

We report a potent pan-RAS–targeting antibody and the corresponding therapeutic strategy against RAS mutant tumors.
Abstract
Oncogenic RAS mutant (RASMUT) proteins have been considered undruggable via conventional antibody regimens owing to the intracellular location restricting conventional-antibody accessibility. Here, we report a pan-RAS–targeting IgG antibody, inRas37, which directly targets the intracellularly activated form of various RASMUT subtypes after tumor cell–specific internalization into the cytosol to block the interactions with effector proteins, thereby suppressing the downstream signaling. Systemic administration of inRas37 exerted a potent antitumor activity in a subset of RASMUT tumor xenografts in mice, but little efficacy in RASMUT tumors with concurrent downstream PI3K mutations, which were overcome by combination with a PI3K inhibitor. The YAP1 protein was up-regulated as an adaptive resistance-inducing response to inRas37 in RASMUT-dependent colorectal tumors; accordingly, a combination of inRas37 with a YAP1 inhibitor manifested synergistic antitumor effects in vitro and in vivo. Our study offers a promising pan-RAS–targeting antibody and the corresponding therapeutic strategy against RASMUT tumors.
INTRODUCTION
Oncogenic mutations in RAS genes (KRAS, NRAS, and HRAS) are found in approximately 30% of human cancers, with the highest incidence in pancreatic (~98%), colorectal (~52%), and lung (~32%) adenocarcinomas (1, 2). RAS proteins are small guanosine triphosphatases functioning as molecular switches on the cytosolic side of the plasma membrane by alternating between a guanosine triphosphate (GTP)–bound active (RAS·GTP) form and a guanosine diphosphate (GDP)–bound inactive (RAS·GDP) form (2, 3). The active RAS·GTP form adopts distinct conformations in the switch I and II regions from those of the inactive form to physically interact with many effector proteins, including RAF and phosphatidylinositol 3-kinase (PI3K), and to initiate downstream signaling (1–3). Oncogenic mutations of RAS, predominantly found at positions G12, G13, and Q61, fix the mutants in a constitutive “on” state due to a defect in cycling off to the resting RAS·GDP form; this situation causes persistent protein-protein interactions (PPIs) with effector proteins, thereby resulting in aberrant activation of RAS-effector signaling pathways that promote tumorigenesis and cancer cell survival and proliferation (1–3).
Thus, oncogenic RAS mutant (RASMUT) proteins are appealing high-priority drug targets for anticancer therapy. However, agents directly targeting RASMUT are not yet clinically available, mainly because of a lack of suitable small-molecule–binding pockets on RAS surface and the intracellular location restricting conventional-antibody accessibility (4). Despite these challenges, diverse approaches were recently designed to directly target RASMUT (5), for example, small-molecule inhibitors (6, 7) and protein-based inhibitors (8–10). In particular, mutation-specific covalent small-molecule inhibitors that specifically target the KRASG12C subtype have shown in vivo antitumor activity and are now in clinical trials (11, 12), but they are limited to KRASG12C-harboring cancers. As for protein-based inhibitors, they have limited practical applications owing to their inability to reach the cytosol of living cells.
Because RAS protein activates more than 11 downstream effector proteins with distinct downstream signaling through PPIs (2, 3), the most effective approach may be the blockade of the PPIs by targeting the highly conserved PPI interfaces, namely, the switch I/II regions, across various RASMUT subtypes to broadly cover numerous RASMUT proteins (13). Given that the PPIs are mediated by large and flat surface interfaces, biologics with a large surface area for interaction with targets may be effective pan-RAS inhibitors with high specificity and affinity (14). Our group recently reported such a pan-RAS–specific antibody, dubbed RT11-i, which, on its own, gains access to the cytosol after cellular internalization and then specifically binds to only the active RAS form by recognizing the activated switch I/II regions, thereby blocking RASMUT–effector PPIs (15). This design of an antibody is termed iMab (15). RT11-i shows broad selectivity against the active form of the three RAS isoforms and their subtype mutants and thereby inhibits in vivo growth of tumors harboring various RASMUT subtypes by attenuating the downstream signaling, such as RAF–MEK [mitogen-activated protein kinase (MAPK) kinase]–ERK (extracellular signal–regulated kinase) and PI3K (phosphatidylinositol 3-kinase)–AKT cascades (15). Thus, RT11-i was categorized as a pan-RAS iMab. Furthermore, RT11-i synergistically increases the antitumor activity of gemcitabine by inhibiting KRASMUT downstream signaling in mouse models of orthotopic pancreatic cancer (16). Nonetheless, RT11-i needs to be further optimized for high potency and developability for practical clinical studies.
Here, we report engineering and characterization of a second-generation pan-RAS iMab, inRas37, with improved biophysical properties and a potent antitumor activity. We show that inRas37 as monotherapy has a substantial antitumor activity toward a subset of RASMUT cell lines and cell line–derived xenograft (CDX) mouse models but little efficacy against other RASMUT tumors with concurrent activating mutations in downstream and/or compensatory signaling pathways. We also present combination strategies to overcome the resistance to inRas37.
RESULTS
Design and preparation of an active RAS-specific iMab, inRas37
The first-generation pan-RAS iMab RT11-i comprises three functional parts in human immunoglobulin G1 (IgG1) format (15): (i) an integrin ανβ3/ανβ5–targeting cyclic peptide (RGD10) fused to the N terminus of the light chain (LC) for tumor cell–specific cellular internalization via receptor-mediated endocytosis, (ii) an LC variable domain (VL) with the capacity for endosomal escape for cytosolic localization, and (iii) a heavy-chain variable domain (VH) with specific binding activity for the active form of RAS. The second-generation pan-RAS iMab, inRas37, was generated here by stepwise engineering of the three components and its Fc region (Fig. 1A), as described in detail in fig. S1. We first replaced the VL of RT11-i with CT03 VL (17), which contains a ~3-fold more effective endosomal escape motif 92WYW94 (TrpL92-TyrL93-TrpL94, where L is the LC with Kabat numbering) in the third complementarity-determining region of VL (VL-CDR3) in comparison with the original 92YYH94 motif (Fig. 1A) (18). Moreover, the CT03 VL has a modified VL-CDR1 sequence to abrogate the nonspecific binding of RT11-i VL to cell surface–expressed heparan sulfate proteoglycan (HSPG) (17). We also coevolved RT11 VH in tandem with CT03 VL for enhanced affinity for the active form of RAS by generating VH-CDR1– and VH-CDR3–modified library based on the yeast surface display technology (15), thereby obtaining RT22 VH (fig. S2A). Given that epithelial cancer cells usually overexpress integrin αvβ5 rather than integrin αvβ3 [overexpressed on tumor-associated endothelial cells (19)], the integrin ανβ3/ανβ5–targeting RGD10 cyclic peptide was engineered using phage display to increase affinity for integrin αvβ5, thus yielding the “in4” cyclic peptide (fig. S2B). Last, we introduced mutations L234A, L235A, and P329G (so-called LALAPG mutation) (20) into the Fc region to improve pharmacokinetics (PK) by minimizing Fcγ receptor–mediated clearance (21). The abovementioned engineered components were integrated into inRas37 in human IgG1 format (Fig. 1A). As a control antibody, inCT37 was generated containing the same components as inRas37 except that inCT37 has the null VH of TMab4 without a RAS-binding activity (fig. S1) (15, 22).
Fig. 1. The engineered active RAS-specific iMab, inRas37, shows target cell–specific cytosol-penetrating activity with improved developability and endosomal escape.
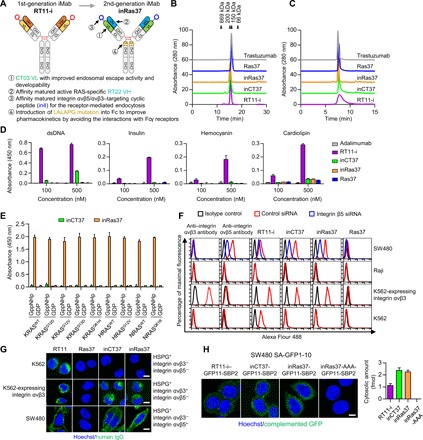
(A) Engineering procedures to develop a second-generation active RAS-specific iMab, inRas37. (B and C) Elution profiles of antibodies purified on a size exclusion Superdex (B) and hydrophobic Zenix (C) column, with monitoring at 280 nm. The arrows indicate the elution positions of molecular weight standards. (D) Evaluation of nonspecific binding of the indicated antibodies to four antigens [double-stranded DNA (dsDNA), insulin, hemocyanin, and cardiolipin], as determined by ELISAs. (E) Selective binding of inRas37 (100 nM) to the GppNHp-bound active form of the indicated RAS proteins in comparison with the inactive GDP-bound form, as determined by an ELISA. (F) Binding specificity of the indicated antibodies (each at 20 nM) to integrin αvβ3 and integrin αvβ5 expressed on the surface of the indicated human cancer cells (flow cytometry data). siRNA, small interfering RNA. (G) Cellular internalization and cytosolic localization of the indicated antibodies (green) in the cells treated with 1 μM antibody for 12 hours before microscopic confocal analysis. Scale bars, 20 μm. (H) Split-GFP complementation assay to determine cytosolic localization of GFP11-SBP2–fused antibodies in SW480-SA-GFP1–GFP10 cells after 6-hour treatment with 1 μM antibody. Right: Quantified amount of cytosolic antibodies. Scale bar, 20 μm.
inRas37 has improved biophysical and biochemical properties
Both inRas37 and inCT37 were expressed predominantly in the correctly assembled monomeric form (≥97%) (Fig. 1B) with purification yields of 70 ± 6 mg and 65 ± 7 mg, respectively, from 1 liter of culture of transiently transfected human embryonic kidney (HEK) 293F cells, much higher than 10 ± 2 mg for RT11-i. To assess colloidal stability, antibodies were analyzed by standup monolayer adsorption chromatography (SMAC) on a Zenix column, where the retention times are inversely related to their colloidal stability, with antibodies prone to precipitation or aggregation being retained longer on the column with broader peaks (23). While parental RT11-i was eluted with a broad asymmetric peak, inRas37 and inCT37 each yielded a sharp symmetrical peak with retention time close to that of a clinically approved antibody, trastuzumab (Herceptin) (Fig. 1C). A multiantigen nonspecificity enzyme-linked immunosorbent assay (ELISA) using four antigens [double-stranded DNA, insulin, hemocyanin, and cardiolipin; (24)] revealed that parental RT11-i substantially bound to all these antigens at a high concentration, but inRas37 and inCT37 did not, and neither did adalimumab (Humira) (Fig. 1D), suggesting the absence of off-target specificity in inRas37 and inCT37. Together, these results meant that inRas37 was successfully engineered to have desirable developability characteristics.
When evaluated for RAS-binding specificity, RT22 VH–integrating inRas37 selectively bound only to the GppNHp (nonhydrolyzable GTP analog)–loaded active form of all three wild-type RAS (RASWT) proteins and their various subtype mutants, with negligible binding to the inactive GDP-loaded form (Fig. 1E and fig. S2C). inRas37 showed approximately twofold stronger affinity only for the active form of various RASMUT subtypes [KD (dissociation constant) ≈ 4 to 12 nM] (table S1) than those of RT11-i (15). These results suggest that inRas37 maintains the binding epitopes of RT11-i, namely, the PPI interfaces of active RAS form (15), thereby retaining the pan-RAS specificity of RT11-i against only the active RAS forms, while showing greater binding affinity than that of RT11-i.
inRas37 reaches the cytosol of cells via better endosomal escape after integrin αvβ3/αvβ5–specific endocytosis
Specificity evaluation of in4 cyclic peptide–fused inRas37 and inCT37 revealed that they specifically bound to cell surface–expressed integrin ανβ3 and integrin ανβ5 (Fig. 1F), thereby showing approximately twofold stronger affinity for integrin αvβ5 (KD ≈ 3.1 to 4.1 nM) but more than twofold weaker affinity for integrin αvβ3 (KD ≈ 1.1 to 4.3 nM) at pH 7.4, as compared with RGD10-fused RT11-i (fig. S2D and table S2). As for antibodies without an LC-fused integrin ανβ3/ανβ5–targeting peptide, the HSPG–cross-reactive parental RT11 got internalized (15), but Ras37 failed to get internalized into HSPG-expressing cells (Fig. 1G), indicating that Ras37 does not undergo nonspecific cellular internalization. Our in4 peptide–fused inRas37/inCT37 reached the cytoplasmic space of the integrin αvβ5– or integrin αvβ3–expressing cells but not integrin αvβ5/αvβ3–deficient cells (Fig. 1G). These results indicated that the in4 peptide was successfully engineered to undergo integrin αvβ5/αvβ3–specific endocytosis with stronger affinity for integrin αvβ5 than that of RGD10.
After cellular internalization, inRas37 needs to be released from the cell surface receptors in the endosomal environment for the subsequent endosomal escape into the cytosol before the lysosomal degradation. The binding affinity values of inRas37 at pH 6.0 for integrin αvβ5 and integrin αvβ3 were ~13- to 20-fold lower than those at pH 7.4 (table S2 and fig. S2D), suggesting that the endosomal acidic pH environment facilitates the dissociation of inRas37 from the receptors for the subsequent endosomal escape (25). Next, when we quantified the endosomal escape efficiency of inRas37 in integrin αvβ5–expressing HeLa and SW480 cells (26), inRas37 showed ~3-fold higher endosomal escape efficiency (~13 to 16%) than did RT11-i (~4 to 6%) (15) at the initial extracellular concentration of 1 μM (table S3). Accordingly, although the cellular uptake efficiency (~3.4 to 4.0%) of inRas37 was lower than that (~4.1 to 5.9%) of HSPG–cross-reactive RT11-i, the enhanced endosomal escape efficiency led to ~2-fold higher cytosolic concentrations (~255 to 531 nM) of inRas37 than those of RT11-i at the initial extracellular concentration of 1 μM (table S3). A split-GFP (green fluorescent protein) complementation assay with one GFP fragment–fused antibody in SW480 cells, which express the other GFP fragment in the cytosol (15, 26), confirmed the improved cytosolic localization of inRas37 and inCT37 over RT11-i (Fig. 1H). The improved endosomal escape ability of inRas37 was ascribed to the newly grafted endosomal escape motif 92WYW94 in VL-CDR3, judging by the loss of the cytosol-penetrating ability of inRas37-AAA with a substitution of 92WYW94 with 92AAA94 (Fig. 1H and fig. S1) (17). Together, these results suggested that inRas37 has greater cytosolic access due to the enhanced endosomal escape efficiency after integrin αvβ3/αvβ5–specific endocytosis.
inRas37 directly targets active RASMUT to block the PPIs with effector proteins
To test whether cytosolic inRas37 can specifically recognize an endogenous active RAS form, we treated integrin ανβ5–expressing human colorectal cancer (CRC) SW480 (KRASG12V) and LoVo (KRASG13D) cells with inRas37 under normal culture conditions. inRas37 colocalized with KRASG12V or KRASG13D on the cytosolic side of the plasma membrane (Fig. 2A). However, in RASWT-harboring CRC HT29 cells, inRas37 accessed the cytosol but did not colocalize with RASWT proteins, in agreement with the observation that RASWT normally resides in the inactive state during quiescence (15, 27). The control inCT37 reached the cytosol of cells but did not colocalize with KRASMUT. Although the endosomal escape motif–deficient inRas37-AAA retained the VH-mediated binding activity toward active RAS (fig. S2E), it failed to show the colocalization with KRASMUT while being entrapped in the endosomes, judging by a mainly punctate fluorescence staining pattern (Fig. 2A).
Fig. 2. inRas37 exerts a potent antiproliferative action on KRASMUT cell lines by inhibiting KRASMUT downstream signaling.
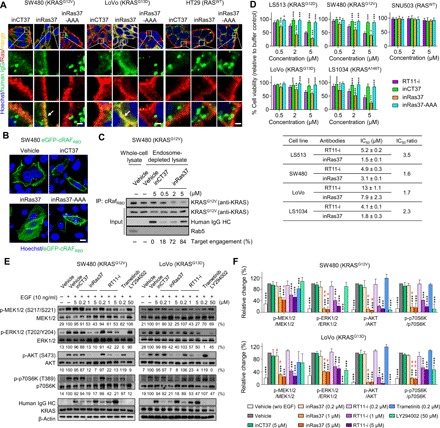
(A) Cellular internalization and colocalization of the indicated antibodies (green) with activated RAS (red) in SW480, LoVo, and HT29 cells treated with 1 μM antibody for 12 hours before microscopic confocal analysis. The arrow indicates the colocalization of inRas37 with activated RAS. Scale bars, 5 μm. (B) Cytoplasmic distribution of the eGFP-cRAFRBD protein (green) in eGFP-cRAFRBD–transformed SW480 cells treated with 1 μM antibody for 12 hours before microscopic confocal analysis. Scale bar, 20 μm. (C) Immunoprecipitation (IP) of active KRASG12V with cRAFRBD from endosome-depleted cell lysates of SW480 cells following treatment with the indicated antibodies at the indicated concentrations for 12 hours. The Rab5 protein was analyzed as an early endosome marker. The number below the panel indicates a percentage of target engagement. (D) Viability of RASWT and various KRASMUT cell lines after treatment every other day (days 0, 2, and 4) with the indicated antibodies at the indicated concentrations for 6 days in three-dimensional spheroid cultures. *P < 0.05, **P < 0.01, and ***P < 0.001 versus the RT11-i–treated group. Bottom: IC50 values for RT11-i and inRas37 toward each cell line. The IC50 ratio was calculated as the IC50 of RT11-i divided by the IC50 of inRas37 for each cell line. (E and F) Representative images (E) and pooled densitometry data (F) of Western blots for SW480 and LoVo cells treated with the indicated antibodies, MEK1/2 inhibitor trametinib, or PI3K-AKT inhibitor LY294002 for 12 hours and then stimulated with EGF (10 ng/ml) for 10 min before cell lysis. The relative band intensity of the phosphorylated proteins toward that of respective total protein was expressed as a percentage of that in the buffer control. The number below the panel indicates the mean (E), and error bars represent means ± SD (F) of at least three independent experiments. ***P < 0.001 for each group versus EGF-stimulated vehicle-treated control; #P < 0.05 and ##P < 0.01 for inRas37 versus RT11-i at each equivalent concentration in each sample (unpaired two-tailed Student’s t test).
We next investigated whether inRas37 can compete with effector proteins for binding to an active RAS form by analyzing the subcellular localization of enhanced GFP–fused cRAF RAS–binding domain (cRAFRBD) (eGFP-cRAFRBD) in eGFP-cRAFRBD–transformed KRASG12V SW480 cells (15). inRas37, but neither inRas37-AAA nor inCT37, displaced eGFP-cRAFRBD from the inner plasma membrane to the cytosol (Fig. 2B), proving that inRas37 orthosterically blocks PPIs between active KRASMUT and effector proteins after cytosolic penetration. To quantify target occupancy of inRas37, SW480 cells pretreated with various concentrations of inRas37 were subjected to immunoprecipitation (IP) of cRAFRBD to selectively pull down the active KRASG12V form. In this assay, the coimmunoprecipitated amount of active KRASG12V was expected to be reduced in proportion to the extent of preexisting shielding of the PPI interfaces of active KRASG12V by inRas37 from cRAFRBD binding. The target engagement of inRas37 was estimated in percentage in comparison with the total precipitated amount of the active KRASG12V form in inCT37-treated cells. inRas37 noticeably engaged KRASG12V with 18, 72, and 84% target engagement at 0.5, 2, and 5 μM, respectively (Fig. 2C), revealing dose-dependent target engagement of inRas37.
inRas37 has an improved antitumor activity toward RASMUT cell lines
The in vitro antiproliferative activities of antibodies for KRASMUT cell lines were examined under spheroid culture conditions (11, 15). Compared with the control antibodies inCT37 and inRas37-AAA, inRas37 significantly inhibited the viability of all KRASMUT cells in a dose-dependent manner, showing activities superior to those of RT11-i but a negligible impact on RASWT cells (Fig. 2D). Judging by the IC50 values (the concentrations causing 50% inhibition of cell viability), inRas37 manifested ~1.6- to 3.5-fold better potency relative to RT11-i, with the potency varying depending on the KRASMUT cell lines (Fig. 2D). We next compared the in vivo antitumor efficacy between RT11-i and inRas37 through intravenous injection twice a week at a dose of 20 mg/kg (mpk) for a total of five doses in athymic nude mice harboring preestablished LoVo (KRASG13D) tumor xenografts. Compared to the inCT37-treated control, inRas37 markedly delayed the tumor growth and showed tumor growth inhibition (TGI; at the end of treatments) of ~75%, notably higher than that (~45%) of RT11-i (fig. S3A), thus manifesting the improved antitumor potency. Treatment with inRas37 did not cause noticeable adverse effects in mice, judging by no significant differences in mouse body weight during treatment and the absence of abnormal histological features of the liver in the mice after the treatment as compared to those of vehicle-treated mice (fig. S3, B to D).
On the basis of the RASMUT–effector PPI–blocking activity of inRas37, we compared the effects on the representative RAS downstream signaling cascades, MAPK (RAF-MEK-ERK) and PI3K [PI3K-AKT-mTOR (mammalian target of rapamycin) (p70S6K)], between inRas37 and RT11-i at three concentrations in KRASG12V SW480 and KRASG13D LoVo cells. Although the MEK1/2 inhibitor trametinib and PI3K inhibitor LY294002 attenuated the targeted signaling, inRas37 and RT11-i inhibited both the MAPK and PI3K pathways, as evidenced by a dose-dependent inhibitory action on growth factor–stimulated formation of phosphorylated MEK1/2, ERK1/2, AKT, and p70S6K (abbreviated as p-MEK1/2, p-ERK1/2, p-AKT, and p-p70S6K, respectively) in the two cell lines (Fig. 2, E and F). Treatment with 1 and 5 μM inRas37 exerted a more potent inhibitory action on the RAS downstream signaling than RT11-i did, judging by the determined mean value of band intensity at each equivalent concentration (Fig. 2E), although significant differences were not observed for all the concentrations (Fig. 2F). The dose-dependent downstream inhibitory activity of inRas37 is in line with the dose-dependent target engagement (Fig. 2C). Together, these data indicated that pan-RAS inRas37 inhibits the proliferation of KRASMUT cell lines by suppressing the downstream signaling via competitive blocking of KRASMUT−effector PPIs; the more efficient PPI inhibition elicited more potent inhibition of downstream signaling, thereby resulting in better growth inhibition.
Systemic administration of inRas37 reveals favorable PK and preferential tumor accumulation
To evaluate the impact of the LALAPG mutation and in4 peptide fusion on plasma PK profiles, the antibodies inRas37 (with LALAPG), inRas07 (without LALAPG), inCT37 (with LALAPG), and Ras37 (with LALAPG, but without in4 peptide fusion; fig. S1) were each intravenously administered at a single dose of 20 mpk to BALB/c athymic nude mice without tumor or with preestablished integrin αvβ5–expressing LoVo tumor xenografts, and then the serum concentrations were measured during a 1-week period. All the antibodies showed the typical biphasic clearance profiles in terms of serum concentration (Fig. 3A). Analysis of PK parameters via the two-compartment model revealed that the terminal serum half-life (T1/2β) of inRas37 was 91.2 ± 8.9 hours and 59.8 ± 2.9 hours for the non–tumor- and tumor-bearing mice, respectively; the corresponding values of Ras37 without integrin αvβ5–targeting cyclic peptide fusion were 90.2 ± 3.7 hours and 86.3 ± 3.5 hours, respectively (Fig. 3A). The much faster clearance of inRas37, but not Ras37, in the tumor-bearing mice than that in the non–tumor-bearing mice was also noted for inRas07 and inCT37, suggesting the presence of a target-mediated elimination of the integrin αvβ5–targeting antibodies, as reported for other tumor-targeting antibodies (28). Of note, LALAPG-harboring antibodies (inRas37 and inCT37) had longer T1/2β than inRas07 did (Fig. 3A), meaning that the LALAPG mutation extends serum half-life, most likely owing to minimization of in vivo depletion by Fcγ receptor–expressing immune cells (21).
Fig. 3. inRas37 has favorable PK and undergoes preferential tumor accumulation.
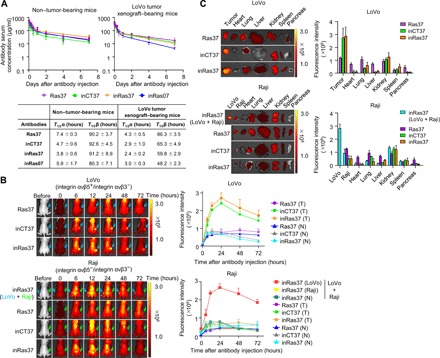
(A) Serum concentration–time profiles of the indicated antibodies following a single intravenous injection at 20 mpk into BALB/c nude mice without tumor (left) or with preestablished LoVo tumor xenografts (initial tumor volume = 120 mm3) (right). Error bars, ±SD (n = 3 per time point). The solid curves represent the fit of a two-compartment PK model to the data to estimate PK parameters: the initial rapid clearance phase (T1/2α) and the latter terminal serum clearance phase (T1/2β), as depicted in the inset table. (B) In vivo tumor-targeting ability of the indicated antibodies, evaluated by intravenous injection of DyLight 755–labeled antibodies (20 μg per mouse) into LoVo (top), Raji, or both LoVo (left flank) and Raji (right flank) tumor xenograft–bearing mice (bottom). Representative images are shown, which were acquired at the indicated time points after injection. Fluorescence intensities in the tumor tissue (T), as indicated by arrows, and normal tissues (N) were quantified by radiant efficiency. (C) Ex vivo analysis of fluorescence intensities quantified by radiant efficiency for excised tumors and normal organs 72 hours after intravenous injection of DyLight 755–labeled antibodies. Tumor tissue and normal organs of one representative mouse from each group are shown. In (B) and (C), error bars represent means ± SD (n = 4 per group).
To determine in vivo targeting specificity of the in4 peptide–fused antibodies, we performed a biodistribution assay with DyLight 755–labeled antibodies in BALB/c athymic nude mice bearing integrin αvβ5–expressing LoVo CDXs or integrin αvβ5–negative Raji CDXs. Compared with Ras37 (without in4 peptide fusion), inRas37 and inCT37 manifested preferential accumulation in LoVo tumors, but not in Raji tumors, as compared to the normal tissues during several days (Fig. 3, B and C). In mice bearing both LoVo tumors in the left thigh and Raji tumors in the right thigh, inRas37 showed ~3-fold higher accumulation only in a LoVo tumor (not in Raji tumors; Fig. 3, B and C). These results validated the in vivo targeting specificity of in4-fused inRas37 and inCT37 antibodies to the integrin αvβ5–expressing tumor tissues.
inRas37 has dose-dependent in vivo antitumor activity with correlations between systemic exposure and target inhibition
To investigate dose-dependent in vivo antitumor efficacy of inRas37 and its relations with PK and pharmacodynamics (PD), inRas37 was intravenously administered twice a week at three doses (5, 10, or 20 mpk) for a total of five doses into mice bearing a preestablished KRASG12V SW480 CDX (Fig. 4A). Antibody administration was nontoxic at all doses according to mouse body weight measurement (fig. S4A). inRas37 noticeably inhibited the tumor growth in a dose-dependent manner with TGI of 16, 55, and 78% at 5, 10 and 20 mpk, respectively, as compared with 20-mpk inCT37-treated controls (Fig. 4B and fig. S4, B and C). Serum concentration–time profiles of inRas37 revealed antibody accumulation in proportion to the injected dose and frequency (Fig. 4C), indicating the dose-proportional plasma PK properties. Western blotting analysis for sampled xenograft explants uncovered a dose-dependent inhibitory activity of inRas37 toward RAS downstream signaling with 10 to 47% down-regulation of p-ERK1/2 and p-AKT at the 5-mpk dose of inRas37 throughout the treatment but maintained almost complete inhibition (>90%) starting from as early as day 3 after 10- and 20-mpk dosing of inRas37 as compared with vehicle-treated controls (Fig. 4D). However, the substantial suppressive effect of the 10- and 20-mpk dosing on the phosphorylation of ERK1/2 and AKT diminished along with tumor growth after day 10 onward, despite the gradual antibody accumulation in serum, probably because of poor tumor tissue penetration of inRas37 in relatively larger tumors (29).
Fig. 4. In vivo antitumor activity of inRas37 correlates with the serum concentrations and target inhibition in the KRASG12V SW480 CDX mouse model.
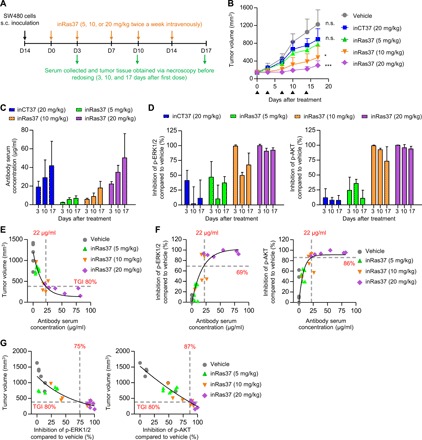
(A) Schematic of dosing and PK and PD assessment of inRas37 in KRASG12V SW480 CDX-bearing BALB/c nude mice. s.c., subcutaneous. (B) Tumor growth curves in response to intravenous injection of the indicated antibodies at the indicated dose twice a week (arrowheads) for a total of five doses (n = 6 per group). *P < 0.05 and ***P < 0.001 versus inCT37; n.s., not significant. (C and D) Serum concentrations of the indicated antibodies (C) and percentage inhibition of p-ERK1/2 (left) and p-AKT (right) formation in tumor tissue lysates (D), as determined before antibody dosing on days 3 and 10 and at the end of treatment on day 17 (n = 3 per group on days 3 and 10 and n = 6 per group on day 17). (E and F) Plot of serum concentrations of inRas37 versus tumor volume (E) and versus percentage down-regulation of p-ERK1/2 (left) and p-AKT (right) (F) on day 17 (n = 6 per group). (G) Plot of tumor volume versus percentage down-regulation of p-ERK1/2 (left) and p-AKT (right) on day 17 (n = 6 per group). In (E) to (G), solid curves indicate the fit to the data via a four-parameter logistic equation.
For the exploratory analysis of the PK-PD relation for inRas37, the serum concentrations of inRas37 were plotted against either tumor volume or down-regulation of p-ERK1/2 and p-AKT as PD markers, all of which were determined at the end of treatment (day 17). The serum concentrations of inRas37 were inversely correlated with tumor volume (Fig. 4E) but well correlated with PD effects (down-regulation of p-ERK1/2 and p-AKT; Fig. 4F and fig. S4D). To achieve 80% TGI, we estimated the required serum concentration of inRas37 to be 22 μg/ml (146 nM; Fig. 4E). This inRas37 serum concentration (146 nM) corresponded to that achieving inhibition of p-ERK1/2 and p-AKT by 69 and 86%, respectively (Fig. 4F). These values roughly matched the observed 75 and 87% down-regulation of p-ERK1/2 and p-AKT, respectively, necessary for 80% TGI (Fig. 4G) (30). These results indicated a direct correlation between the serum concentrations of inRas37 and its PD effects, as well as TGI activity, suggesting that the in vivo antitumor activity of inRas37 was mediated by inhibition of KRASMUT signaling in the SW480 CDX mouse model.
In vitro profiles of sensitivity of RASMUT cancer cells to inRas37
Some reports about RNA interference (RNAi)–based knockdown approaches indicate that RASMUT tumors show heterogeneous dependence of cell survival on RASMUT expression, classifying RASMUT cells into RAS dependent (i.e., RAS addicted) and RAS independent (27, 31, 32). Using inRas37 as a pharmacological tool, we next sought to profile the responses to inRas37 (2 μM) in spheroid culture of 36 cell lines harboring various RASMUT subtypes and 8 cell lines with RASWT from mainly CRC and non–small cell lung cancer (NSCLC) (table S4). Most cell lines were found to express integrin αvβ5 and/or integrin αvβ3 on the cell surface (fig. S5) (19). Again, inRas37 did not show significant cytotoxicity toward RASWT cells (Fig. 5A). However, RASMUT cells manifested varying responses, with a subset of cells showing high sensitivity to inRas37 and other cells putting up moderate to high resistance to inRas37 (Fig. 5A). The percentages of cell viability in the presence of inRas37 were roughly correlated with each RAS gene dependence score (ATARiS) from the Project DRIVE RNAi screen (Fig. 5B) (27). These data suggested that blocking PPIs of RASMUT at the protein level by inRas37 elicits effects similar to those mediated by a RASMUT knockdown. The varying responses were not correlated with particular RASMUT subtypes or the expression levels of integrin αvβ5 and/or integrin αvβ3 on the cell surface (fig. S5), suggesting that the differential sensitivity of RASMUT cells to inRas37 reflected different dependences of cellular proliferation on RASMUT signaling in each cellular context. In keeping with this rationale, we categorized RASMUT cells into two groups: inRas37-sensitive cells (<65% cell viability) and inRas37-resistant cells (>65% cell viability) according to the threshold of 65% cell viability after inRas37 treatment (Fig. 5A).
Fig. 5. In vitro responses of various RASMUT cell lines to inRas37 correlate with the in vivo responses.
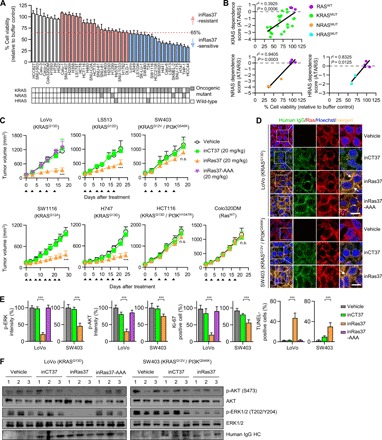
(A) Viability of various RASMUT and RASWT cell lines after treatment every other day (days 0, 2, and 4) with inRas37 (2 μM) for 6 days as compared with buffer-treated control (n = 3). Bottom: Mutation status of RAS genes in the cells. RASMUT cells are colored according to the inRas37 sensitivity threshold at 65% cell viability. (B) Cell viability under the action of inRas37 treatment plotted versus KRAS, NRAS, or HRAS dependence scores (ATARiS) from Project DRIVE. Statistical analysis was performed using linear regression to determine r2 and P values. (C) Tumor growth curves in response to intravenous injection of the indicated antibodies at 20 mpk twice a week (arrowheads) into BALB/c nude mice harboring the indicated tumor xenografts. Error bars, ±SD (n = 6 per group, except for LoVo, n = 7 per group). ***P < 0.001 versus the inCT37 group. (D) IHC analysis of the indicated antibodies (green), with activated RAS (red) in KRASMUT LoVo and SW403 tumor tissues prepared 24 hours after the last treatment. The arrows indicate the colocalization of inRas37 with activated RAS. Scale bars, 10 μm. (E) IHC analysis of p-ERK1/2 (green), p-AKT (green), and Ki-67 (red) and TUNEL (green) staining levels in LoVo- and SW403-derived tumor tissues, as shown in fig. S6B. The panels show the percentage of relative fluorescence intensity compared to that in the vehicle-treated control and the percentage of Ki-67–positive and TUNEL-positive cells compared to the number of Hoechst 33342–stained cells in each sample. Error bars represent means ± SD of five random visual fields for each sample (two tumors per group). ***P < 0.001. (F) Western blot analysis of the indicated proteins in LoVo and SW403 tumor tissue lysates. In (D) to (F), tumor tissues were excised from mice 24 hours after the last treatment, as shown in (C).
In vivo antitumor activity of inRas37 in a mouse CDX model
To validate the in vitro sensitivity as an in vivo response, we assessed antitumor efficacy of inRas37 in athymic nude mice bearing preestablished inRas37-sensitive (LoVo, H747, SW1116, and LS513), inRas37-resistant (SW403 and HCT116), and RASWT (Colo320DM) colorectal CDX tumors. Compared with inCT37 or vehicle control, inRas37 substantially inhibited the in vivo tumor growth with ~54 to 81% TGI for inRas37-sensitive CDXs but did not for inRas37-resistant CDXs (Fig. 5C and fig. S6, A and B). However, inRas37-AAA did not exert any antitumor action even on inRas37-sensitive LoVo CDXs, confirming the necessity of endosomal escape of inRas37 for the blockade of cytosolic RASMUT. In the case of RASWT Colo320DM CDX, inRas37 did not show any significant antitumor activity. Accordingly, the in vivo responsiveness of KRASMUT CDXs to inRas37 correlated well with the in vitro observations.
Immunohistochemical (IHC) examination of excised tumor tissues at the end of treatment showed colocalization of inRas37 with KRASMUT at the inner plasma membrane of tumor cells, whereas inCT37 was observed only in the cytoplasmic regions without colocalization with KRASMUT (Fig. 5D and fig. S7A). This result revealed in vivo KRASMUT target occupancy of inRas37 inside the cytosol of tumor cells after systemic administration. Compared to inCT37- and inRas37-AAA–treated tumors, inRas37-treated tumors featured significantly lower levels of p-ERK1/2 and p-AKT and decreased staining for the cell proliferation marker Ki-67, as well as increased apoptosis as measured by terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) staining for inRas37-sensitive tumors (Fig. 5, E and F, and fig. S7, B to D). However, inRas37-resistant SW403 and HCT116 tumors with PIK3CA mutation (PI3KMUT) showed a substantial decrease in p-ERK1/2 amounts but marginal down-regulation of p-AKT (Fig. 5, E and F, and fig. S7, B to D), suggesting that PI3KMUT-driven downstream activation contributes to the resistance to inRas37.
Activating mutations of PI3K or β-catenin mediate intrinsic resistance to inRas37
To gain insight into the molecular mechanism(s) underlying the differences in sensitivity of RASMUT cell lines to inRas37, we retrieved the genetic background of all the tested cell lines from databases Cancer Cell Line Encyclopedia (CCLE) and Catalogue of Somatic Mutations in Cancer (COSMIC) (33, 34), with a focus on genes associated with RAS downstream compensatory and parallel pathways, which can potentially nullify effects of RASMUT blockade. Consistent with their CRC and NSCLC origins, almost all RASMUT cell lines harbored alterations in other genes such as APC and TP53 (table S4). We found that among the gene mutations examined, gain-of-function activating mutations mainly in PIK3CA (PI3K) and/or, to a lesser extent, in CTNNB1 (β-catenin) were present in most inRas37-resistant RASMUT cell lines (table S4).
To identify pharmacological combinations that overcome the primary resistance to inRas37, we chose five targeted pharmacological inhibitors. They are either clinically approved or in clinical trials and have shown synergistic antitumor effects on KRASMUT tumors—when combined with inhibitors of MEK or RAF (downstream of RAS), such as a PI3K inhibitor (copanlisib) (35), a β-catenin inhibitor (iCRT14) (36), an epidermal growth factor (EGF) receptor (EGFR)/ErbB2 dual inhibitor (afatinib) (37), a fibroblast growth factor receptor 1 (FGFR1) inhibitor (ponatinib) (38), and a Yes-associated protein 1 (YAP1) inhibitor (verteporfin) (39). We measured each combinatorial effect on the viability of six KRASMUT CRC cell lines (three inRas37-sensitive and three inRas37-resistant cell lines) in spheroid culture. We calculated the combination index (CI) for each combination based on IC50 values (40). We regarded a CI below 0.75 as synergistic, between 0.75 and 1.25 as additive, and above 1.25 as antagonistic.
Combined treatment with inRas37 and the PI3K inhibitor (inRas37 + PI3Ki) showed substantial synergy in inRas37-resistant cells with concomitant PIK3CA mutation (PI3KMUT) compared with the inCT37 + PI3Ki combination, but not in inRas37-sensitive cells with wild-type PIK3CA (PI3KWT) (Fig. 6, A and B, and fig. S8A). Likewise, a combination of inRas37 with the β-catenin inhibitor exerted synergistic effects only on the HCT116 cell lines with both PI3KMUT and β-cateninMUT. The other combinations showed only additive effects on the inRas37-resistant cells examined (Fig. 6A). When evaluated in mouse CDX models by means of the Bliss independence model (41), consistent with the in vitro results, the combination of inRas37 and PI3Ki synergized to inhibit the in vivo growth of PI3KMUT-carrying inRas37-resistant CDXs, but not PI3KWT inRas37-sensitive CDXs, when compared with either single agent alone (Fig. 6, C and D). Notably, the combination inRas37 + PI3Ki had a more pronounced effect only on PI3KMUT-carrying SW403 tumors with ~104% TGI in comparison with PI3KMUT/β-cateninMUT double mutant HCT116 tumors (~83% TGI). These findings suggested that β-cateninMUT signaling contributes to the residual growth of HCT116 tumors during KRASMUT/PI3KMUT blocking. Western blotting of the combination-treated tumors uncovered a decrease in both p-ERK and p-AKT amounts in KRASMUT/PI3KMUT double mutant CDXs, as compared with those treated by either agent alone (Fig. 6E). A lack of significant differences in body weight between drug-treated groups meant negligible systemic toxicity of the combination therapy (fig. S8B). These results indicated that constitutive activation of PI3KMUT and/or β-cateninMUT is associated with the primary resistance to KRASMUT blockade by inRas37, but this resistance can be circumvented by the combination of inRas37 with a pharmacological inhibitor of the druggable mutant.
Fig. 6. Combination treatment identifies synergistic drug targets for inRas37 and some pharmacological inhibitors in RASMUT cell lines.
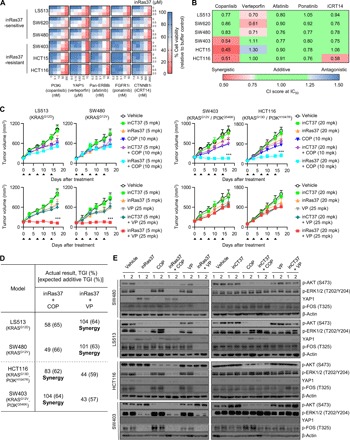
(A) Dose-response effects of combined treatment with inRas37 and one of the five pharmacological inhibitors (“i” in the label) on the viability of KRASMUT cell lines after combined treatment every other day (days 0, 2, and 4) at the indicated concentrations for 6 days in three-dimensional spheroid cultures. Heat maps show percentages of cell viability relative to a buffer-treated control (n = 3). (B) CI at IC50 for the combined treatment shown in (A), analyzed in the CompuSyn software. CI < 0.75 means a synergistic effect, 0.75 ≤ CI ≤ 1.25 denotes an additive effect, and CI > 1.25 means an antagonistic effect. (C) Tumor growth curves in response to the indicated treatments of BALB/c nude mice harboring the indicated tumor xenografts. The arrowheads indicate twice a week intravenous injection of the antibody and COP (PI3Ki copanlisib; top) or intraperitoneal injection of VP (YAP1i verteporfin; bottom) or the combined treatments at the indicated doses. Error bars indicate means ± SD (n = 6 per group). ***P < 0.001 versus group “inCT37 + COP” or “inCT37 + VP.” (D) Percentage of observed TGI and expected additive TGI, calculated via the Bliss independence model. The observed TGI by combination treatment that was higher than the expected additive TGI was considered synergistic. (E) Western blot analysis of the indicated proteins in SW480-, LS513-, HCT116-, and SW403-derived tumor tissues excised from mice 24 hours after the last treatment, as shown in (C).
A combination of inRas37 with the YAP1 inhibitor shows synergy in KRASMUT-dependent tumors
Of note, we found that the combination of inRas37 at a low dose of 5 mpk with the YAP1 inhibitor (YAP1i) synergized only in the inRas37-sensitive CRC cells, almost completely suppressing the in vivo tumor growth (~104 and ~101% TGI for LS513 and SW480 cells, respectively), but not the growth of inRas37-resistant tumors (Fig. 6, A to D). In contrast to inCT37- and vehicle-treated controls, inRas37 as monotherapy up-regulated the YAP1 protein in the inRas37-sensitive cells, but not in the inRas37-resistant cells, both in vitro and in vivo (Fig. 6E and fig. S8C). However, the combination inRas37 + YAP1i abrogated the overexpression of YAP1. These results suggested that YAP1 overexpression is an adaptive bypass signal transduction counteracting the KRASMUT-blocking effect of inRas37 in the tumors (42, 43). Because YAP1 activates transcription factor FOS to induce tumor survival in the absence of KRAS signaling in colon and lung cancer (43), we examined the FOS activation by monitoring the phosphorylation of FOS. Combined treatment inRas37 + YAP1i, but not each single agent, abrogated the phosphorylation of FOS in inRas37-sensitive cells and in CDX tumor tissues (Fig. 6E and fig. S8C). As for inRas37-resistant tumors, inRas37 alone was enough to suppress the phosphorylation of FOS, indicating that FOS is activated by YAP1 up-regulation upon KRASMUT blockage only in KRASMUT-dependent tumors (43). These results suggested that combination therapy inRas37 + YAP1i can synergize the antitumor therapeutic effects on KRASMUT-dependent CRC tumors.
DISCUSSION
Oncogenic RASMUT signaling is mediated by PPIs through the highly conserved switch I/II interfaces, with many effector proteins eliciting distinct downstream signaling. Therefore, blocking PPIs of RASMUT with effectors as far upstream as possible may offer a more effective therapeutic strategy in comparison with separate inhibition of individual downstream targets. In this study, we engineered a target cell– or tissue-specific cytosol-penetrating RASMUT–effector PPI blocking pan-RAS iMab, called inRas37, with improved antitumor potency and developability for practical clinical studies. The engineered inRas37 showed suitable biophysical and biological properties for high expression in cultured mammalian cells, tumor-specific cellular internalization through integrin αvβ3/αvβ5–mediated endocytosis, followed by efficient delivery to the cytosol with a good endosomal escape ability for RASMUT target engagement. inRas37 selectively recognized the active RAS form, but not the inactive form, regardless of RASMUT isoforms and subtypes examined, and thereby orthosterically blocked the PPIs of RASMUT with effector proteins. This PPI blockade by inRas37 led to inhibition of RASMUT downstream signaling and consequent suppression of in vitro and in vivo growth of a subset of cancer cell lines harboring various oncogenic RASMUT proteins.
For targeting a cytosolic protein by the iMab technology, the most challenging task is to achieve sufficient cytosolic access necessary to inactivate the target. inRas37 with an engineered CT03 VL with a strong endosomal escape motif reached the cytosol of cells in proportion to the extracellular concentrations (table S3), resulting in ~72 and ~84% RASMUT target engagement after treatment with 2 and 5 μM inRas37, respectively, in KRASG12V SW480 cells (Fig. 2C). Assuming that the total membrane-anchored concentrations of active RASMUT were 0.4 to 1.6 μM in a cell (44), the high target engagement efficiency indicated that most of the cytosolically localized inRas37 with two arms for target binding engages RASMUT at one-to-two (bivalent) stoichiometry. The bivalent IgG format of inRas37 with single-digit nanomolar affinity seems to contribute to high target engagement efficiency because of the avidity effect, which also explains the strong blocking activity of inRas37 toward PPIs between RAS and effector proteins, including RAF and PI3K with double-digit nanomolar-to-micromolar affinity for active RAS (15). Several recent studies proved that RASMUT may function as a dimer or a high-order oligomer at the inner plasma membrane for the activation of its effectors, and the dimerization is critical for the oncogenic signaling through both RAF-MAPK and PI3K-AKT pathways (45, 46). This finding further supports the importance of bivalent engagement of RASMUT for a blocking agent such as inRas37. The absence of bivalence in RASMUT-targeting proteins may explain the negligible anti-RAS activity (9).
Because there are more than 130 different missense mutations in the RAS oncogene in various cancers (2), a pan-RASMUT blocking agent, rather than an isoform-selective RASMUT inhibitor, is essential for effective treatment of diverse RASMUT cancer types and cells (5). Owing to the pan-RASMUT blocking activity, inRas37 manifested an antitumor activity in a subset of various RASMUT tumors in vitro and in vivo by inhibiting the most prominent downstream signaling—both MAPK and PI3K pathways—thereby highlighting the effectiveness of the RASMUT–effector PPI blockade strategy for RASMUT-dependent tumors. Among small-molecule inhibitors directly targeting RASMUT proteins, KRASG12C-specific inhibitor ARS-1620 (11) and pan-RAS inhibitor 3144 (47) showed in vivo antitumor potency. However, the covalent KRASG12C-specific inhibitors are limited only to patients with KRASG12C tumors, e.g., ~14 and ~7% of KRASMUT NSCLC and CRC cases, respectively (48). Although the pan-RAS 3144 compound bound to various RASMUT subtypes to inhibit the PPIs with RAF and PI3K, it did not strictly distinguish between the active and inactive forms of RASMUT and accompanied off-target binding activity, causing serious in vivo systemic toxicity (47). In contrast, the specificity of inRas37 for the active RAS form may account for the insignificant effects on RASWT tumors, which also means sparing the mouse CDX models from the systemic toxicity of inRas37. Accordingly, pan-RASMUT iMab inRas37 may have a broad therapeutic potential across tumor types harboring various RASMUT without toxicity to RASWT tissues.
By analyzing large panels of RASMUT cell lines and several CDXs, we noted that tumors dependent on RASMUT signaling without alternative activation pathways were strongly sensitive to inRas37. By contrast, RASMUT tumors with secondary mutations in downstream (e.g., PI3KMUT) or parallel (e.g., β-cateninMUT) pathways were not appreciably affected by inRas37, despite significant inhibition of p-ERK1/2 at a level similar to that of inRas37-sensitive KRASMUT tumors. This observation is in line with reports showing primary resistance of RASMUT tumors to a MEK inhibitor because of concurrent mutations in PIK3CA (PI3K) and/or CTNNB1 (β-catenin) (30, 49, 50). The intrinsic resistance of PI3KMUT and/or β-cateninMUT CRC cells to inRas37 was overcome by combined treatment with a PI3K or β-catenin inhibitor, respectively. However, the combination did not show any synergistic effects on inRas37-sensitive cells. These results point to a therapeutic strategy for KRASMUT/PI3KMUT or KRASMUT/β-cateninMUT double mutant tumors, which represent ~5 to 10% and ~1% of all CRC, respectively (51).
Intriguingly, we found that inRas37 provoked up-regulation of transcriptional coactivator YAP1 as a compensatory response only in inRas37-sensitive tumors, but not in inRas37-resistant tumors. YAP1 suppression by a YAP1i substantially synergized with a low dose of inRas37 (5 mpk), even slightly regressing the inRas37-sensitive CDXs. These findings suggest that YAP1 overexpression is the adaptive resistance-inducing response of the KRASMUT-dependent tumor to the KRASMUT blockade by inRas37, consistent with previous studies suggesting that KRASMUT depletion by a knockdown causes overexpression of YAP1 to bypass the loss of KRASMUT signaling in KRASMUT-dependent CRC and pancreatic cancer cells (42, 43). KRAS and YAP1 signaling independently activates transcription factor FOS to regulate tumor survival and epithelial-mesenchymal transition in CRC and NSCLC (43). We demonstrated that the combination of inRas37 and YAP1i, but neither inRas37 nor YAP1i monotherapy, substantially inhibited FOS activation in CRC tumors, suggesting that simultaneous blocking of both KRASMUT-FOS and YAP1-FOS signaling is necessary for effective treatment of KRASMUT-dependent CRC tumors.
In summary, we engineered a pan-RAS inRas37 iMab that effectively inhibits in vitro and in vivo growth of RASMUT-dependent tumors by blocking the PPIs with effector proteins and thereby suppressing downstream signaling. The significant antitumor effect of inRas37 as a single agent in RASMUT-dependent CRC tumors suggests that the blockade of RASMUT–effector PPIs may be an effective therapeutic strategy against RASMUT-dependent CRC. Nonetheless, inRas37 alone was not sufficient for many other RASMUT tumors with alternative oncogenic mutations in the downstream and/or compensatory pathways; this problem can be overcome by the combination with a pharmacological inhibitor of the druggable mutant. Our results provide a promising pan-RAS–blocking antibody, inRas37, and the corresponding therapeutic strategy for RASMUT tumors.
MATERIALS AND METHODS
Study design
No statistical method was used to predetermine sample size, but sample size was similar to sample sizes routinely used in the field. The investigators were not blinded to allocation during experiments. No samples or animals were excluded.
Reagents and antibodies
Chemical and protein reagents and antibodies used as reagents in this study are listed in table S5.
Protein expression and purification
Bacterial expression plasmids for KRASWT (amino acid residues 1 to 188), HRASWT (residues 1 to 189), NRASWT (residues 1 to 189), and their oncogenic mutants, subcloned in-frame into the pGEX-3X vector (GE Healthcare) to be expressed in the N-terminal glutathione S-transferase (GST)–fused form, were described before (15). The GST-fused proteins were expressed in Escherichia coli BL21 (DE3)plysE and purified on glutathione-Sepharose resin (GE Healthcare) as described elsewhere (15). The purified GST-fused RAS proteins were used in the ELISA and surface plasmon resonance (SPR) analysis. The N-terminal 8× His- and Avi-tag (GLNDIFEAQKIEWHE)–fused KRASG12D (residues 1 to 169) were prepared as described before (15) and were used for antibody library screening by fluorescence-activated cell sorting (FACS). Protein concentrations were determined with the Bicinchoninic Acid (BCA) Kit (Thermo Fisher Scientific) and by measuring absorbance at 280 nm. The RAS protein was loaded with the nonhydrolyzable GTP analog GppNHp (Sigma-Aldrich, G0635) or GDP (Millipore, 20-177), as described previously (15).
Generation of RT22 VH
To increase the affinity of the VH for the active Ras·GTP form, we constructed heavy-chain (HC) libraries (VH library–CH1) with randomization of exposed residues of VH-CDR1 (residues 31 to 33) and VH-CDR3 (residues 95 to 97) of the RT11-i VH in single-chain Fab format by pairing with a fixed LC (CT03 VL-Cκ) using the yeast surface display technology, as described before (15). The targeted residues 31, 32, and 33 in VH-CDR1 were diversified using RNC (AG/ACGT/C), THC (T/ACT/C), and KSG (GT/CG/G) degenerate codons, respectively. The targeted residues 95, 96, and 97 of VH-CDR3 were randomized by means of hand-mixed spiked oligonucleotides, which were designed to randomly mutate each residue while retaining the parental amino acids at a level of ~50% at each residue (15). The randomized HC (VH library–CH1) fragment was assembled with the LC (CT03 VL-Cκ) via a G4S-based 63–amino acid linker by serial overlap extension polymerase chain reaction (PCR), generating a single-chain Fab library with a diversity of approximately 2.0 × 108. The library was screened against a biotinylated GppNHp-loaded KRASG12D∙(KRASG12D·GppNHp) active form in the presence of a 10-fold molar excess of a nonbiotinylated GDP-loaded KRASG12D (KRASG12D·GDP) inactive form as a soluble competitor on a FACSAria III instrument (BD Biosciences). We lastly isolated the Ras03 Fab with the engineered RT22 VH and CT03 VL (fig. S1).
Isolation of in4 cyclic peptide from a cyclic-peptide phage library
The phage library was generated on the basis of the RGD10 peptide fused to the N terminus of the CT03 VL–g3p protein in the pComb3X phagemid vector using a primer containing a degenerate codon of NNK (ACGT/ACGT/GT), ARR (A/AG/AG), and GAN (G/A/ACGT) for randomization of the residues surrounding RGD by overlap extension PCRs, as previously described (52) and illustrated in fig. S2B. Polyacrylamide gel electrophoresis–purified primers supplied by Integrated DNA Technology (USA) were used in the PCR. For phage library panning, an immunotube (SPL Life Sciences, 43005) was coated with a soluble antigen (integrin αvβ5; R&D Systems) for 2 hours at 25°C and blocked with 3% skim milk in tris-buffered saline (TBS) [50 mM tris-HCl (pH 7.4) and 137 mM NaCl] for 1 hour at 25°C. After washing with wash buffer [TBS (pH 7.4), supplemented with 2 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2, and 0.05% Tween 20], 1011 plaque-forming units of phages preincubated with a binding solution [TBS (pH 7.4), supplemented with 2 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2, and 3% skim milk] were applied to the immunotube for 1-hour incubation at 25°C. After washing, bound phage particles were eluted with 0.1 M glycine buffer (pH 2.2) and neutralized with 1 M tris buffer (pH 9.0). These eluted phages were used to infect mid-log phase E. coli XL1-Blue cells and were reamplified by phage rescue. During the five rounds of panning, the immobilized integrin αvβ5 amount was reduced (5, 3, 2, 1, and 1 μg), and washing frequency was increased (3, 5, 10, 15, and 25 times). To evaluate affinity for human integrin αvβ5, individual clones, in cyclic peptide–fused CT03 VL format, were inoculated into each well of a 96-well tissue culture plate and grown at 37°C and 250 rpm for 5 hours. Isopropyl-β-d-1-thiogalactopyranoside (1 mM final concentration) was added into each well and incubated at 30°C and 120 rpm for 16 hours. The supernatant containing cyclic peptide–fused CT03 VL was obtained via three cycles of freezing and thawing and was directly subjected to an ELISA. A horseradish peroxidase–conjugated anti-hemagglutinin antibody (Roche) was used for the detection of bound clones using a 3,3′,5,5′-tetramethylbenzidine–ELISA solution (Thermo Fisher Scientific). Each clone was compared with a parent clone (carrying RGD10), and the in4 peptide was eventually selected in terms of the best binding affinity and specificity, as presented in fig. S2B.
Construction of antibody-expressing plasmids
The isolated RT22 VH gene was subcloned in-frame, without additional amino acids, at Not I/Apa I sites of pcDNA 3.4–RT11-HC carrying the human IgG1 constant-domain sequence (CH1-hinge-CH2-CH3) with a mutation of CH3 at three sites (L234A, L235A, and P329G) for HC expression. For LC expression, the CT03 VL gene was subcloned in-frame, without additional amino acids, at Not I/Bsi WI sites of pcDNA 3.4–TMab4-LC carrying the human κ constant domain sequence (residues 108 to 214). For LC expression of integrin αvβ5/αvβ3–targeting cyclic peptide–fused antibodies, the synthesized DNA encoding the cyclic peptides (RGD10 and in4) and a six–amino acid linker (MGSSSN) was subcloned in-frame, without additional amino acids, at the N terminus of CT03 VL via the Not I/Bsi WI sites of pcDNA 3.4–CT03-LC. All the constructs were confirmed by sequencing (Macrogen, Korea).
Expression and purification of antibodies
The plasmids encoding HC and LC were transiently cotransfected in pairs, at equivalent molar ratios, into cultured HEK293F cells in the FreeStyle 293F medium (Invitrogen) following the standard protocol (22). Culture supernatants were collected after 6 to 7 days by centrifugation and filtration (0.22 μm, polyethersulfone; Corning, CL S43118). Antibodies were purified from the culture supernatants on a protein A–agarose chromatographic column (GE Healthcare) and were extensively dialyzed to achieve the final composition of histidine buffer [25 mM histidine (pH 6.5) and 150 mM NaCl]. The anti-HER2 monoclonal antibody trastuzumab (Herceptin) and anti–tumor necrosis factor–α monoclonal antibody adalimumab (Humira) were produced and purified. Before cell treatments, the antibodies were sterilized by means of a cellulose acetate membrane filter (0.22 μm; Corning) and Mustang Q membrane filter (0.8 μm; Pall, MSTG25Q6). Antibody concentrations were determined with the BCA Kit and by measuring the absorbance at 280 nm (22).
High-performance liquid chromatography
Size exclusion chromatography (SEC) analyses were performed on the Agilent 1100 high-performance liquid chromatography (HPLC) system with a Superdex 200 10/300 GC column (10 mm × 300 mm; GE Healthcare) and by SMAC on a Zenix SEC-300 (7.8-mm internal diameter × 300 mm) HPLC column (Sepax Technologies). Unless specified otherwise, 20 μg of an antibody (20 μl, 1 mg/ml) was injected into the Superdex 200 column equilibrated with 12 mM sodium phosphate buffer (pH 7.4; containing 500 mM NaCl and 2.7 mM KCl) as running buffer at a flow rate of 0.75 ml/min (Superdex 200) or into the Zenix SEC-300 column equilibrated with 150 mM sodium phosphate buffer (pH 7.0) as running buffer at a flow rate of 1 ml/min. Chromatograms were obtained by monitoring absorbance at 280 nm. In SEC analysis on the Superdex 200 column, the molecular mass of antibodies was estimated by calculating elution time from a calibration curve built using standard molecular mass markers (thyroglobulin, 669 kDa; β-amylase, 200 kDa; alcohol dehydrogenase, 150 kDa; bovine serum albumin, 66 kDa; Sigma-Aldrich, MWGF1000). In SMAC analysis on the Zenix SEC-300 column, trastuzumab served as a control.
Surface plasmon resonance
Kinetic interactions between antibodies and RAS were measured at 25°C on a Biacore 2000 SPR instrument (GE Healthcare) exactly as described before (15). The binding data were normalized by subtracting the response of a blank cell and then were globally fitted in the BIAevaluation software to obtain kinetic interaction parameters.
Biolayer interferometry
The kinetic interactions of either integrin ανβ5 or integrin ανβ3 with either RGD10- or in4-fused antibodies were monitored at pH 7.4 or 6.0 using the Octet QKe System (ForteBio). All kinetic experiments were conducted at 30°C with orbital shaking at 1000 rpm in 200 μl in 96-well black flat-bottom plates (Greiner Bio-One). All the reagents were diluted with pH 7.4 binding buffer [TBS (pH 7.4), supplemented with 2 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2, and 0.02% Tween 20] or pH 6.0 binding buffer [25 mM histidine (pH 6.0), supplemented with 2 mM CaCl2, 1 mM MgCl2, 1 mM MnCl2, and 0.02% Tween 20]. Anti-human IgG Fc capture biosensors (ForteBio) were equilibrated for 10 min in binding buffer. After an equilibrium was reached, the biosensors were loaded for 1 min with RGD10- or in4-fused antibodies (2 μg/ml; RT11-i, inRas37, or inCT37). Then, a 30-s baseline in binding buffer was established before association for 5 min in a range of indicated concentrations of integrin αvβ3 or integrin αvβ5. Dissociation of the binding was then monitored for 8 min for integrin αvβ3 and for 15 min for integrin αvβ5 in binding buffer with the respective pH. For all the experiments, an empty reference sensor without integrin ανβ3 or integrin ανβ5 was used to take into account nonspecific binding of the analyte to the sensor. All the data were globally fitted via the 1:1 Langmuir binding model in the Octet Data Analysis Software, version 11.0 (ForteBio).
Enzyme-linked immunosorbent assay
Binding specificity of antibodies to antigens [GST-fused protein (RAS wild-type or mutants) loaded with GppNHp or GDP] was determined by ELISA, as described before (15).
Cell lines
The human cell lines, colorectal carcinoma H747, LS513, LS1034, LS174T, SW403, SW480, SW620, SW1116, DLD-1, LoVo, SNU-61, SNU-81, SNU-175, SNU-407, SNU-503, SNU1033, SNU-C2A, HCT8, HCT15, HCT116, Colo320DM, Caco-2, and HT29 cells; lung carcinoma Calu-1, Calu-3, Calu-6, H358, H441, H460, H522, H1299, H1650, H1792, H2009, H2030, HCC44, HCC827, HCC2108, SK-LU-1, and A549 cells; melanoma SK-MEL-2 cells; bladder carcinoma T24 cells; acute monocytic leukemia THP-1 cells; Burkitt’s lymphoma Raji cells; and chronic myeloid leukemia K562 cells were purchased from the Korean Cell Line Bank and maintained in the RPMI 1640 (HyClone). Human breast Hs578T cell lines from the Korean Cell Line Bank were maintained in Dulbecco’s modified Eagle’s medium (HyClone). All cells were cultured in a growth medium that was supplemented with 10% of heat-inactivated fetal bovine serum (FBS) (HyClone, Seoul, Korea), penicillin (100 U/ml), streptomycin (100 μg/ml), and amphotericin B (0.25 μg/ml; HyClone). All cell lines were authenticated by DNA short tandem repeat profiling (ABION CRO, Korea) and used within 10 passages. All cell lines were maintained at 37°C in a humidified 5% CO2 incubator and were routinely screened for Mycoplasma contamination (CellSafe, Suwon, Korea). Genetic alterations in cancer cell lines were analyzed using public databases, including COSMIC (34) and CCLE (33).
Flow cytometry
Cell surface expression of integrin αvβ3 and integrin αvβ5 was determined by flow cytometry after indirect immunofluorescent labeling of cells with an anti-mouse IgG antibody conjugated with Alexa Fluor 488 and either the mouse anti-human integrin αvβ3 antibody (R&D Systems) or the mouse anti-human integrin αvβ5 antibody (R&D Systems). To examine the binding of cyclic peptide–fused antibodies to integrin-expressing cells, the cells were incubated with one of the antibodies (20 nM) in a blocking solution [TBS (pH 7.4), supplemented with 2 mM, CaCl2, 1 mM MgCl2, 1 mM MnCl2, and 1% FBS] for 1 hour at 4°C and then stained with the Alexa Fluor 488–conjugated goat anti-human IgG antibody for 30 min at 4°C. After washing with the ice-cold blocking solution, the cells were analyzed on the FACSCalibur flow cytometer (Becton Dickinson). Data analysis—to quantify changes in the surface expression levels of the respective receptors—was performed in the FlowJo v10 software (Tree Star). Relative expression levels were calculated as follows: relative expression = (MFI of integrin ανβ3 or integrin ανβ5)/(MFI of isotype control), where MFI is the mean fluorescence intensity. An integrin β5 knockdown was implemented with two small interfering RNAs specific for integrin β5; they were synthesized by Bioneer Co. (Korea). Cells (5 × 105 per well in a six-well plate) were transfected with a mixture of integrin β5 small interfering RNAs (100 nM) using Lipofectamine RNAiMAX (Invitrogen) and incubated for 48 hours before analysis (53).
Western blotting
To evaluate the inhibitory effect of inRas37 on RAS downstream signaling, various cells (105 cells per well) were seeded in 24-well ultralow attachment plates (Corning) in media containing 10% FBS. The cells were cultured for 24 to 48 hours until they formed spheroids with a diameter of 100 to 150 μm. Then, the spheroids were starved in media containing 1% FBS for 6 hours at 37°C, treated with the various agents (an antibody and/or the pharmacological inhibitor) for another 12 hours at 37°C, and then were stimulated with EGF (10 ng/ml) for 10 min before cell lysis. To evaluate the inhibitory effect of the combination of inRas37 with the PI3K or YAP1i, spheroids were treated twice (at 0 and 48 hours) with inRas37 (2 μM), inCT37 (2 μM), copanlisib (50 nM), or verteporfin (2.5 μM) as a single agent or as the indicated combination during 54-hour cultivation in media containing 1% FBS. The resultant spheroids were washed and then stimulated with EGF (10 ng/ml) for 10 min. Western blotting was performed with specific antibodies following a standard procedure (see table S5 for primary and secondary antibodies) (22, 54). For quantification of the Western blotting data, band intensities were determined in the ImageJ software (National Institutes of Health, MD) and normalized to values of the corresponding β-actin loading control. The phosphorylation levels of proteins were normalized to the total levels of each protein, equivalently loaded on gels for SDS–polyacrylamide gel electrophoresis (53). Relative band intensity was expressed as a percentage of the value from the corresponding control (53). To quantify the internalized amount and cellular uptake efficiency of an antibody (inRas37 and RT11-i), quantitative Western blotting was performed essentially as described before (15, 18, 26), and the details are also described in table S3.
Cytosolic split-GFP complementation assay
To estimate the cytosolic localization of antibodies, an enhanced split-GFP complementation assay was performed as previously described (18, 26). RT11-i-GFP11-SBP2, inCT37-GFP11-SBP2, inRas37-GFP11-SBP2, and inRas37-AAA-GFP11-SBP2 antibodies were prepared by coexpression of the respective GFP11 (residues 215 to 230)–SBP2 [streptavidin (SA)–binding peptide 2]–fused HC and its cognate LC in HEK293F cells, as described above. HeLa and SW480 cells stably expressing SA-fused GFP1 to GFP10 (residues 1 to 214) were generated as described before (18, 26) and designated as HeLa-SA-GFP1–GFP10 and SW480-SA-GFP1–GFP10 cells, respectively. These cells (2 × 104 cells per well), grown in black 96-well culture plates (SPL Life Sciences, 30296), were incubated at 37°C for 6 hours with one of the antibodies (0.1, 0.5, or 1 μM) in 50 μl of the medium. After washing with phosphate-buffered saline (PBS) to get rid of the autofluorescence of phenol red from the culture media, 50 μl of PBS was added into each well. The complemented GFP signals from cells were measured on a Cytation 3 multimode reader (BioTek, USA). The intact GFP was purified from E. coli, as described before (26). To construct a GFP fluorescence standard plot with known amounts of intact GFP, various amounts (0 to 0.5 ng) of the purified intact GFP were diluted twofold and added into wells of the same 96-well plate in 50 μl of PBS. GFP fluorescence intensity was read at 528 nm after excitation at 485 nm. The cytosolic amount and cytosolic concentration of iMabs were estimated via the formula described in detail elsewhere (26).
Confocal immunofluorescence microscopy
Cellular internalization and localization of antibodies in cultured cells were studied by confocal microscopy (22, 26). Briefly, cells (5 × 104) that were grown on coverslips with a 12-mm diameter in 24-well culture plates were treated with various antibodies, as specified in the figure legends. Nonadherent cell lines (2 × 105 cells) were grown on the coverslips with the 12-mm diameter that were precoated with poly-l-lysine (Sigma-Aldrich, P8920). After two washes with PBS, the cells were washed two times for 30 s at 25°C with low-pH glycine buffer [200 mM glycine and 150 mM NaCl (pH 2.5)], followed by two additional washes with PBS to remove nonspecifically surface-bound antibodies (22). After fixation with 4% paraformaldehyde in PBS for 10 min at 25°C, permeabilization with 0.1% Triton X-100 in PBS was allowed to proceed for 10 min at 25°C, followed by blocking with 2% bovine serum albumin in PBS for 1 hour at 25°C. Internalized antibodies were detected with the Alexa Fluor 488–conjugated goat anti-human IgG antibody (Invitrogen) for 1 hour at 25°C. RAS proteins were detected with the rabbit anti-RAS antibody (Abcam) and, subsequently, with the tetramethylrhodamine-conjugated anti-rabbit antibody (Sigma-Aldrich) for 1 hour at 25°C. The nuclei were stained with Hoechst 33342 in PBS for 5 min at 25°C. After mounting of the coverslips onto glass slides with the Fluorescence Mounting Medium (Dako), center-focused single–z-section images were captured on a Zeiss LSM 710 system with ZEN software (Carl Zeiss).
IP and target engagement assay
IP experiments were conducted with endosome-depleted cell lysates to exclude any possibility of IP by antibodies released from endosomes during cell lysis, as described before (15). SW480 cells (108 per well in a 100-mm dish) were treated with PBS or various antibodies at 2 μM for 12 hours at 37°C. The endosome-depleted fractions were prepared by the removal of early and late endosomes from the cell lysates by density gradient centrifugation (15). The removal of endosomal fractions from the whole-cell lysates was assessed by monitoring Rab5, an early endosome marker (22). To measure target engagement, the endosome-depleted cell lysates were then subjected to IP for 2 hours at 4°C with cRAFRBD-immobilized agarose beads (Merck Millipore) to pull down active GTP-bound RAS proteins. The complexes were next washed with lysis buffer, and equal amounts of the precipitates were analyzed by Western blotting (53, 54). The percentage of target engagement was calculated according to the following formula: TE (%) = 100 × [1 − (KRASipinRas37/KRASinputinRas37)/(KRASipinCT37/KRASinputinCT37)], where KRASip is the KRAS band intensity after the pulldown with cRAFRBD and KRASinput is the KRAS band intensity of input control in the inRas37- or inCT37-treated group.
Spheroid proliferation assay
Various cells (103 per well) were seeded in 96-well ultralow attachment round-bottom plates (Corning, 7007) in media containing 1% FBS. The cells were cultured for 24 hours to form spheroids (typically ~100 to 120 μm in diameter) and then treated three times (at 0, 48, and 96 hours) with various concentrations of an antibody and/or the pharmacological inhibitor (copanlisib, afatinib, ponatinib, iCRT14, or verteporfin) during 6-day cultures in media containing 1% FBS, as specified in the figure legend. Cell viability was measured in the CellTiter-Glo assay (Promega). The luminescence was measured by means of the Cytation 3 multimode reader (BioTek). IC50 values were estimated by fitting the normalized dose-response data to a nonlinear sigmoidal curve in GraphPad Prism 5 software (40).
Intracellular KRASG12V-cRAFRBD interaction inhibition assay
The ability of inRas37 to interrupt physical interactions between activated KRAS and effector proteins in live cells was evaluated by analyzing the subcellular localization of the eGFP-tagged cRAFRBD protein in KRASG12V-harboring SW480 cells, as described elsewhere (15). To construct a plasmid encoding the eGFP-fused cRAFRBD protein, cRAFRBD (residues 51 to 220) was inserted at the C terminus of eGFP in pEGFP-C2 (Clontech). SW480 cells were transfected with the eGFP-cRAFRBD–expressing plasmid using Lipofectamine 3000 (Invitrogen). After three passages involving selection with G418 (3 mg/ml), the cells were treated with antibodies at the various doses for 12 hours at 37°C in serum-free media. After two washes with PBS, the cells were fixed with 4% paraformaldehyde in PBS for 10 min at 25°C. After staining of nuclei with Hoechst 33342 in PBS for 5 min at 25°C, center-focused single–z-section images were captured by confocal microscopy, as described above (26, 54).
Animal experiments
All animal experiments were approved by the Animal and Ethics Review Committee of Ajou University and were performed in accordance with the guidelines established by the Institutional Animal Care and Use Committee. The approval ID for the use of the animals was no. 2017-0007 (the Animal Core Facility of Ajou University). For tumor xenograft models, 4-week-old female BALB/c athymic nude mice (Orient Bio, Korea) weighing 15 to 17 g were subcutaneously injected into the right thigh with SW480 (5 × 106 cells per mouse), LoVo (2 × 106 cells per mouse), LS513 (107 cells per mouse), SW1116 (107 cells per mouse), H747 (107 cells per mouse), SW403 (107 cells per mouse), HCT116 (5 × 106 cells per mouse), or Colo320DM cells (5 × 106 cells per mouse) in 150 μl of a 1:1 mixture of PBS and Matrigel (Corning, 354234). When the mean tumor volume reached approximately 100 to 120 mm3, the mice were randomly assigned to treatment groups, and an antibody or a vehicle control {Histidine buffer [25 mM histidine (pH 6.5) and 150 mM NaCl]}, as specified in the figure legend, was administered via the tail vein, in a dose/weight-matched manner. Copanlisib (AdooQ BioScience) was dissolved in 5% mannitol (vehicle) and administered via the tail vein at 10 mpk twice a week. Verteporfin (Sigma-Aldrich) was dissolved in PBS containing 10% dimethyl sulfoxide and was administered intraperitoneally at 25 mpk twice a week. Tumor volume and body weight were recorded at regular intervals until tumors reached approximately 1000 mm3, at which point the mice were euthanized. Tumor volume (V) was evaluated using digital calipers and was estimated by means of the formula V = L × W2/2, where L and W are the long and short dimensions of a tumor, respectively (54). TGI by inRas37 compared to that by inCT37 was determined on the last day of the study according to the formula TGI (%) = [100 − (VfinRas37 − ViinRas37)/(VfinCT37 − ViinCT37) × 100], where Vi is the initial mean tumor volume in the inRas37 or inCT37 treatment group and Vf is the final mean tumor volume in the inRas37 or inCT37 treatment group, as indicated by the superscripts (15). The mice were euthanized by CO2 asphyxiation, and some tumors and livers were excised for histological analysis, as described before (15). Hematoxylin and eosin staining of liver tissues was performed, as described previously (16). PK experiments on mice and biodistribution imaging in vivo were performed, as previously described (15, 54).
PK experiments on mice
Female BALB/c nude mice (~4 weeks old) without tumor or with LoVo tumor xenografts (initial tumor volume = 120 mm3) received a single injection (at 20 mpk) of various antibodies in a total volume of 200 μl via the tail vein. At the time points 30 min and 1, 4, 8, 12, and 24 hours and on days 2, 4, and 7, blood samples were taken from the tail vein or retro-orbital sinus of CO2-anesthetized mice (n = 3 per time point). Concentrations of the antibody in the serum samples were determined by an ELISA, as described elsewhere (15). For comparison, the first value (at 30 min) was set to 100%. PK parameters [distribution phase serum half-life (T1/2α) and elimination phase serum half-life (T1/2β)] were calculated by two-phasic nonlinear regression analysis in the GraphPad Prism software (15, 54). For the analysis of the PK-PD relation in SW480 xenograft tumor–bearing mice, inRas37 serum concentration and the percentage down-regulation of p-ERK1/2 and p-AKT at TGI 80% were interpolated by means of a four-parameter logistic equation in GraphPad Prism.
Biodistribution imaging in vivo
Antibodies were conjugated with DyLight 755 and purified with the DyLight 755 Antibody Labeling Kit (Thermo Fisher Scientific) in accordance with the manufacturer’s specifications (15). LoVo (2 × 106 cells per mouse) and/or Raji (107 cells per mouse) cells in 150 μl of a 1:1 mixture of PBS and Matrigel were injected subcutaneously into the thigh of BALB/c athymic nude mice. When the mean tumor volume reached approximately 200 mm3, DyLight 755–labeled antibodies (20 μg) were injected into the mice through the tail vein. Before imaging, the mice were anesthetized with 1.5 to 2.5% isoflurane (Piramal Critical Care). The whole-body distribution profiles of the antibodies were determined via in vivo fluorescence using an IVIS Lumina XRMS Series III instrument (PerkinElmer) at various time points. After the final scan, tumor tissues and normal organs were excised and imaged ex vivo. To reduce the effects of a tissue autofluorescence background, manual spectral unmixing was performed, and the fluorescence intensity of an identically sized region of interest was then quantified by radiant efficiency [photons/(s·cm2·steradian) per μW/cm2] in the Living Image Software (PerkinElmer).
Combination synergy analysis
For in vitro combination synergy analysis, the CI from the Chou-Talalay method was calculated in the CompuSyn software (ComboSyn) (55). The CIs at IC50 during either single or combination treatment were used. CI < 0.75 meant a synergistic effect, 0.75 ≤ CI ≤ 1 denoted an additive effect, and CI > 1.25 meant an antagonistic effect. In vivo combination synergy analysis was carried out via the Bliss independence model (40, 56). The expected additive response tumor volume (VExp) for the combination group was defined as follows: VExp = (V1 × V2)/VC, where V1 and V2 are the mean tumor volumes in the single-agent groups and VC is the mean volume in the control group. The control group tumor volume change from baseline was defined as ΔV = VC − V0. Then, an additive range around the VExp was defined, via upper and lower limits, as VExpU = min(2 × VExp, VExp + 0.15 × ΔV) and VExpL = max(VExp/2, VExp − 0.15 × ΔV). VObs < VExpL meant a synergistic effect, VExpL ≤ VObs ≤ VExpU indicated an additive effect, and VObs > VExpU or VObs > V1 and V2 denoted an antagonistic effect, where VObs is the observed mean tumor volume in the combination group.
Immunofluorescence microscopy of tumor tissues
This analysis of the tumors excised from mice was performed using a Zeiss LSM 710 system (Carl Zeiss), as described before (15). The primary and secondary antibodies for this experiment are listed in table S5. Apoptotic cells were detected by a standard TUNEL assay using the DeadEnd Fluorometric TUNEL System (Promega). In the fluorescence images acquired from each tissue sample, the fluorescence intensity and number of positively stained cells were quantified using the ImageJ software and presented as relative staining (%) compared to that of the PBS-treated control.
Statistical analysis
Data are represented as the means ± SD of triplicate samples from one representative experiment from at least three independent experiments, unless otherwise specified. Comparisons of data from tests and controls were analyzed for statistical significance by unpaired two-tailed Student’s t test. One-way analysis of variance (ANOVA) with the Newman-Keuls multiple-comparisons post hoc test was performed by means of GraphPad Prism to determine the significance of data from in vivo tumor growth experiments. Data with a P value of less than 0.05 were considered significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Pioneer Research Center Program (grant number 2014M3C1A3051470) and the Priority Research Center Program (2019R1A6A1A11051471) from the National Research Foundation funded by the Korean government. Author contributions: S.-M.S., J.-S.K., and Y.-S.K. conceived the idea. S.-W.P. and D.-K.C. performed antibody affinity maturation and purified and characterized antibodies and recombinant proteins. S.-Y.J. generated and characterized the in4 cyclic peptide. S.-M.S., J.-S.K., S.-W.P., S.-Y.J., and H.-J.K. carried out the in vitro cellular and mouse xenograft experiments. D.L. performed the liver histological analysis. S.-M.S., Y.B.C., and Y.-S.K. designed and interpreted the combination experiments. All authors analyzed and interpreted the data. Y.-S.K. supervised the research. S.-M.S., J.-S.K., and Y.-S.K. wrote the manuscript with input from all coauthors. All authors approved the final version of the manuscript. Competing interests: Y.-S.K., S.-M.S., J.-S.K., S.-W.P., S.-Y.J., and D.-K.C. are listed as inventors on pending patents [U.S. 62,688,339 (filed on 21 June 2018) and PCT/KR/2018/014110 (filed on 16 November 2018)] related to the technology described in this work. D.-K.C. is currently an employee of Orum Therapeutics. D.-K.C. and Y.-S.K. have equity in the company. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The inCT37 and inRas37 antibodies can be provided by Orum Therapeutics Inc. pending scientific review and a completed material transfer agreement. Requests for the antibodies should be submitted on the Orum website (www.orumrx.com/contact). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/3/eaay2174/DC1
Fig. S1. Procedures of stepwise engineering to generate the second-generation active RAS-specific iMab, inRas37.
Fig. S2. Preparation of the RT22 VH and in4 cyclic peptide and biochemical characterization of inRas37.
Fig. S3. inRas37 has more potent antitumor efficacy in vivo than RT11-i does, without noticeable toxic side effects.
Fig. S4. inRas37 causes dose-dependent inhibition of in vivo growth of KRASG12V SW480 tumor xenografts in mice.
Fig. S5. Comparison of cell surface expression levels of integrin ανβ5 and integrin ανβ3 in various RASWT and RASMUT cell lines.
Fig. S6. inRas37 suppresses the in vivo growth of various RASMUT tumor xenografts in mice without noticeable systemic toxicity.
Fig. S7. IHC and Western blot analyses of tumor tissues excised from mice after treatment.
Fig. S8. Combined treatment of KRASMUT cell lines with a pharmacological inhibitor and either inRas37 or inCT37.
Table S1. Binding constants for the interactions of inRas37 with GppNHp-loaded active forms of RAS, as determined by SPR analysis.
Table S2. Binding constants for the interactions of inRas37 with integrin ανβ5 and integrin ανβ3, as determined at pH 7.4 and/or pH 6.0 by biolayer interferometry.
Table S3. Quantitative assessment of the cellular uptake and cytosolic concentrations of RT11-i and inRas37 in HeLa and SW480 cells.
Table S4. CRC driver mutations from the CCLE and COSMIC datasets.
Table S5. List of resources (antibodies, recombinant proteins, and chemicals) used in this study.
REFERENCES AND NOTES
- 1.Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., Der C. J., Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 13, 828–851 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hobbs G. A., Der C. J., Rossman K. L., RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 129, 1287–1292 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephen A. G., Esposito D., Bagni R. K., McCormick F., Dragging ras back in the ring. Cancer Cell 25, 272–281 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Spencer-Smith R., O'Bryan J. P., Direct inhibition of RAS: Quest for the Holy Grail? Semin. Cancer Biol. 54, 138–148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan M. B., Corcoran R. B., Therapeutic strategies to target RAS-mutant cancers. Nat. Rev. Clin. Oncol. 15, 709–720 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Athuluri-Divakar S. K., Vasquez-Del Carpio R., Dutta K., Baker S. J., Cosenza S. C., Basu I., Gupta Y. K., Reddy M. V., Ueno L., Hart J. R., Vogt P. K., Mulholland D., Guha C., Aggarwal A. K., Reddy E. P., A small molecule RAS-mimetic disrupts RAS association with effector proteins to block signaling. Cell 165, 643–655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quevedo C. E., Cruz-Migoni A., Bery N., Miller A., Tanaka T., Petch D., Bataille C. J. R., Lee L. Y. W., Fallon P. S., Tulmin H., Ehebauer M. T., Fernandez-Fuentes N., Russell A. J., Carr S. B., Phillips S. E. V., Rabbitts T. H., Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat. Commun. 9, 3169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGee J. H., Shim S. Y., Lee S.-J., Swanson P. K., Jiang S. Y., Durney M. A., Verdine G. L., Exceptionally high-affinity Ras binders that remodel its effector domain. J. Biol. Chem. 293, 3265–3280 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kauke M. J., Tisdale A. W., Kelly R. L., Braun C. J., Hemann M. T., Wittrup K. D., A Raf-competitive K-Ras binder can fail to functionally antagonize signaling. Mol. Cancer Ther. 17, 1773–1780 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Guillard S., Kolasinska-Zwierz P., Debreczeni J., Breed J., Zhang J., Bery N., Marwood R., Tart J., Overman R., Stocki P., Mistry B., Phillips C., Rabbitts T., Jackson R., Minter R., Structural and functional characterization of a DARPin which inhibits Ras nucleotide exchange. Nat. Commun. 8, 16111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janes M. R., Zhang J., Li L.-S., Hansen R., Peters U., Guo X., Chen Y., Babbar A., Firdaus S. J., Darjania L., Feng J., Chen J. H., Li S., Li S., Long Y. O., Thach C., Liu Y., Zarieh A., Ely T., Kucharski J. M., Kessler L. V., Wu T., Yu K., Wang Y., Yao Y., Deng X., Zarrinkar P. P., Brehmer D., Dhanak D., Lorenzi M. V., Hu-Lowe D., Patricelli M. P., Ren P., Liu Y., Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Lito P., Solomon M., Li L.-S., Hansen R., Rosen N., Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science 351, 604–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keeton A. B., Salter E. A., Piazza G. A., The RAS–effector interaction as a drug target. Cancer Res. 77, 221–226 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei D., Chen K., Liao H., Targeting Ras with macromolecules. Cold Spring Harb. Perspect. Med. 8, a031476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shin S.-M., Choi D.-K., Jung K., Bae J., Kim J.-s., Park S.-w., Song K.-H., Kim Y.-S., Antibody targeting intracellular oncogenic Ras mutants exerts anti-tumour effects after systemic administration. Nat. Commun. 8, 15090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang Y. W., Lee J. E., Jung K. H., Son M. K., Shin S.-M., Kim S. J., Fang Z., Yan H. H., Park J. H., Han B., Cheon M. J., Woo M. G., Lim J. H., Kim Y.-S., Hong S.-S., KRAS targeting antibody synergizes anti-cancer activity of gemcitabine against pancreatic cancer. Cancer Lett. 438, 174–186 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Kim J.-s., Park J.-Y., Shin S.-M., Park S.-W., Jun S.-Y., Hong J.-S., Choi D.-K., Kim Y.-S., Engineering of a tumor cell-specific, cytosol-penetrating antibody with high endosomal escape efficacy. Biochem. Biophys. Res. Commun. 503, 2510–2516 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Kim J.-S., Choi D.-K., Shin J.-Y., Shin S.-M., Park S.-W., Cho H.-S., Kim Y.-S., Endosomal acidic pH-induced conformational changes of a cytosol-penetrating antibody mediate endosomal escape. J. Control. Release 235, 165–175 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Nieberler M., Reuning U., Reichart F., Notni J., Wester H. J., Schwaiger M., Weinmuller M., Rader A., Steiger K., Kessler H., Exploring the role of RGD-recognizing integrins in cancer. Cancers 9, 116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schlothauer T., Herter S., Koller C. F., Grau-Richards S., Steinhart V., Spick C., Kubbies M., Klein C., Umaña P., Mössner E., Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 29, 457–466 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Tzeng A., Kwan B. H., Opel C. F., Navaratna T., Wittrup K. D., Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc. Natl. Acad. Sci. U.S.A. 112, 3320–3325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi D.-K., Bae J., Shin S.-M., Shin J.-Y., Kim S., Kim Y.-S., A general strategy for generating intact, full-length IgG antibodies that penetrate into the cytosol of living cells. MAbs 6, 1402–1414 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohli N., Jain N., Geddie M. L., Razlog M., Xu L., Lugovskoy A. A., A novel screening method to assess developability of antibody-like molecules. MAbs 7, 752–758 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain T., Sun T., Durand S., Hall A., Houston N. R., Nett J. H., Sharkey B., Bobrowicz B., Caffry I., Yu Y., Cao Y., Lynaugh H., Brown M., Baruah H., Gray L. T., Krauland E. M., Xu Y., Vasquez M., Wittrup K. D., Biophysical properties of the clinical-stage antibody landscape. Proc. Natl. Acad. Sci. U.S.A. 114, 944–949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Igawa T., Ishii S., Tachibana T., Maeda A., Higuchi Y., Shimaoka S., Moriyama C., Watanabe T., Takubo R., Doi Y., Wakabayashi T., Hayasaka A., Kadono S., Miyazaki T., Haraya K., Sekimori Y., Kojima T., Nabuchi Y., Aso Y., Kawabe Y., Hattori K., Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat. Biotechnol. 28, 1203–1207 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Kim J.-s., Choi D.-K., Park S.-w., Shin S.-M., Bae J., Kim D.-M., Yoo T. H., Kim Y.-S., Quantitative assessment of cellular uptake and cytosolic access of antibody in living cells by an enhanced split GFP complementation assay. Biochem. Biophys. Res. Commun. 467, 771–777 (2015). [DOI] [PubMed] [Google Scholar]
- 27.McDonald E. R. III, de Weck A., Schlabach M. R., Billy E., Mavrakis K. J., Hoffman G. R., Belur D., Castelletti D., Frias E., Gampa K., Golji J., Kao I., Li L., Megel P., Perkins T. A., Ramadan N., Ruddy D. A., Silver S. J., Sovath S., Stump M., Weber O., Widmer R., Yu J., Yu K., Yue Y., Abramowski D., Ackley E., Barrett R., Berger J., Bernard J. L., Billig R., Brachmann S. M., Buxton F., Caothien R., Caushi J. X., Chung F. S., Cortes-Cros M., deBeaumont R. S., Delaunay C., Desplat A., Duong W., Dwoske D. A., Eldridge R. S., Farsidjani A., Feng F., Feng J., Flemming D., Forrester W., Galli G. G., Gao Z., Gauter F., Gibaja V., Haas K., Hattenberger M., Hood T., Hurov K. E., Jagani Z., Jenal M., Johnson J. A., Jones M. D., Kapoor A., Korn J., Liu J., Liu Q., Liu S., Liu Y., Loo A. T., Macchi K. J., Martin T., McAllister G., Meyer A., Molle S., Pagliarini R. A., Phadke T., Repko B., Schouwey T., Shanahan F., Shen Q., Stamm C., Stephan C., Stucke V. M., Tiedt R., Varadarajan M., Venkatesan K., Vitari A. C., Wallroth M., Weiler J., Zhang J., Mickanin C., Myer V. E., Porter J. A., Lai A., Bitter H., Lees E., Keen N., Kauffmann A., Stegmeier F., Hofmann F., Schmelzle T., Sellers W. R., Project DRIVE: A compendium of cancer dependencies and synthetic lethal relationships uncovered by large-scale, deep RNAi screening. Cell 170, 577–592.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Urva S. R., Balthasar J. P., Target mediated disposition of T84.66, a monoclonal anti-CEA antibody: Application in the detection of colorectal cancer xenografts. MAbs 2, 67–72 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shin T.-H., Sung E.-S., Kim Y.-J., Kim K.-S., Kim S.-H., Kim S.-K., Lee Y.-D., Kim Y.-S., Enhancement of the tumor penetration of monoclonal antibody by fusion of a neuropilin-targeting peptide improves the antitumor efficacy. Mol. Cancer Ther. 13, 651–661 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Misale S., Fatherree J. P., Cortez E., Li C., Bilton S., Timonina D., Myers D. T., Lee D., Gomez-Caraballo M., Greenberg M., Nangia V., Greninger P., Egan R. K., McClanaghan J., Stein G. T., Murchie E., Zarrinkar P. P., Janes M. R., Li L.-S., Liu Y., Hata A. N., Benes C. H., KRAS G12C NSCLC models are sensitive to direct targeting of KRAS in combination with PI3K inhibition. Clin. Cancer Res. 25, 796–807 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Singh A., Greninger P., Rhodes D., Koopman L., Violette S., Bardeesy N., Settleman J., A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 15, 489–500 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh A., Sweeney M. F., Yu M., Burger A., Greninger P., Benes C., Haber D. A., Settleman J., TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 148, 639–650 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A. A., Kim S., Wilson C. J., Lehar J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M. F., Monahan J. E., Morais P., Meltzer J., Korejwa A., Jane-Valbuena J., Mapa F. A., Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I. H., Cheng J., Yu G. K., Yu J., Aspesi P. Jr., de Silva M., Jagtap K., Jones M. D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R. C., Liefeld T., MacConaill L., Winckler W., Reich M., Li N., Mesirov J. P., Gabriel S. B., Getz G., Ardlie K., Chan V., Myer V. E., Weber B. L., Porter J., Warmuth M., Finan P., Harris J. L., Meyerson M., Golub T. R., Morrissey M. P., Sellers W. R., Schlegel R., Garraway L. A., The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sondka Z., Bamford S., Cole C. G., Ward S. A., Dunham I., Forbes S. A., The COSMIC cancer gene census: Describing genetic dysfunction across all human cancers. Nat. Rev. Cancer 18, 696–705 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Will M., Qin A. C. R., Toy W., Yao Z., Rodrik-Outmezguine V., Schneider C., Huang X., Monian P., Jiang X., de Stanchina E., Baselga J., Liu N., Chandarlapaty S., Rosen N., Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS–ERK signaling. Cancer Discov. 4, 334–347 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonsalves F. C., Klein K., Carson B. B., Katz S., Ekas L. A., Evans S., Nagourney R., Cardozo T., Brown A. M. C., DasGupta R., An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 5954–5963 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun C., Hobor S., Bertotti A., Zecchin D., Huang S., Galimi F., Cottino F., Prahallad A., Grernrum W., Tzani A., Schlicker A., Wessels L. F., Smit E. F., Thunnissen E., Halonen P., Lieftink C., Beijersbergen R. L., Di Nicolantonio F., Bardelli A., Trusolino L., Bernards R., Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep. 7, 86–93 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Manchado E., Weissmueller S., Morris J. P., Chen C.-C., Wullenkord R., Lujambio A., de Stanchina E., Poirier J. T., Gainor J. F., Corcoran R. B., Engelman J. A., Rudin C. M., Rosen N., Lowe S. W., A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 534, 647–651 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao X., Wang X., Fang L., Lan C., Zheng X., Wang Y., Zhang Y., Han X., Liu S., Cheng K., Zhao Y., Shi J., Guo J., Hao J., Ren H., Nie G., A combinatorial strategy using YAP and pan-RAF inhibitors for treating KRAS-mutant pancreatic cancer. Cancer Lett. 402, 61–70 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Miller M. L., Molinelli E. J., Nair J. S., Sheikh T., Samy R., Jing X., He Q., Korkut A., Crago A. M., Singer S., Schwartz G. K., Sander C., Drug synergy screen and network modeling in dedifferentiated liposarcoma identifies CDK4 and IGF1R as synergistic drug targets. Sci. Signal. 6, ra85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen G., Gao C., Gao X., Zhang D. H., Kuan S.-F., Burns T. F., Hu J., Wnt/β-catenin pathway activation mediates adaptive resistance to BRAF inhibition in colorectal cancer. Mol. Cancer Ther. 17, 806–813 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kapoor A., Yao W., Ying H., Hua S., Liewen A., Wang Q., Zhong Y., Wu C. J., Sadanandam A., Hu B., Chang Q., Chu G. C., Al-Khalil R., Jiang S., Xia H., Fletcher-Sananikone E., Lim C., Horwitz G. I., Viale A., Pettazzoni P., Sanchez N., Wang H., Protopopov A., Zhang J., Heffernan T., Johnson R. L., Chin L., Wang Y. A., Draetta G., DePinho R. A., Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shao D. D., Xue W., Krall E. B., Bhutkar A., Piccioni F., Wang X., Schinzel A. C., Sood S., Rosenbluh J., Kim J. W., Zwang Y., Roberts T. M., Root D. E., Jacks T., Hahn W. C., KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 158, 171–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujioka A., Terai K., Itoh R. E., Aoki K., Nakamura T., Kuroda S., Nishida E., Matsuda M., Dynamics of the Ras/ERK MAPK cascade as monitored by fluorescent probes. J. Biol. Chem. 281, 8917–8926 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Ambrogio C., Köhler J., Zhou Z.-W., Wang H., Paranal R., Li J., Capelletti M., Caffarra C., Li S., Lv Q., Gondi S., Hunter J. C., Lu J., Chiarle R., Santamaría D., Westover K. D., Jänne P. A., KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell 172, 857–868.e15 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Nan X., Tamgüney T. M., Collisson E. A., Lin L.-J., Pitt C., Galeas J., Lewis S., Gray J. W., McCormick F., Chu S., Ras-GTP dimers activate the mitogen-activated protein kinase (MAPK) pathway. Proc. Natl. Acad. Sci. U.S.A. 112, 7996–8001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Welsch M. E., Kaplan A., Chambers J. M., Stokes M. E., Bos P. H., Zask A., Zhang Y., Sanchez-Martin M., Badgley M. A., Huang C. S., Tran T. H., Akkiraju H., Brown L. M., Nandakumar R., Cremers S., Yang W. S., Tong L., Olive K. P., Ferrando A., Stockwell B. R., Multivalent small-molecule pan-RAS inhibitors. Cell 168, 878–889.e29 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Forbes S. A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C. G., Ward S., Dawson E., Ponting L., Stefancsik R., Harsha B., Kok C. Y., Jia M., Jubb H., Sondka Z., Thompson S., De T., Campbell P. J., COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Engelman J. A., Chen L., Tan X., Crosby K., Guimaraes A. R., Upadhyay R., Maira M., McNamara K., Perera S. A., Song Y., Chirieac L. R., Kaur R., Lightbown A., Simendinger J., Li T., Padera R. F., García-Echeverría C., Weissleder R., Mahmood U., Cantley L. C., Wong K.-K., Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 14, 1351–1356 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mologni L., Brussolo S., Ceccon M., Gambacorti-Passerini C., Synergistic effects of combined Wnt/KRAS inhibition in colorectal cancer cells. PLOS ONE 7, e51449 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cancer Genome Atlas Network , Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang H. Y., Kang K. J., Chung J. E., Shim H., Construction of a large synthetic human scFv library with six diversified CDRs and high functional diversity. Mol. Cells 27, 225–235 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Kim Y.-J., Jung K., Baek D.-S., Hong S.-S., Kim Y.-S., Co-targeting of EGF receptor and neuropilin-1 overcomes cetuximab resistance in pancreatic ductal adenocarcinoma with integrin β1-driven Src-Akt bypass signaling. Oncogene 36, 2543–2552 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Kim Y.-J., Bae J., Shin T.-H., Kang S. H., Jeong M., Han Y., Park J.-H., Kim S.-K., Kim Y.-S., Immunoglobulin Fc-fused, neuropilin-1-specific peptide shows efficient tumor tissue penetration and inhibits tumor growth via anti-angiogenesis. J. Control. Release 216, 56–68 (2015). [DOI] [PubMed] [Google Scholar]
- 55.Chou T.-C., Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 58, 621–681 (2006). [DOI] [PubMed] [Google Scholar]
- 56.Chen S.-H., Gong X., Zhang Y., Van Horn R. D., Yin T., Huber L., Burke T. F., Manro J., Iversen P. W., Wu W., Bhagwat S. V., Beckmann R. P., Tiu R. V., Buchanan S. G., Peng S.-B., RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 37, 821–832 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/3/eaay2174/DC1
Fig. S1. Procedures of stepwise engineering to generate the second-generation active RAS-specific iMab, inRas37.
Fig. S2. Preparation of the RT22 VH and in4 cyclic peptide and biochemical characterization of inRas37.
Fig. S3. inRas37 has more potent antitumor efficacy in vivo than RT11-i does, without noticeable toxic side effects.
Fig. S4. inRas37 causes dose-dependent inhibition of in vivo growth of KRASG12V SW480 tumor xenografts in mice.
Fig. S5. Comparison of cell surface expression levels of integrin ανβ5 and integrin ανβ3 in various RASWT and RASMUT cell lines.
Fig. S6. inRas37 suppresses the in vivo growth of various RASMUT tumor xenografts in mice without noticeable systemic toxicity.
Fig. S7. IHC and Western blot analyses of tumor tissues excised from mice after treatment.
Fig. S8. Combined treatment of KRASMUT cell lines with a pharmacological inhibitor and either inRas37 or inCT37.
Table S1. Binding constants for the interactions of inRas37 with GppNHp-loaded active forms of RAS, as determined by SPR analysis.
Table S2. Binding constants for the interactions of inRas37 with integrin ανβ5 and integrin ανβ3, as determined at pH 7.4 and/or pH 6.0 by biolayer interferometry.
Table S3. Quantitative assessment of the cellular uptake and cytosolic concentrations of RT11-i and inRas37 in HeLa and SW480 cells.
Table S4. CRC driver mutations from the CCLE and COSMIC datasets.
Table S5. List of resources (antibodies, recombinant proteins, and chemicals) used in this study.