

Abstract
The developmental regulator SOX9 is linked to cancer progression mainly as a result of its role in the regulation of cancer stem cells (CSCs). However, its activity in the differentiated cells that constitute the heterogeneous tumor bulk has not been extensively studied. In this work, we addressed this aspect in gastric cancer, glioblastoma and pancreatic adenocarcinoma. SOX9 silencing studies revealed that SOX9 is required for cancer cell survival, proliferation and evasion of senescence in vitro and tumor growth in vivo. Gain of-SOX9 function showed that high levels of SOX9 promote tumor cell proliferation in vitro and in vivo. Mechanistically, the modulation of SOX9 changed the expression of the transcriptional repressor BMI1 in the same direction in the three types of cancer, and the expression of the tumor suppressor p21CIP in the opposite direction. In agreement with this, SOX9 expression positively correlated with BMI1 levels and inversely with p21CIP in clinical samples of the different cancers. Moreover, BMI1 re-establishment in SOX9-silenced tumor cells restored cell viability and proliferation as well as decreased p21CIP in vitro and tumor growth in vivo. These results indicate that BMI1 is a critical effector of the pro-tumoral activity of SOX9 in tumor bulk cells through the repression of p21CIP. Our results highlight the relevance of the SOX9-BMI1-p21CIP axis in tumor progression, shedding novel opportunities for therapeutic development.
Subject terms: Mechanisms of disease, Tumour heterogeneity
Introduction
Some types of cancer exhibit a dismal prognosis, mainly due to its late diagnosis, its aggressive progression and the resistance to the available chemotherapy, in which inter and intra-tumoral heterogeneity play a major role1,2. One of these types of cancer is pancreatic ductal adenocarcinoma (PDAC), with a 5-year survival rate of less than 6%3 and very similar rates of incidence and mortality (458,918 new diagnoses and 432,242 deaths worldwide in 2018)4. Glioblastoma (GBM), the most malignant primary brain tumor, also represents a challenge in oncology despite having a low incidence (less than 5 cases per 100,000 people), as exhibits a 5-year survival rate of 5.6%5 and a median survival of 14.6 months6. For its part, gastric cancer (GC) is the third leading cause of cancer deaths worldwide, with an estimated 783,000 deaths in 20184.
SOX9 [SRY (Sex determining region Y)-box 9] is a member of the SOX family of transcription factors, which are developmental regulators characterized by a conserved high mobility group (HMG) DNA-binding domain7. SOX9 regulates stem cell maintenance and instruction of cell fate, and exerts relevant roles during development, such as sex determination, neural crest development, chondrogenesis or pancreas development8–10. During embryogenesis, SOX9 is expressed and regulates progenitor proliferation and differentiation, being required for maintaining tissue identity in different contexts, but mainly in the brain and gastrointestinal system10–12. Likewise, in the adulthood, SOX9 also plays a relevant role in the maintenance of the homeostasis of these tissues through the regulation of the residing populations of adult stem cells11,13, although not exclusively, as its expression has also been linked to several differentiated cells within different contexts and tissues8,14.
In cancer, several studies demonstrated the involvement of SOX9 in cancer formation, as the elevation of its levels favors transformation of stem cells. For example in pancreas, where SOX9 regulates pancreatic progenitor cells during pancreas development and maintains ductal integrity in mature pancreas15,16, it is essential during acinar to ductal metaplasia (ADM) initiation17 and has been shown indispensable for the formation of intraepithelial neoplasias (PanINs) induced by oncogenic KRAS18. Similar observations have been observed in the brain, breast or skin19–22. Moreover, several studies have linked SOX9 to cancer initiation and the regulation of the population of cancer stem cells (CSCs) in multiples tissues19,21,23, promoting processes associated to this specific population such as metastasis or chemoresistance24–27.
In clinical samples, SOX9 expression is elevated in glioblastoma, pancreatic ductal adenocarcinoma, gastric cancer, colon, skin or breast cancer samples respect to adjacent normal tissue28–30. Furthermore, high levels of SOX9 have been associated with tumor grade, dismal prognosis and poor patient survival in patients of those types of cancer28,31,32. Thus, SOX9 expression and function are altered in diverse human cancers, acting as an oncogene in a wide range of them, mainly through the regulation of CSCs activity, and as a tumor suppressor in specific situations26. Besides, it has been shown that Sox9 is able to promote proliferation and induce neoplastic transformation of primary fibroblasts33, indicative that SOX9 is relevant in cancer beyond its initiation and its role in CSC activity. In this regard, its activity in cells of tumor bulk and the underlying molecular mechanisms remain poorly understood. Therefore, in this study we elucidated the functional role of SOX9 in critical processes for cancer progression such as survival, proliferation and senescence in tumor differentiated cells, and deciphered its molecular mechanism, providing new knowledge regarding the role and molecular pathway of this critical stem cell factor on cancer progression and heterogeneity.
Results
SOX9 is required for tumor cell survival and proliferation
In order to assess the impact of SOX9 in tumor cell survival, we knocked down SOX9 expression in different cancer cell lines. In particular, we silenced SOX9 expression in cell lines of gastric cancer (AGS and MKN45), pancreatic cancer (Panc-1 and RWP-1) and glioblastoma (U373 and U251), which exhibit high SOX9 expression levels. After confirming the successful reduction of SOX9 levels in these cell lines (Fig. 1A and Fig. Suppl), we determined cell viability by cell count experiments. In these analyses, we observed a significantly reduced number of cells in SOX9-silenced cultures respect to control cells 5 days after the seeding (Fig. 1B), indicating that SOX9 silencing compromises the viability of tumor cells.
Figure 1.

SOX9 silencing impairs tumor cell survival, induces senescence and abrogates proliferation in cancer cells. (A) Representative Western blots of SOX9 protein expression in MKN45 and AGS GC cell lines, Panc-1 and RWP-1 PDAC cell lines, and U373 and U251 GBM cell lines lentivirally transduced with a specific shRNA against SOX9 (sh1) or the corresponding control vector (pLKO) (n ≥ 4). β-actin levels were used as a loading control. (B) Cell growth at day 5 (percentage of cells) in SOX9-silenced (sh1) cells respect to control (pLKO) cells (n ≥ 4). (C) Apoptosis represented by the percentage of active Caspase-3 positive cells in SOX9-silenced (sh1) cells and the corresponding control cells (pLKO) determined by immunofluorescence staining (n ≥ 4). (D) Representative image of active Caspase-3 positive cells in RWP-1 control (pLKO) and SOX9-silenced (sh1) cells. (E) Apoptosis represented by the percentage of cleaved PARP1 positive cells in SOX9-silenced (sh1) cells and the corresponding control cells (pLKO) determined by immunofluorescence staining (n ≥ 4). (F) Representative image of cleaved PARP1 in RWP-1 control (pLKO) and SOX9-silenced (sh1) cells. (G) Senescence represented as the quantification of the percentage of β-Galactosidase (SA β-Gal) positive cells in SOX9-silenced (sh1) cells and the corresponding control cells (pLKO) (n ≥ 4). (H) Representative images of SA β-Gal staining in RWP-1 (left) and U251 (right) control (pLKO) and SOX9-silenced (sh1) cells. (I) Proliferative capacity represented by the percentage of phospho-histone H3 (p-H3) positive cells (n ≥ 4) determined by immunofluorescence staining in control (pLKO) and SOX9-silenced (sh1) cells. (J) Representative image of p-H3 in RWP-1 control (pLKO) and SOX9-silenced (sh1) cells. Asterisks (*, ** and ***) indicate statistical significance (p < 0.05, p < 0.01, and p < 0.001, respectively).
In order to evaluate the role of SOX9 in cancer cell survival, we studied apoptosis. For this, we analyzed the activation of Caspase-3 and the proteolytic inactivation of PARP1 through immunofluorescence staining. These experiments revealed the presence of a significantly higher number of apoptotic cells in cultures with SOX9 silencing, with a marked increase of over 10 fold in both active Caspase-3 (Fig. 1C,D) and cleaved PARP1-positive cells (Fig. 1E,F) in SOX9-silenced cells respect to controls. Thus, SOX9 is required for cancer cell survival, wherein exerts an antiapoptotic role.
Given that cellular senescence constitutes a tumor-suppressive mechanism that restricts tumor progression, we explored whether SOX9 would mediate senescence evasion in cancer cells. To assess this aspect, we analyzed the senescence associated β-galactosidase activity in SOX9-silenced cells. We detected a significant increase in the number of β-galactosidase-positive cells in the different cancer types (Fig. 1G,H), revealing that SOX9 silencing promotes the induction of senescence in cancer cells.
Next, we measured cell proliferation through the evaluation of the percentage of cells positive for the marker of mitosis phospho-Histone H3 (p-H3). Our results revealed a marked and significant decrease in mitotic cells in SOX9-silenced cancer cells respect to cells transduced with empty vector (Fig. 1I,J). On the contrary, ectopic upregulation of SOX9 in cancer cell lines (Fig. 2A) resulted in a significant increase in the percentage of p-H3 positive cells in cultures from the 3 types of cancer (Fig. 2B), as well as increased cell count (Fig. 2C). In line with this, tumors derived from MKN45 gastric cancer cells and U373 glioma cells with overexpression of SOX9 presented a markedly higher number of Ki67 positive cells than those tumors formed by control cells in vivo (Fig. 2D), together demonstrating that SOX9 regulates cancer cell proliferation.
Figure 2.

SOX9 ectopic upregulation enhances tumor cell proliferation. (A) Representative Western blots of SOX9 protein expression in IMIMPC-2 and BxPC-3 PDAC cell lines, and U373 and U87 GBM cell lines lentivirally transduced with plasmids harboring GFP (GFP) or SOX9 (SOX9) coding sequences (n ≥ 3). (B) Quantification of the number of p-H3 positive cells in SOX9 transduced cells compared to control cells (GFP) (n ≥ 4). (C) Cell growth at day 5 (percentage of cells) comparing control (GFP) and SOX9 overexpressing U373 and U87 GBM cells (n ≥ 3). (D) Representative images of SOX9, BMI1 and Ki67 protein expression determined by immunohistochemistry in subcutaneous tumors generated in nude mice by injection of SOX9 overexpressing (SOX9) and control (GFP) MKN45 (left) and U373 (right) cells. Asterisks (*, ** and ***) indicate statistical significance (p < 0.05, p < 0.01, and p < 0.001, respectively).
SOX9 expression regulates BMI1 and p21CIP levels
Next, we wanted to unravel the molecular mechanism underlying the role of SOX9 in cell survival and proliferation. Since we had previously found that Sox9 promotes proliferation and facilitates neoplastic transformation of primary fibroblasts via the transcriptional repressor Bmi133, and other groups have shown that SOX9 induces cancer cell proliferation through downregulation of the tumor suppressor p21CIP 34,35, we studied their expression.
We first investigated the effect of SOX9 silencing in their expression in the different tumor cell lines of various origins. Our results revealed that BMI1 protein expression was reduced in SOX9-silenced cells of PDAC, GBM and GC (Fig. 3A), whereas p21CIP levels were elevated (Fig. 3A). Similar effect was also observed at transcriptional level (Fig. 3B,C). On the contrary, cells with ectopic SOX9 overexpression displayed elevated levels of BMI1 and lower p21CIP expression (Fig. 3D,E). These results show that SOX9 regulates the expression of BMI1 and p21CIP at transcriptional level in cancer cells in vitro and this might influence tumor cell survival and proliferation.
Figure 3.

SOX9 modulation impacts on BMI1 and p21CIP expression in cancer cells. (A) Representative Western blots of SOX9, BMI1 and p21CIP protein expression in MKN45 and AGS GC cell lines, Panc-1 and RWP-1 PDAC cell lines, and U373 and U251 GBM cell lines lentivirally transduced with a specific shRNA against SOX9 (sh1) or the corresponding control vector (pLKO) (n ≥ 3). β-actin levels were used as a loading control. (B) BMI1 mRNA levels in control (pLKO) and SOX9-silenced (sh1) cells in the indicated GC, PDAC and GBM cell lines (n ≥ 2). (B) p21CIP mRNA levels in control (pLKO) and SOX9-silenced (sh1) cells in the indicated GC, PDAC and GBM cell lines (n ≥ 2). (D) Representative Western blots of the indicated proteins in AGS GC cell line, IMIMPC-2 and BxPC-3 PDAC cell lines, and U87 and U373 GBM cell lines lentivirally transduced with plasmids harboring GFP (GFP) or SOX9 (SOX9) coding sequences (n ≥ 3). β-actin levels were used as a loading control. (E) p21CIP mRNA levels in SOX9 overexpressing (SOX9) or control (GFP) cells in the indicated PDAC and GBM cell lines (n ≥ 2). (F) Representative images of SOX9, BMI1, p21CIP and Ki67 protein expression determined by immunohistochemistry in subcutaneous tumors generated in nude mice by injection of MKN45 (upper panel) and Panc-1 (bottom panel) control (pLKO) and SOX9-silenced (sh1) cells. Asterisks (*, ** and ***) indicate statistical significance (p < 0.05, p < 0.01, and p < 0.001, respectively).
To further characterize the link between SOX9 expression with BMI1 and p21CIP, we stained tumors derived from SOX9 silenced cells and controls with antibodies for BMI1 and p21CIP, as well as for SOX9 and Ki67. Thus, confirming previous results revealing that SOX9 inhibition reduces tumor growth25,31, immunohistochemistry analysis showed lower staining of SOX9 as well as reduction in Ki67 positive cells in gastric and pancreatic tumors with reduced SOX9 (Fig. 3F). In these contexts, BMI1 staining was lower, whereas p21CIP was increased in tumors derived from SOX9 knockdown cells (Fig. 3F). These results show that SOX9 modulates the expression of BMI1 and p21CIP in different cancer types in vivo.
Correlation between SOX9, BMI1 and p21CIP in clinical samples
Next, we wondered whether the association between SOX9, BMI1 and p21CIP levels could be translated to clinical samples. Therefore, we checked the relationship between their expression using previously described cohorts25 and available datasets from all genome-wide expression profiling arrays of glioblastoma, gastric and pancreatic cancer cohorts of patients. First, we found that SOX9 and BMI1 were increased by more than 5 and 2.5-fold respectively, while p21CIP was not significantly altered in GBM compared to normal brain tissue (Fig. 4A). In line with this, 80% and 60% of the GBMs showed high levels of SOX9 and BMI1 respectively (fold change higher than 2 respect to normal brain), whereas 83% showed low or moderate expression of p21CIP (Fig. 4B). Moreover, majority of the biopsies with high SOX9 expression also presented increased levels of BMI1 and moderate or low p21CIP levels (Fig. 4C).
Figure 4.

SOX9 expression correlates positively with BMI1 expression and negatively with p21CIP expression in cancer patient samples. (A) SOX9, BMI1 and p21CIP mRNA expression levels in glioblastoma samples (GBM) from Hospital Donostia patients relative to the expression in normal brain samples (healthy tissue). (B) Frecuency of SOX9, BMI1 and p21CIP high and low mRNA expressing GBM samples from Hospital Donostia patients. (C) Association of high SOX9 mRNA expression with high BMI1 and low p21CIP expression in human GBM samples from Hospital Donostia patients. (D) Graphs representing the correlation between SOX9 and BMI1 mRNA expression, and SOX9 and p21CIP (CDKN1A) mRNA expression in gastric adenocarcinoma (GC) and GBM patients from The Cancer Genome Atlas (TCGA) cohort. GBM (upper panel): positive correlation between SOX9 and BMI1 mRNA expression (Spearman’s correlation = 0.22, p-value = 8.31e-3, n = 147); and positive correlation between SOX9 and p21CIP mRNA expression (Spearman’s correlation = 0.27, p-value = 1.119e-3, n = 147). Data obtained from https://www.cancer.gov/tcga. GC (bottom panel): positive correlation between SOX9 and BMI1 mRNA expression (r-value = 0.086, p-value = 0.080, n = 450). Data obtained from R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl); negative correlation between SOX9 and p21CIP mRNA expression (Spearman’s correlation = −0.123, p-value = 0.0478, n = 258). Data obtained from https://www.cancer.gov/tcga. (E) Violin plots representing the expression of SOX9, BMI1 and p21CIP analyzed by microarrays in normal (N) and tumor (T) samples of three gastric cancer cohorts (datasets GSE13911, GSE79973 and GSE19826), two pancreatic adenocarcinoma cohorts (datasets GSE16515 and GSE15471) and one GBM cohort (dataset GSE50161). The black dots represent the individual expression values and the symbol + denote the mean values of the distributions. (F) Heatmap representing the expression of SOX9, BMI1, p21CIP (CDKN1A) and p16Ink4a (CDKN2A) in normal and tumor tissue samples belonging to the datasets represented in (E).
Next, we checked the available information obtained by The Cancer Genome Atlas (TCGA) project and found a significant correlation between SOX9 and BMI1 in GBM (Fig. 4D), detecting also that the expression of SOX9 correlated positively with BMI1 expression and negatively with p21CIPexpression in gastric cancer (Fig. 4D). Finally, we analyzed the expression of these markers in 3 cohorts of gastric tumor-normal samples (n = 60), 2 of pancreatic tumor-normal samples (n = 124), and additional one of GBM and normal brain samples (n = 47). The comparison between normal and tumor tissue revealed frequent overexpression of SOX9 and BMI1, whereas p21CIP levels were similar or decreased in tumor samples (Fig. 4E). Noteworthy, the heatmap representation revealed that the expression of SOX9 correlated positively with BMI1 expression and negatively with p21CIP in samples from the different types of cancer (Fig. 4F). Similarly, there was an inverse correlation between SOX9 and p16INK4a levels in the same samples (Fig. 4F). Importantly, these results translate the association of SOX9-BMI1-p21CIP from tumor cells in vitro and in vivo tumors to clinical biopsies.
BMI1 restoration in SOX9-silenced cells rescues the malignant phenotype of tumor cells
The fact that SOX9 modulation impacted on the expression of BMI1 in tumor cells in vitro and in vivo, suggested that BMI1 could constitute an effector of the pro-tumoral activity of SOX9. To further test this idea, we lentivirally transduced a construct encoding BMI1 gene in MKN45 GC cells and Panc-1 and RWP-1 pancreatic cancer cells with SOX9 knockdown or controls. The cells transduced with BMI1 encoding construct presented BMI1 overexpression in the case of control cells or restoration of BMI1 levels in SOX9 silenced cells (Fig. 5A). Moreover, at molecular level, BMI1 restoration reduced the expression of p21CIP respect to the levels found in SOX9-silenced cells (Fig. 5A).
Figure 5.

BMI1 re-expression in cancer cells with SOX9 silencing restores the aggressive phenotype conferred by SOX9 in vitro. (A) Representative Western blots of SOX9, BMI1, p21CIP and GFP protein expression in MKN45, Panc-1 and RWP-1 control (pLKO) and SOX9-silenced cells (sh1) lentivirally transduced with BMI1 (pLKO BMI1 and sh1 BMI1) or GFP (pLKO GFP and sh1 GFP)(n ≥ 4). β-actin levels were used as a loading control. (B) Relative cell growth determined by cell count experiments in RWP-1 pLKO and SOX9-silenced PDAC cell line (sh1) lentivirally transduced with BMI1 (pLKO BMI1 and sh1 BMI1) or GFP (pLKO GFP and sh1 GFP) (n ≥ 3). (C) Apoptosis represented by the percentage of active Caspase-3 positive cells determined by immunofluorescence staining in MKN45, Panc-1 and RWP-1 pLKO and SOX9-silenced cells (sh1) lentivirally transduced with BMI1 (pLKO BMI1 and sh1 BMI1) or GFP (pLKO GFP and sh1 GFP) (n ≥ 4). (D) Apoptosis represented by the percentage of cleaved PARP1 positive cells determined by immunofluorescence staining in MKN45, Panc-1 and RWP-1 pLKO and SOX9-silenced cells (sh1) lentivirally transduced with BMI1 (pLKO BMI1 and sh1 BMI1) or GFP (pLKO GFP and sh1 GFP) (n ≥ 4). (E) Cellular senescence represented by the percentage of β-Galactosidase (SA β-Gal) positive cells in Panc-1 and RWP-1 pLKO and SOX9-silenced cells (sh1) lentivirally transduced with BMI1 (pLKO BMI1 and sh1 BMI1) or GFP (pLKO GFP and sh1 GFP) (n ≥ 4). (F) Proliferative capacity represented by the percentage of phospho-histone H3 (p-H3) positive cells analyzed by immunosfluorescence staining in MKN45, Panc-1 and RWP-1 pLKO and SOX9-silenced cells (sh1) lentivirally transduced with BMI1 (pLKO BMI1 and sh1 BMI1) or GFP (pLKO GFP and sh1 GFP) (n ≥ 4). Asterisks (*, ** and ***) indicate statistical significance (p < 0.05, p < 0.01, and p < 0.001, respectively).
Then, we evaluated cellular phenotypes in terms of viability, apoptosis, proliferation and senescence in vitro. First, we noted that ectopic expression of BMI1 increased the number of tumor cells or restored the number counted in SOX9-silenced cultures (Fig. 5B). Accordingly, ectopic BMI1 also abrogated significantly the induction of apoptosis promoted by SOX9 silencing, reducing by over 50% the percentage of cells with active Caspase-3 and proteolyzed PARP1 in gastric and pancreatic cancer cells (Fig. 5C,D). Similarly, senescence induction by SOX9 silencing was significantly mitigated (over 50% reduction) by BMI1 restoration in tumor cells (Fig. 5E). Regarding cell proliferation, our results showed that BMI1 overexpression significantly increased the number of p-H3 positive cells compared to SOX9 knockdown cells (Fig. 5F). This indicates that BMI1 re-establishes the proliferative capacity of cancer cells abrogated by SOX9 knockdown.
Finally, we tested whether BMI1 is necessary for SOX9 pro-tumoral activity in vivo. For this, we inoculated either control (pLKO) or SOX9-silenced (sh1) MKN45 or Panc-1 cells, which were also transduced with BMI1 or empty vector, to immunodeficient mice. While MKN45 control cell derived tumors grew to almost 200 mm3, those derived from sh1 cells only grew to 50 mm3 (Fig. 6A,B). Interestingly, BMI1 restoration completely abrogated the reduction of tumor growth elicited by SOX9 knockdown (Fig. 6A,B). Similarly, BMI1 restoration also increased the tumor growth capacity of SOX9 knockdown Panc-1 cells (Fig. 6C). Accordingly, immunohistochemistry of tumors derived from MKN45 cells showed increased number of Ki67 positive cells in sh1 with BMI1 restoration compared to sh1 alone (Fig. 6D). Furthermore, this technique also revealed reduction in the staining of p21CIP in tumors derived form cells with BMI1 restoration (Fig. 6D). Overall, these results show that the ectopic restoration of BMI1 in SOX9-silenced cells recovers the aggressive phenotype of tumor cells.
Figure 6.

BMI1 re-expression in cancer cells with SOX9 knockdown restores tumor growth in vivo. (A) Volume at the indicated time points of the tumors generated after subcutaneous injection of 1 × 105 MKN45 pLKO GFP, sh1 GFP or sh1 BMI1 cells in nude mice (n = 12 injections/ condition). (B) Picture of representative tumors generated in (A). (C) Average volume of the subcutaneous tumors generated by the injection of 0.5 × 106 Panc-1 cells with SOX9 silencing (sh1 GFP) and Panc-1 cells with SOX9 silencing and BMI1 restoration (sh1 BMI1) in nude mice (n = 8 injections/condition). (D) Representative images of the hematoxilin-eosin (HE) staining and immunohistochemical staining of SOX9, BMI1, p21CIP and Ki67 in tumors from (A). Asterisks (*, ** and ***) indicate statistical significance (p < 0.05, p < 0.01, and p < 0.001, respectively).
Discussion
In this study, we show that SOX9 plays a role in cancer progression, not only regulating the activity of CSCs, but also modulating the function of the heterogeneous tumor cells that constitute the tumor bulk. In this context, SOX9 affects a broad plethora of cellular processes that contribute to tumor progression. In particular, SOX9 maintains cell viability, is relevant for cell survival and senescence evasion and promotes proliferation. At the mechanistic level, the transcriptional repressor BMI1 is an important effector of SOX9 in those processes through the negative regulation of p21CIP.
We found that SOX9 activity impacts on cell viability and influences cell proliferation in differentiated pancreatic, glioblastoma and gastric cancer cells. Furthermore, our data reveal that SOX9 silencing promotes that tumor cells undergo senescence and apoptosis. In agreement with our findings, it has been previously observed that high levels of Sox9 were sufficient to bypass cellular senescence33 and prevented apoptosis in non-tumor cells36. Our results highlight the function of SOX9 in controlling multiple processes associated to cancer and, hence explaining, how SOX9 potentiates tumor progression not only regulating the activity of CSCs.
BMI1 is an important epigenetic regulator, which restricts cell proliferation and mediates senescence and apoptosis during homeostasis37. In cancer, its levels are commonly elevated and it plays a potent oncogenic role in multiple types of cancer including pancreatic, glioblastoma and gastric cancer38–40. In this work, we found that modulation of SOX9 levels affects BMI1 expression in multiple cancer cell lines in vitro and in vivo. These results are in agreement with previous studies that linked SOX9 and BMI1 in primary fibroblasts and colorectal CSCs23,33. This regulation is likely to be direct since chromatin immunoprecipitation experiments have shown that SOX9 binds to the promoter of BMI1 in different types of cells and contexts33,41. Moreover, our results show that there is also a positive correlation between SOX9 and BMI1 expression, and negative between SOX9 and p21CIP in clinical samples. In line with this, previous studies analyzed the correlation between SOX9, BMI1 and p21CIP in different types of cancers and showed independently that SOX9 regulates proliferation through a positive correlation with BMI1 and an inverse correlation with p21CIP expression23,31,33–35. Together, these results highlight that BMI1 is a relevant mediator of SOX9 in promoting tumor malignancy and show the importance of this axis in cancer progression, thus providing new therapeutic possibilities, such as BMI1 inhibition, a strategy that is being currently evaluated in different cancer clinical trials. Thus, the trial NCT03761095 is aimed at evaluating the safety of the orally active BMI1 inhibitor PTC596 (PTC Therapeutics) in combination with dacarbazine for the treatment of advanced leiomyosarcoma. Moreover, also the trials NCT03206645 and NCT03605550 are currently evaluating this inhibitor. The first is a dose-escalation study oriented to evaluate its safety, tolerability and pharmacokinetics in combination with conventional chemotherapy for the treatment of ovarian and fallopian tube cancer or primary peritoneal cancer. The latter tries to determine the safe dose of PTC596 for its administration in combination with radiation in children with newly diagnosed diffuse intrinsic pontine glioma and high-grade glioma.
BMI1 represses the transcription of multiple genes including relevant tumor suppressor genes such as p21CIP 42–44, as well as the Ink4a/Arf locus, encoding p14Arf and p16INK4a, being the latter the best-known target in cell growth arrest and senescence37. Indeed, we have previously shown that Sox9 promotes proliferation and favors neoplastic transformation of primary cells through Bmi1, whose upregulation consequently represses the expression of Ink4a and Arf genes33. Ink4a/Arf locus is frequently mutated and inactivated in human cancers45, as it is the case also of many tumor cell lines including the ones used in this study. Thus, the action of SOX9-BMI1 axis in driving processes associated to tumor progression is not attributable to the repression of p16INK4a and/or p14Arf. Indeed, our results show that this axis promotes tumor cell survival and proliferation via p21CIP, since the restoration of BMI1 levels in SOX9 silenced cells, also modulated, in this case inhibited, the expression of this tumor suppressor. Thus, we postulate that the axis SOX9-BMI1 plays a relevant role in the different stages of cancer initiation and progression, in which they inhibit the expression of tumor suppressors sequentially. The repression of Ink4a/Arf locus is required at early stages of neoplastic transformation and tumor formation, whereas the repression of p21CIP is necessary in the action exerted by the axis SOX9-BMI1 for cancer progression (Fig. 7).
Figure 7.
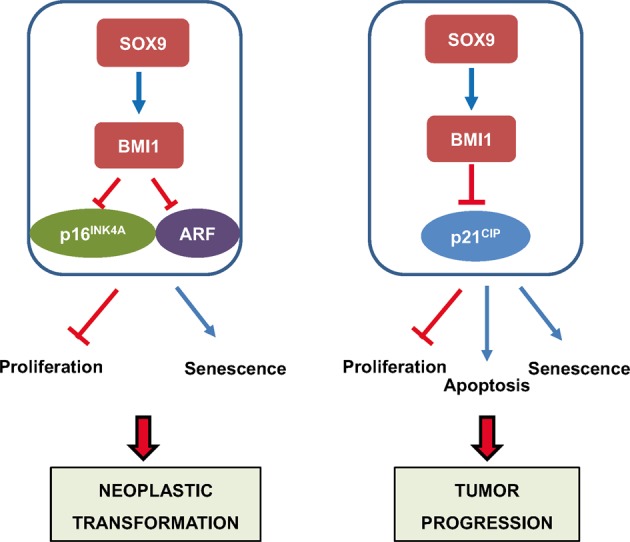
Pro-tumoral action and molecular mechanism of SOX9 in cancer. In the context of non-tumor cells (left panel), SOX9 is able to promote neoplastic transformation through the induction of cell proliferation and the evasion of senescence. At the molecular level, this transformation takes place through the induction of BMI1 expression, which represses the expression of the tumor suppressors p16INK4A and ARF (previously described by Matheu et al., 2012). In the context of tumor cells (right panel), generally deficient in p16INK4A and ARF expression, SOX9 enhances tumor progression through the promotion of cell proliferation and the evasion of apoptosis and senescence. In this context, the molecular mechanism involves the repression of the tumor suppressor p21CIP.
In summary, our findings show that SOX9 is highly relevant in the survival of population of cells constituting the tumor bulk in multiple types of cancer contributing to evasion of apoptosis. Moreover, we reveal that SOX9 controls the proliferative capacity of tumor cells and facilitates evading senescence. Mechanistically, SOX9 exerts these pro-tumoral actions through BMI1- p21CIP, providing novel knowledge regarding the molecular events leading to cancer progression.
Experimental Procedures
Human subjects
Clinical information of glioblastoma patients was obtained from the Donostia University Hospital. Human glioma samples were provided by the Basque Biobank for Research-OEHUN (http://www.biobancovasco.org). All study participants signed informed consent form approved by the Institutional Ethical Committee. The study was approved by the ethic committee of Euskadi (PI2016151). All methods were performed in accordance with the relevant guidelines and regulations.
Cell culture
Pancreatic carcinoma cell lines (Panc-1, RWP-1, IMIMPC-2 and BxPC-3) were kindly provided by Dr. Real (CNIO) and Dr. Navarro (IMIM-Medical Research Institute). Glioblastoma cell lines U251MG (U251), U87MG (U87) and U373MG (U373) were purchased from the ATCC (American Type Culture Collection). The human gastric adenocarcinoma AGS and MKN45 cell lines were gifts from Dr. Haas (Ludwig-Maximilians-Universität München, München, Germany). All cell lines were mycoplasma free. Cells were cultured as adherent monolayers at 37 °C and 5% CO2 in DMEM medium (Gibco) (MKN45 cells in RPMI) supplemented with 10% FBS (Gibco), L-glutamine (2 mM), 100 U/ml penicillin and 100 µg/ml streptomycin.
Gene silencing and overexpression
For SOX9 silencing by shRNA, cells were infected with lentivirus harboring the shSOX9 plasmid #40644 and the corresponding pLKO.1 puro control plasmid #8453 from Addgene, gifts from Dr. Bob Weinberg. Transduced cells were selected 48 hours later in the presence of 2 μg/mL puromycin for 48–72 hours. For lentiviral SOX9 overexpression the pWXL-SOX9 plasmid #36979 from Addgene, gift from Bob Weinberg, was used. The pWXL plasmid #12257 from Addgene, gift from Didier Trono, was used as control. For BMI1 upregulation cells were infected with lentivirus harboring the plasmid pLenti CMV GFP Puro-Bmi1 (gift from Jacqueline Lees). Lentiviral infections were performed as previously described31. All infections were performed at a MOI of 10 for 6 hours.
Western blot
Immunoblots were performed following standard procedures. Primary antibodies used were: SOX9 (AB5535, Millipore), BMI1 (05-637, Millipore), p21CIP (sc-397-G, Santa Cruz Biotechnology), GFP (ab6673, abcam) and β-actin (AC-15, Sigma). Primary antibodies were detected with HRP-linked secondary antibodies: anti-rabbit (7074S, Cell Signaling Technology), anti-mouse (7076S, Cell Signaling Technology) and anti-goat (sc-2020, Santa Cruz Biotechnology). Protein bands were detected using the ECL system (NOVEX® ECL, Invitrogen).
Immunofluorescence
For immunofluorescent detections, cells were seeded in Lab-Tek II Chamber Slides (ThermoFisher Scientific) and fixed with 4% paraformaldehyde for 15 min at RT. Then, cells were incubated with PBS supplemented with 0.3% Triton X-100 and 5% FBS for 1 hour at RT. Cells were incubated overnight at 4 °C with different primary antibodies: phospho-histone H3 (phospho S10) (ab14955, Abcam), Cleaved PARP1 (ab32064, Abcam) and Active Caspase 3 (AF835, R&D Systems). Secondary antibodies conjugated to fluorochromes were incubated for 1 hour at RT and chromatin staining was performed with Hoechst 33342 (Molecular Probes). Slides were mounted with Fluoro-Gel mounting medium (Electron Microscopy Sciences) and preparations were visualized and documented using a Nikon Eclipse 80i microscope.
β-Galactosidase activity
To analyze cellular senescence, β-Galactosidase activity was measured in cells using the Senescence β-Galactosidase Staining Kit of Cell Signaling Technology (#9860), according to the manufacturer’s protocol. Briefly, cells were fixed and incubated overnight at 37 °C in a dry incubator with a staining solution containing X-Gal. Cells were observed in an inverted light microscope and different views were captured randomly to calculate the positive staining rate for each experimental condition.
mRNA expression analysis
Total RNA was extracted with trizol (Life Technologies). Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher) according to the manufacturer’s guidelines. Quantitative real-time PCR was performed in an ABI PRISM 7300 thermocycler (Applied Biosystems) using Power SYBR® Green Master Mix (ThermoFisher), 10 mmol/L of primers and 20 ng of cDNA. GAPDH was used as housekeeping gene. The ΔΔCT method was used for relative quantification.
Computational biology analysis
TCGA data were obtained from https://www.cancer.gov/tcga and https://r2.amc.nl. All the software and graphs for transcriptomic analysis were developed using in-house code developed in MATLAB.
In vivo carcinogenesis assays
All animal handling and protocols were approved by the animal care ethic committee of Biodonostia Institute and were conducted in conformity with the EU guidelines and regulations for animal experimentation. For subcutaneous injection, cells were harvested with trypsin/EDTA and resuspended in PBS. 1 × 105 MKN45 cells and 0.5 × 106 Panc-1 cells were injected into both flanks of 8 week-old Foxn1nu/Foxn1nu mice. External calipers were used to measure tumor size, from which tumor volume was estimated by V = L*W2*0.5, where L is the tumor length and W is the tumor width.
Hematoxylin and eosin staining and immunohistochemistry
Tumors generated in mice were dissected, fixed in 10% formalin for 48 hours and embedded in paraffin. 4 µm thick sections were stained with hematoxylin and eosin (H&E). For immunohistochemistry, sections were rehydrated and heated in citrate buffer pH 6 for antigen retrieval. Antibodies used for detections included SOX9 (AB5535, Millipore), BMI1 (05-637, Millipore), p21CIP (sc-6246, Santa Cruz Biotechnology) and Ki67 (ab15580, Abcam). The stainings were developed with 3,3′ Diaminobenzidine (DAB) and nuclei were counterstained with hematoxylin.
Data evaluation
Data are presented as mean values ± standard error (SEM), with the number of experiments (n) in parentheses. Unless otherwise indicated, statistical significance (p-values) was calculated using the Student´s t -test. Asterisks (*, ** and ***) indicate statistical significance (p < 0.05, p < 0.01, and p < 0.001, respectively).
Supplementary information
Acknowledgements
P.A., M.O.-U., A.S.-A. and M.G.-P. were recipient of predoctoral fellowships from the AECC foundation, FPU grant, pFIS and University of the Basque Country (PIF 15/245). E.C.-G. was a recipient of a Stop Fuga de Cerebros postdoctoral fellowship. We thank the Histology Platform of the Biodonostia Health Research Institute for their help. A.M. lab is supported by grants from Instituto de Salud Carlos III and FEDER Funds (CP16/00039, DTS16/00184, PI16/01580, DTS18/00181, PI18/01612, CP19/00085), Diputacion Guipuzcoa (112/18), REFBIO II and Industry and Health Departments of the Basque Country.
Author contributions
E.C.-G., P.A., M.O.-U., A.S.-A. and M.G.-P. performed all the cellular and molecular experiments; M.M.-V. and N.S. helped with clinical analysis. D.G. and M.J.A.-B. performed computational studies and biostatistical analysis. J.M.F. performed immunohistochemistry. All of them revised the manuscript; E.C.-G. wrote the manuscript, designed the research, and obtained funds. A.M. designed the research, directed the project, obtained funds and wrote the manuscript.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Paula Aldaz and Maddalen Otaegi-Ugartemendia.
Contributor Information
Ander Matheu, Email: ander.matheu@biodonostia.org.
Estefania Carrasco-Garcia, Email: estefania.carrasco@biodonostia.org.
Supplementary information
is available for this paper at 10.1038/s41598-019-57047-w.
References
- 1.McGranahan N, Swanton C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell. 2017;168:613–628. doi: 10.1016/j.cell.2017.01.018. [DOI] [PubMed] [Google Scholar]
- 2.Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017;16:41. doi: 10.1186/s12943-017-0600-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 4.Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 5.Ostrom QT, et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro Oncol. 2018;20:iv1–iv86. doi: 10.1093/neuonc/noy131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koshy M, et al. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J Neurooncol. 2012;107:207–212. doi: 10.1007/s11060-011-0738-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sarkar A, Hochedlinger K. The sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15–30. doi: 10.1016/j.stem.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pritchett, J., Athwal, V., Roberts, N., Hanley, N. A. & Hanley, K. P. Understanding the role of SOX9 in acquired diseases: lessons from development. Trends in molecular medicine. (2010). [DOI] [PubMed]
- 9.Cheung M, Briscoe J. Neural crest development is regulated by the transcription factor Sox9. Development. 2003;130:5681–5693. doi: 10.1242/dev.00808. [DOI] [PubMed] [Google Scholar]
- 10.Kawaguchi Y. Sox9 and programming of liver and pancreatic progenitors. J Clin Invest. 2013;123:1881–1886. doi: 10.1172/JCI66022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furuyama K, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet. 2011;43:34–41. doi: 10.1038/ng.722. [DOI] [PubMed] [Google Scholar]
- 12.Belo J, Krishnamurthy M, Oakie A, Wang R. The role of SOX9 transcription factor in pancreatic and duodenal development. Stem Cells Dev. 2013;22:2935–2943. doi: 10.1089/scd.2013.0106. [DOI] [PubMed] [Google Scholar]
- 13.Scott CE, et al. SOX9 induces and maintains neural stem cells. Nat Neurosci. 2010;13:1181–1189. doi: 10.1038/nn.2646. [DOI] [PubMed] [Google Scholar]
- 14.Sun W, et al. SOX9 Is an Astrocyte-Specific Nuclear Marker in the Adult Brain Outside the Neurogenic Regions. J Neurosci. 2017;37:4493–4507. doi: 10.1523/JNEUROSCI.3199-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seymour PA. Sox9: a master regulator of the pancreatic program. Rev Diabet Stud. 2014;11:51–83. doi: 10.1900/RDS.2014.11.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shih HP, et al. A Notch-dependent molecular circuitry initiates pancreatic endocrine and ductal cell differentiation. Development. 2012;139:2488–2499. doi: 10.1242/dev.078634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hessmann E, et al. NFATc4 Regulates Sox9 Gene Expression in Acinar Cell Plasticity and Pancreatic Cancer Initiation. Stem Cells Int. 2016;2016:5272498. doi: 10.1155/2016/5272498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kopp JL, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:737–750. doi: 10.1016/j.ccr.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Swartling FJ, et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell. 2012;21:601–613. doi: 10.1016/j.ccr.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Larsimont JC, et al. Sox9 Controls Self-Renewal of Oncogene Targeted Cells and Links Tumor Initiation and Invasion. Cell Stem Cell. 2015;17:60–73. doi: 10.1016/j.stem.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Ye X, et al. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature. 2015;525:256–260. doi: 10.1038/nature14897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo W, et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell. 2012;148:1015–1028. doi: 10.1016/j.cell.2012.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrasco-Garcia E, et al. SOX9-regulated cell plasticity in colorectal metastasis is attenuated by rapamycin. Sci Rep. 2016;6:32350. doi: 10.1038/srep32350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higashihara T, et al. Sex Determining Region Y Box 9 Induces Chemoresistance in Pancreatic Cancer Cells by Induction of Putative Cancer Stem Cell Characteristics and Its High Expression Predicts Poor Prognosis. Pancreas. 2017;46:1296–1304. doi: 10.1097/MPA.0000000000000945. [DOI] [PubMed] [Google Scholar]
- 25.Garros-Regulez L, et al. mTOR inhibition decreases SOX2-SOX9 mediated glioma stem cell activity and temozolomide resistance. Expert Opin Ther Targets. 2016;20:393–405. doi: 10.1517/14728222.2016.1151002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimm, D. et al. The role of SOX family members in solid tumours and metastasis. Semin Cancer Biol. (2019). [DOI] [PubMed]
- 27.Malladi S, et al. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell. 2016;165:45–60. doi: 10.1016/j.cell.2016.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia S, et al. Clinical implication of Sox9 and activated Akt expression in pancreatic ductal adenocarcinoma. Med Oncol. 2015;32:358. doi: 10.1007/s12032-014-0358-0. [DOI] [PubMed] [Google Scholar]
- 29.Grimont A, et al. SOX9 regulates ERBB signalling in pancreatic cancer development. Gut. 2015;64:1790–1799. doi: 10.1136/gutjnl-2014-307075. [DOI] [PubMed] [Google Scholar]
- 30.Gnerlich JL, et al. Increased SOX9 Expression in Premalignant and Malignant Pancreatic Neoplasms. Ann Surg Oncol. 2019;26:628–634. doi: 10.1245/s10434-018-6925-4. [DOI] [PubMed] [Google Scholar]
- 31.Santos JC, et al. SOX9 Elevation Acts with Canonical WNT Signaling to Drive Gastric Cancer Progression. Cancer Res. 2016;76:6735–6746. doi: 10.1158/0008-5472.CAN-16-1120. [DOI] [PubMed] [Google Scholar]
- 32.Ruan H, Hu S, Zhang H, Du G, Li X. Upregulated SOX9 expression indicates worse prognosis in solid tumors: a systematic review and meta-analysis. Oncotarget. 2017;8:113163–113173. doi: 10.18632/oncotarget.22635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matheu A, et al. Oncogenicity of the developmental transcription factor Sox9. Cancer Res. 2012;72:1301–1315. doi: 10.1158/0008-5472.CAN-11-3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang SS, et al. Upregulation of SOX9 in lung adenocarcinoma and its involvement in the regulation of cell growth and tumorigenicity. Clin Cancer Res. 2010;16:4363–4373. doi: 10.1158/1078-0432.CCR-10-0138. [DOI] [PubMed] [Google Scholar]
- 35.Saegusa M, Hashimura M, Suzuki E, Yoshida T, Kuwata T. Transcriptional up-regulation of Sox9 by NF-kappaB in endometrial carcinoma cells, modulating cell proliferation through alteration in the p14(ARF)/p53/p21(WAF1) pathway. Am J Pathol. 2012;181:684–692. doi: 10.1016/j.ajpath.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 36.Roche KC, et al. SOX9 maintains reserve stem cells and preserves radioresistance in mouse small intestine. Gastroenterology. 2015;149:1553–1563 e1510. doi: 10.1053/j.gastro.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 38.Song W, et al. Bmi-1 is related to proliferation, survival and poor prognosis in pancreatic cancer. Cancer Sci. 2010;101:1754–1760. doi: 10.1111/j.1349-7006.2010.01577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdouh M, et al. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci. 2009;29:8884–8896. doi: 10.1523/JNEUROSCI.0968-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kreso A, et al. Self-renewal as a therapeutic target in human colorectal cancer. Nat Med. 2014;20:29–36. doi: 10.1038/nm.3418. [DOI] [PubMed] [Google Scholar]
- 41.Shi Z, et al. Context-specific role of SOX9 in NF-Y mediated gene regulation in colorectal cancer cells. Nucleic acids research. 2015;43:6257–6269. doi: 10.1093/nar/gkv568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fasano CA, et al. shRNA knockdown of Bmi-1 reveals a critical role for p21-Rb pathway in NSC self-renewal during development. Cell Stem Cell. 2007;1:87–99. doi: 10.1016/j.stem.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 43.Subkhankulova T, Zhang X, Leung C, Marino S. Bmi1 directly represses p21Waf1/Cip1 in Shh-induced proliferation of cerebellar granule cell progenitors. Molecular and cellular neurosciences. 2010;45:151–162. doi: 10.1016/j.mcn.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 44.Karimian A, Ahmadi Y, Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst). 2016;42:63–71. doi: 10.1016/j.dnarep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 45.Sherr CJ. The INK4a/ARF network in tumour suppression. Nature reviews. 2001;2:731–737. doi: 10.1038/35096061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.