, and figures were generated using PyMOL.
Molecular modeling
The initial model of WT APJ (18-327) in complex with JN241 was built via homology modeling in Discovery Studio (Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Systèmes, 2016) using APJ-JN241 cocrystal structure as template. The ICL3 loop of APJ (A229-R244) and a part of CDR3 loop of JN241 (K110-G117) were optimized with Prime Refine Loops in Schrödinger (Schrödinger Release 2017-3: Prime, Schrödinger, LLC, New York, NY, 2017) with implicit membrane placed as aligned APJ structure (PDB ID 5VBL) from the Orientations of Proteins in Membranes database (40). The resultant model was used as template to build APJ–JN241-9 complex model by homology modeling. The two models were embedded in POPC membrane with the TIP3P explicit water model and counterions under the OPLS3 force field. Sodium and chloride ions were added with a concentration of 150 mM. Molecular dynamics simulations were performed on the systems with Desmond in Schrödinger (Schrödinger Release 2017-3: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2017. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY). The systems were relaxed before simulation with default protocol. Two hundred forty–nanosecond simulations were performed in the isothermal-isobaric ensemble (constant temperature and constant pressure ensemble) at a temperature of 300 K and a pressure of 1 bar.
Supplementary Material
Acknowledgments
We thank M. Zhang for supporting this study and for guidance and insightful discussions; H. Yang and X. Chen for technical support; C. King, C. Murawsky, and B. Ason for critical reading of the manuscript; and P. Tagari for a very helpful discussion. Funding: The study was funded by Amgen Inc., and the authors were employees of Amgen at the time the research was conducted. Author contributions: Yanbin Ma designed, optimized, and characterized APJ constructs, performed cocrystallization of APJ-JN241 and data collection and processing, assisted in structural determination, and designed JN241 conversion mutants. Y.D. reconstituted APJ463 micelles in nanodiscs and liposomes, performed panning and screening of the APJ camel immune phage–displayed sdAb library, and produced soluble JN241 for cocrystallization and JN241 site mutants fused to Fc for paratope characterization. X.S. solved APJ-JN241 cocrystal structure and performed structure refinement and analysis. X.M. performed Biacore analysis, receptor endocytosis study using confocal microscope, and flow cytometry with APJ site mutants for epitope characterization. X.L. performed molecular modeling and MD simulation analysis and designed site mutants for epitope and paratope characterization. N.Z. performed flow cytometry with JN241 and other APJ sdAbs for epitope localization and produced JN241 conversion mutants fused to human Fc. Y.So., Y.Su., and Y.Sh. contributed to radioligand binding, cAMP, and β-arrestin assays. W.Z. and L.A.H. assisted in study design, data interpretation, and manuscript preparation. Yingli Ma contributed to APJ-JN241 cocrystal data collection and assisted in study design, data interpretation, and manuscript preparation. M.-Y.Z. conceived and supervised the project, designed experiments, reviewed data, and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Coordinates and structure factors are deposited in the PDB with ID 6KNM.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/3/eaax7379/DC1
Fig. S1. Binding and thermo-stabilizing effect of JN241 to APJ and formation of stable APJ-JN241 complex and co-crystals.
Fig. S2. Simple omit maps of JN241 CDR1, CDR2 and CDR3.
Fig. S3. Characterization of JN241 epitope and identification of the critical residue E174 in APJ ECL2 for its binding and function.
Fig. S4. Amino acid sequence alignment of JN241 and its nine mutants.
Fig. S5. Comparison of the receptor-ligand interactions.
Table S1. Data collection and structure refinement statistics.
Table S2. Interaction residues on APJ-JN241 interface according to the cocrystal structure.
Table S3. EC50 and IC50 values of JN241 and its mutants fused to human Fc in APJ cAMP and β-arrestin assays.
Table S4. Conservation of WT APJ, AT1R, and AT2R residues critical for ligand binding.
REFERENCES AND NOTES
- 1.Hutchings C. J., Koglin M., Olson W. C., Marshall F. H., Opportunities for therapeutic antibodies directed at G-protein-coupled receptors. Nat. Rev. Drug Discov. 16, 787–810 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Hauser A. S., Attwood M. M., Rask-Andersen M., Schiöth H. B., Gloriam D. E., Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayoub M. A., Crépieux P., Koglin M., Parmentier M., Pin J.-P., Poupon A., Reiter E., Smit M., Steyaert J., Watier H., Wilkinson T., Antibodies targeting G protein-coupled receptors: Recent advances and therapeutic challenges. MAbs 9, 735–741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dolgin E., First GPCR-directed antibody passes approval milestone. Nat. Rev. Drug Discov. 17, 457–459 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Mullard A., FDA approves second GPCR-targeted antibody. Nat. Rev. Drug Discov. 17, 613 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Dodd R. B., Wilkinson T., Schofield D. J., Therapeutic monoclonal antibodies to complex membrane protein targets: Antigen generation and antibody discovery strategies. BioDrugs 32, 339–355 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Koth C. M., Murray J. M., Mukund S., Madjidi A., Minn A., Clarke H. J., Wong T., Chiang V., Luis E., Estevez A., Rondon J., Zhang Y., Hötzel I., Allan B. B., Molecular basis for negative regulation of the glucagon receptor. Proc. Natl. Acad. Sci. U.S.A. 109, 14393–14398 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang L., Yang D., de Graaf C., Moeller A., West G. M., Dharmarajan V., Wang C., Siu F. Y., Song G., Reedtz-Runge S., Pascal B. D., Wu B., Potter C. S., Zhou H., Griffin P. R., Carragher B., Yang H., Wang M.-W., Stevens R. C., Jiang H., Conformational states of the full-length glucagon receptor. Nat. Commun. 6, 7859 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ravn P., Madhurantakam C., Kunze S., Matthews E., Priest C., O’Brien S., Collinson A., Papworth M., Fritsch-Fredin M., Jermutus L., Benthem L., Gruetter M., Jackson R. H., Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose-dependent insulinotropic polypeptide receptor. J. Biol. Chem. 288, 19760–19772 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hennen S., Kodra J. T., Soroka V., Krogh B. O., Wu X., Kaastrup P., Ørskov C., Rønn S. G., Schluckebier G., Barbateskovic S., Gandhi P. S., Reedtz-Runge S., Structural insight into antibody-mediated antagonism of the glucagon-like peptide-1 receptor. Sci. Rep. 6, 26236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain S., Yuan H., Spare N., Silberstein S. D., Erenumab in the treatment of migraine. Pain Manag 8, 415–426 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Mumaw M. M., de la Fuente M., Arachiche A., Wahl J. K. III, Nieman M. T., Development and characterization of monoclonal antibodies against protease activated receptor 4 (PAR4). Thromb. Res. 135, 1165–1171 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olson W. C., Rabut G. E. E., Nagashima K. A., Tran D. N. H., Anselma D. J., Monard S. P., Segal J. P., Thompson D. A. D., Kajumo F., Guo Y., Moore J. P., Maddon P. J., Dragic T., Differential inhibition of human immunodeficiency virus type 1 fusion, gp120 binding, and CC-chemokine activity by monoclonal antibodies to CCR5. J. Virol. 73, 4145–4155 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trkola A., Ketas T. J., Nagashima K. A., Zhao L., Cilliers T., Morris L., Moore J. P., Maddon P. J., Olson W. C., Potent, broad-spectrum inhibition of human immunodeficiency virus type 1 by the CCR5 monoclonal antibody PRO 140. J. Virol. 75, 579–588 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rossant C. J., Carroll D., Huang L., Elvin J., Neal F., Walker E., Benschop J. J., Kim E. E., Barry S. T., Vaughan T. J., Phage display and hybridoma generation of antibodies to human CXCR2 yields antibodies with distinct mechanisms and epitopes. MAbs 6, 1425–1438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng L., Damschroder M. M., Cook K. E., Wu H., Dall’Acqua W. F., Molecular basis for the antagonistic activity of an anti-CXCR4 antibody. MAbs 8, 163–175 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douthwaite J. A., Sridharan S., Huntington C., Hammersley J., Marwood R., Hakulinen J. K., Ek M., Sjögren T., Rider D., Privezentzev C., Seaman J. C., Cariuk P., Knights V., Young J., Wilkinson T., Sleeman M., Finch D. K., Lowe D. C., Vaughan T. J., Affinity maturation of a novel antagonistic human monoclonal antibody with a long VH CDR3 targeting the Class A GPCR formyl-peptide receptor 1. MAbs 7, 152–166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X., Duan X., Yang K., Zhang W., Zhang C., Fu L., Ren Z., Wang C., Wu J., Lu R., Ye Y., He M., Nie C., Yang N., Wang J., Yang H., Liu X., Tan W., Comparative analysis of immune repertoires between Bactrian camel’s conventional and heavy-chain antibodies. PLOS ONE 11, e0161801 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradley M. E., Dombrecht B., Manini J., Willis J., Vlerick D., De Taeye S., Van den Heede K., Roobrouck A., Grot E., Kent T. C., Laeremans T., Steffensen S., Van Heeke G., Brown Z., Charlton S. J., Cromie K. D., Potent and efficacious inhibition of CXCR2 signaling by biparatopic nanobodies combining two distinct modes of action. Mol. Pharmacol. 87, 251–262 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Maussang D., Mujić-Delić A., Descamps F. J., Stortelers C., Vanlandschoot P., Stigter-van Walsum M., Vischer H. F., van Roy M., Vosjan M., Gonzalez-Pajuelo M., van Dongen G. A. M. S., Merchiers P., van Rompaey P., Smit M. J., Llama-derived single variable domains (nanobodies) directed against chemokine receptor CXCR7 reduce head and neck cancer cell growth in vivo. J. Biol. Chem. 288, 29562–29572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Griffiths K., Dolezal O., Cao B., Nilsson S. K., See H. B., Pfleger K. D. G., Roche M., Gorry P. R., Pow A., Viduka K., Lim K., Lu B. G. C., Chang D. H. C., Murray-Rust T., Kvansakul M., Perugini M. A., Dogovski C., Doerflinger M., Zhang Y., Parisi K., Casey J. L., Nuttall S. D., Foley M., i-bodies, human single domain antibodies that antagonize chemokine receptor CXCR4. J. Biol. Chem. 291, 12641–12657 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang P., Read C., Kuc R. E., Buonincontri G., Southwood M., Torella R., Upton P. D., Crosby A., Sawiak S. J., Carpenter T. A., Glen R. C., Morrell N. W., Maguire J. J., Davenport A. P., Elabela/Toddler is an endogenous agonist of the apelin APJ receptor in the adult cardiovascular system, and exogenous administration of the peptide compensates for the downregulation of its expression in pulmonary arterial hypertension. Circulation 135, 1160–1173 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuba K., Sato T., Imai Y., Yamaguchi T., Apelin and Elabela/Toddler; double ligands for APJ/apelin receptor in heart development, physiology, and pathology. Peptides 111, 62–70 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Ashley E. A., Powers J., Chen M., Kundu R., Finsterbach T., Caffarelli A., Deng A., Eichhorn J., Mahajan R., Agrawal R., Greve J., Robbins R., Patterson A. J., Bernstein D., Quertermous T., The endogenous peptide apelin potently improves cardiac contractility and reduces cardiac loading in vivo. Cardiovasc. Res. 65, 73–82 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Japp A. G., Cruden N. L., Barnes G., van Gemeren N., Mathews J., Adamson J., Johnston N. R., Denvir M. A., Megson I. L., Flapan A. D., Newby D. E., Acute cardiovascular effects of apelin in humans: Potential role in patients with chronic heart failure. Circulation 121, 1818–1827 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Japp A. G., Cruden N. L., Amer D. A. B., Li V. K. Y., Goudie E. B., Johnston N. R., Sharma S., Neilson I., Webb D. J., Megson I. L., Flapan A. D., Newby D. E., Vascular effects of apelin in vivo in man. J. Am. Coll. Cardiol. 52, 908–913 (2008). [DOI] [PubMed] [Google Scholar]
- 27.Warne T., Serrano-Vega M. J., Baker J. G., Moukhametzianov R., Edwards P. C., Henderson R., Leslie A. G. W., Tate C. G., Schertler G. F. X., Structure of a β1-adrenergic G-protein-coupled receptor. Nature 454, 486–491 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dore A. S., Robertson N., Errey J. C., Ng I., Hollenstein K., Tehan B., Hurrell E., Bennett K., Congreve M., Magnani F., Tate C. G., Weir M., Marshall F. H., Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure 19, 1283–1293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Y., Yue Y., Ma Y., Zhang Q., Zhou Q., Song Y., Shen Y., Li X., Ma X., Li C., Hanson M. A., Han G. W., Sickmier E. A., Swaminath G., Zhao S., Stevens R. C., Hu L. A., Zhong W., Zhang M., Xu F., Structural basis for apelin control of the human apelin receptor. Structure 25, 858–866.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Asada H., Horita S., Hirata K., Shiroishi M., Shiimura Y., Iwanari H., Hamakubo T., Shimamura T., Nomura N., Kusano-Arai O., Uemura T., Suno C., Kobayashi T., Iwata S., Crystal structure of the human angiotensin II type 2 receptor bound to an angiotensin II analog. Nat. Struct. Mol. Biol. 25, 570–576 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Wingler L. M., McMahon C., Staus D. P., Lefkowitz R. J., Kruse A. C., Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 176, 479–490.e12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shihoya W., Nishizawa T., Okuta A., Tani K., Dohmae N., Fujiyoshi Y., Nureki O., Doi T., Activation mechanism of endothelin ETB receptor by endothelin-1. Nature 537, 363–368 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Iturrioz X., Gerbier R., Leroux V., Alvear-Perez R., Maigret B., Llorens-Cortes C., By interacting with the C-terminal Phe of apelin, Phe255 and Trp259 in helix VI of the apelin receptor are critical for internalization. J. Biol. Chem. 285, 32627–32637 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murza A., Parent A., Besserer-Offroy E., Tremblay H., Karadereye F., Beaudet N., Leduc R., Sarret P., Marsault É., Elucidation of the structure-activity relationships of apelin: Influence of unnatural amino acids on binding, signaling, and plasma stability. ChemMedChem 7, 318–325 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Soave M., Cseke G., Hutchings C. J., Brown A. J. H., Woolard J., Hill S. J., A monoclonal antibody raised against a thermo-stabilised β1-adrenoceptor interacts with extracellular loop 2 and acts as a negative allosteric modulator of a sub-set of β1-adrenoceptors expressed in stable cell lines. Biochem. Pharmacol. 147, 38–54 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishchenko A., Wacker D., Kapoor M., Zhang A., Han G. W., Basu S., Patel N., Messerschmidt M., Weierstall U., Liu W., Katritch V., Roth B. L., Stevens R. C., Cherezov V., Structural insights into the extracellular recognition of the human serotonin 2B receptor by an antibody. Proc. Natl. Acad. Sci. U.S.A. 114, 8223–8228 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McCoy A. J., Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Adams P. D., Grosse-Kunstleve R. W., Hung L.-W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C., PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Lomize M. A., Pogozheva I. D., Joo H., Mosbery H. I., Lomize A. L., OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/3/eaax7379/DC1
Fig. S1. Binding and thermo-stabilizing effect of JN241 to APJ and formation of stable APJ-JN241 complex and co-crystals.
Fig. S2. Simple omit maps of JN241 CDR1, CDR2 and CDR3.
Fig. S3. Characterization of JN241 epitope and identification of the critical residue E174 in APJ ECL2 for its binding and function.
Fig. S4. Amino acid sequence alignment of JN241 and its nine mutants.
Fig. S5. Comparison of the receptor-ligand interactions.
Table S1. Data collection and structure refinement statistics.
Table S2. Interaction residues on APJ-JN241 interface according to the cocrystal structure.
Table S3. EC50 and IC50 values of JN241 and its mutants fused to human Fc in APJ cAMP and β-arrestin assays.
Table S4. Conservation of WT APJ, AT1R, and AT2R residues critical for ligand binding.