

Abstract
Breast cancer patients are commonly treated with taxane (e.g. docetaxel) chemotherapy, despite poor outcomes and eventual disease relapse. We previously identified the Bcl-2-associated death promoter (BAD) as a prognostic indicator of good outcome in taxane-treated breast cancer patients. We also demonstrated that BAD expression in human breast carcinoma cells generated larger tumors in mouse xenograft models. These paradoxical results suggest that BAD-expressing tumors are differentially sensitive to taxane treatment. We validated this here and show that docetaxel therapy preferentially reduced growth of BAD-expressing xenograft tumors. We next explored the cellular mechanism whereby BAD sensitizes cells to docetaxel. Taxanes are microtubule inhibiting agents that cause cell cycle arrest in mitosis whereupon the cells either die in mitosis or aberrantly exit (mitotic slippage) and survive as polyploid cells. In response to docetaxel, BAD-expressing cells had lengthened mitotic arrest with a higher proportion of cells undergoing death in mitosis with decreased mitotic slippage. Death in mitosis was non-apoptotic and not dependent on Bcl-XL interaction or caspase activation. Instead, cell death was necroptotic, and dependent on ROS. These results suggest that BAD is prognostic for favourable outcome in response to taxane chemotherapy by enhancing necroptotic cell death and inhibiting the production of potentially chemoresistant polyploid cells.
Subject terms: Cancer therapeutic resistance, Chemotherapy, Tumour biomarkers, Breast cancer
Introduction
Triple-negative breast cancer patients receive taxane chemotherapy, such as docetaxel (Taxotere®), as standard first-line treatment despite an overall poor prognosis, high rate of relapse, and adverse effects1. While multiple causes of cellular taxane resistance are known, these have not yet provided clinical markers to guide taxane therapy decisions2–4. Understanding the molecular mechanisms that mediate outcome to taxane therapy may identify predictive biomarkers and novel therapeutic targets. The Bcl-2 family member BAD (Bcl-2-associated death promoter) is a prognostic indicator for good clinical outcome of taxane-treated breast cancer patients5. BAD modulates breast cancer cell proliferation and tumor progression by regulating cell cycle progression6,7. Thus, understanding how BAD better predicts patient outcome could aid in understanding docetaxel chemoresistance.
Taxanes are anti-mitotic drugs that perturb microtubule dynamics, leading to chronic activation of the spindle assembly checkpoint and inhibition of the anaphase promoting complex that delays the degradation of cyclin B1 and inhibits mitotic exit8. Ideally, this aberrant mitotic arrest initiates cell death in mitosis by facilitating the accumulation of a caspase-dependent death signal9. Often, however, cells degrade sufficient cyclin B1 prior to full activation of apoptotic caspases, and cells slip out of mitosis in the absence of cytokinesis and enter G1 as polyploid cells. These polyploid cells have differential fates of G1 arrest, post-mitotic death, or continued cell cycle progression10,11. The survival and expansion of these polyploid cells is postulated to generate aggressive clones that are resistant to therapy12,13.
Programmed necrosis, termed necroptosis, is a form of caspase-independent cell death that is activated in response to many anticancer drugs14. Cells can activate necroptosis in the absence of functional apoptosis15. The cell morphology of necroptotic death is similar to that of necrosis, as it includes loss of plasma membrane integrity, mitochondrial dysfunction, oxidative stress, and absence of nuclear fragmentation16. Recent evidence suggests necroptosis can be exploited for cancer therapy, in particular, for apoptosis-resistant cancers17. Taxane treatment can promote necroptosis, although the mechanism remains unclear18.
In the current study, we demonstrated that BAD increases sensitivity of breast cancer tumor xenografts to docetaxel treatment in vivo. BAD-expressing cells prolonged mitotic arrest, and enhanced cell death. Cell death was not dependent on caspases or Bcl-XL indicating a non-apoptotic pathway. Instead, cell death had morphological hallmarks of necrosis and was dependent on reactive oxygen species (ROS) and the necroptotic kinase MLKL. Thus, we identified a novel role for BAD in enhancing necroptosis during taxane-induced mitotic arrest. Our results provide a potential cellular mechanism wherein BAD is prognostic for clinical docetaxel chemotherapy.
Results
BAD increases sensitivity of breast cancer cells to docetaxel
We had previously shown BAD-dependent taxane sensitivity in the breast carcinoma cell lines MCF-7 (luminal B), SKBR-3 (HER2) and MDA-MB-468 (TNBC)5. To study the structure/function relationship of BAD in docetaxel-treated breast cancer cells, we utilized MDA-MB-231 cells stably expressing ectopic BAD7. We performed a longitudinal cell death assay over 5 days of docetaxel treatment to determine docetaxel sensitivity (Fig. 1a). At the earliest measured time point BAD protected cells from docetaxel-induced cell death. This was transient, however, as with increasing time BAD sensitized cells to docetaxel-toxicity. To examine the in vivo relevance of these effects, we performed orthotopic mammary fat pad xenografts in nude mice. Mice were treated with docetaxel on the days indicated by the red arrows (Fig. 1b) and tumor volume was measured. Similar to what we had reported previously, BAD tumors grew significantly larger than vector tumors due to increased cell proliferation and survival signalling7. Tumor growth of BAD expressing cells was significantly decreased in response to docetaxel treatment (Fig. 1c,d). On the other hand, there was no change in tumor size in docetaxel-treated vector control tumors. Additionally, overall survival of mice with BAD tumors treated with docetaxel was increased relative to untreated BAD tumors (Fig. 1e). Altogether, these results indicate BAD expression increases tumor volume, however, these cells are more sensitive to docetaxel treatment with enhanced cell death and decreased tumor size.
Figure 1.

BAD increases sensitivity to docetaxel. (a) MDA-MB-231 cells expressing vector or BAD were treated with 125 nM docetaxel for 5 days. Cells were stained with Annexin V-647 and PI and analyzed via flow cytometry daily. Cell death in control group were subtracted from the docetaxel treated group. Annexin V+/PI+ population is depicted. Student’s t-test; n = 3. (b) MDA-MB-231 cells expressing vector or BAD were injected into the mammary fat pads of Taconic nude mice. Red arrows indicate docetaxel or vehicle injection time points. Tumor volume was measured weekly. One-way ANOVA with Dunnett’s post-hoc test; Vector + vehicle = 8, vector + docetaxel = 6, BAD + vehicle = 7, BAD + docetaxel = 7. (c) Scatter plot of tumor volume at day 49. (d) Representative images of tumors in the mammary gland of nude mice. Arrows indicate tumor location. (e) Kaplan-Meier survival curve of mice treated with vehicle or docetaxel.
BAD increases length in docetaxel-mediated mitotic arrest to promote cell death over mitotic slippage
Docetaxel binds to tubulin and disrupts microtubule dynamics, induces prolonged mitotic arrest, and can lead to cell death in mitosis19. Cells that do not die in mitosis ‘slip’ out of mitosis and re-enter G1 without cytokinesis. This phenomenon is referred to as ‘mitotic slippage’20. To understand how BAD sensitized cells to docetaxel, we used time-lapse live imaging to characterize cell morphology in response to docetaxel. Representative cell fates of docetaxel-treated cells are shown (Fig. 2a). We defined death in mitosis as cells with mitotic morphology that eventually ceased all cell movement. This cell death had a non-apoptotic morphology, indicated by cell swelling, granulation of the cytoplasm, and lack of cellular blebbing. Mitotic slipped cells were defined as cells with mitotic morphology that then transitioned to become flat adherent cells. These slipped cells remained viable and maintained subcellular movements. Mitotic slippage at imaging endpoint was confirmed by DAPI-staining, which revealed large multinucleated cells (Fig. 2b), typical of cells that have undergone mitotic exit in the absence of cytokinesis21.
Figure 2.

BAD increases length in mitotic arrest with docetaxel treatment. (a) Representative cell fates of MDA-MB-231 cells expressing vector or BAD treated with 125 nM docetaxel for 72 hours. Scale bar = 20 μM. (b) Immunofluorescence images taken 48 hours after docetaxel treatment and stained with DAPI and α-Tubulin. Scale bar = 20 μM. (c) Cell fates of cells treated with docetaxel for 72 hours. Each horizontal line represents an individual cell. Line endpoint represents the time at which the indicated cell fate occurred. (d) Scatter-plot representation of individual cell which either slipped or died in mitosis, and the corresponding hours of mitotic arrest. Median and interquartile range are shown. Mann-Whitney statistical test. (e) Length of mitotic arrest for all cell fates. Mann-Whitney statistical test. (f) Cells were treated with 125 nM docetaxel for 16, 24, or 48 hours. DMSO control treatment was for 48 hours. Right: Quantification of cyclin B1 levels normalized to tubulin. Student’s t-test; n = 3. (g) Cell cycle phases of docetaxel treated cells were analyzed daily for 5 days with PI staining and flow cytometry. Student’s t-test; n = 3.
To quantify differences in cell fates in vector versus BAD cells, we generated cell fate maps over 72 hours of drug treatment (Fig. 2c). The bar length indicates the time each cell spent in mitotic arrest, and the bar color indicates the subsequent cell fate until the experimental endpoint. Cells that entered mitosis (Fig. 2c) either remained in mitosis (blue), died in mitosis (green) or slipped out of mitotic arrest (purple). The average length a cell spent in mitotic arrest that culminated in death in mitosis was longer than for the cells that underwent slippage, (Fig. 2d), supporting a model of competing pathways between cell death and mitotic exit22,23. Additionally, BAD cells showed significantly longer times in mitotic arrest than vector control cells, irrespective of cell fate (Fig. 2e). Degradation of cyclin B1 is critical for determining cell fate, as the premature attenuation of cyclin B1 prior to accumulation of a sufficient cell death signal ultimately leads to mitotic slippage24. BAD cells retained higher levels of cyclin B1, suggesting that enhanced cyclin B1 stability lengthened mitotic arrest thus enabling the cells to accumulate a cell death signal (Fig. 2f). In support of this, DNA content-based cell cycle analysis revealed a greater proportion of BAD cells in mitotic arrest (G2/M; days 1–2), that then is followed by a higher amount of cell death (sub-G1 population; days 4–5) (Fig. 2g). Taken together, these results reveal BAD increases length in mitotic arrest upon docetaxel treatment in association with inhibited cyclin B1 degradation. This may allow the cells to accumulate a greater cell death signal, favouring cell death in mitosis versus mitotic slippage. These data therefore suggest BAD sensitizes cells to docetaxel by facilitating mitotic arrest-dependent non-apoptotic cell death.
The BAD and Bcl-XL interaction is dispensable for docetaxel cell death
The anti-apoptotic Bcl-2 family members Mcl-1, Bcl-2, and Bcl-XL regulate death in mitosis and mitotic slippage. Mcl-1 degradation is required for death in mitosis25. Additionally, phosphorylation of Bcl-2, and Bcl-XL by CDK1 upon taxane treatment inactivates these anti-apoptotic proteins and enables mitotic death26,27, whereas lack of Bcl-XL phosphorylation is associated with mitotic slippage and cell survival26. Thus, we examined Mcl-1, Bcl-2, and Bcl-XL after docetaxel addition to determine if BAD cells elevated death in mitosis due to increased degradation or phosphorylation of these anti-apoptotic Bcl-2 family members. We found no difference in Mcl-1 and Bcl-2 levels between vector and BAD cells (Fig. 3a). However, Bcl-XL levels were higher with more phosphorylation on Ser62 in BAD cells compared to vector cells (Fig. 3a), confirming a higher proportion of BAD-expressing cells with active CDK1 in mitotic arrest. Furthermore, phosphorylation of Ser62 attenuates the anti-apoptotic activity of Bcl-XL by inhibiting binding to BAX28, suggesting cells are sensitized to undergo apoptosis. Since BAD binds to Bcl-XL to displace BAX and stimulate cell death29, we tested whether the BAD and Bcl-XL interaction was necessary for docetaxel-mediated cell death. Immunoprecipitation of BAD protein at 24 hours of docetaxel treatment revealed a strong interaction between BAD and phospho-Bcl-XL (Ser62) (Fig. 3b). Surprisingly, we did not see a Bcl-XL and BAX interaction under any conditions (Fig. 3c), which was inconsistent with an apoptotic “primed to die” phenotype. Therefore, we functionally tested the requirement of the BAD and Bcl-XL interaction for docetaxel-mediated cell death using a cell line expressing BAD-L114A that abrogates BAD:Bcl-XL binding30. The BAD L114A mutant did not attenuate docetaxel cell death, indicating the BAD and Bcl-XL interaction is not required for cell death (Fig. 3d). Thus, these results indicate that BAD-potentiation of docetaxel sensitivity is not dependent on binding to Bcl-XL.
Figure 3.

BAD and Bcl-XL binding is not required for docetaxel killing. (a) Left: MDA-MB-231 cells stably expressing vector or BAD were treated with 125 nM docetaxel for the indicated time points. Control is DMSO for 48 hours. Right: Quantification of protein band density for pBcl-XL-Ser62 over total Bcl-XL. Student’s t-test; n = 3. (b) Immunoprecipitation (IP) of BAD antibody after 24 hours of 125 nM docetaxel treatment. GST antibody was used as a negative control. (c) Immunoprecipitation with Bcl-XL antibody after 125 nM docetaxel for 5 days. Vimentin antibody was used as a negative control. Right: The flow throughs (FT) from the IPs were retained and subjected to western blot. (d) Annexin V+/PI+ staining and flow cytometry analysis of 125 nM docetaxel treated cells after 5 days. One-way ANOVA with Dunnett’s post-hoc test; n = 5.
Docetaxel can induce necroptotic cell death
The observation that Bcl-XL does not regulate the ability of BAD to enhance docetaxel-induced cell death, suggests that BAD modulates a non-apoptotic mechanism. To assess apoptosis, we examined the apoptotic markers of cleaved caspase-3, cleaved PARP and BAX activation. Docetaxel treatment induced minimal caspase cleavage and detectable PARP cleavage that was similar between vector and BAD-expressing cells (Fig. 4a). BAX activation was not induced by docetaxel treatment (Fig. 4b). On the other hand, these markers were positive for apoptosis induced by a known apoptotic inducer, staurosporine (Supplementary Fig. 1)31. Therefore, despite having a competent apoptotic machinery, apoptosis is minimally activated with docetaxel. Instead, the majority of cells appeared necrotic with compromised plasma membranes (Annexin-V positive/PI positive) (Fig. 1a). In contrast, apoptotic inducers trigger initial phosphatidylserine externalization followed by secondary necrosis (Annexin-V positive/PI negative) (Fig. 4c and Supplementary Fig. 1). Thus, docetaxel caused concurrent plasma membrane damage and phosphatidylserine exposure, consistent with necrotic-like cell death.
Figure 4.

Docetaxel is killing the cells through a non-apoptotic mechanism. (a) MDA-MB-231 cells stably expressing vector or BAD were treated with DMSO control (left) or 125 nM docetaxel (middle) and subjected to western blot. Right: Protein band quantification was performed. Student’s t-test; no statistical significance. (b) Cells were treated with 125 nM docetaxel for 3 days prior to immunoprecipitation (IP) with 6A7 BAX antibody. Vimentin antibody was used as a negative control. Input lanes were also probed with cleaved caspase-3 antibody (right). (c) Cells were treated with 125 nM docetaxel for 5 days and stained with Annexin V-647 and PI daily and analyzed via flow cytometry. The Annexin V+/PI− population is graphed. Student’s t-test; no statistical significance between vector and BAD. Right: Dot plots from flow cytometric analysis. Annexin V positive cells are on the x-axis, PI positive cells are on the y-axis. Time, in days, is increasing to the right.
Necroptosis is a regulated form of necrotic cell death that compensates when the apoptotic pathway is blocked32. The necrosome is the core executioner complex that consists of RIP3 oligomers and MLKL and is activated in a RIP1-dependent and independent manner32,33. Docetaxel has previously been shown to initiate both necrosis and apoptosis in MDA-MB-231 cells34, so we assessed whether necroptosis was triggered in our studies. Inhibition of caspases did not attenuate cell death (Fig. 5), consistent with necroptotic signaling. Inhibition of necroptosis with the MLKL inhibitor necrosulfonamide35 significantly decreased cell death in BAD cells only, indicating that BAD stimulates docetaxel-induced necroptosis. The addition of both a pan-caspase inhibitor, z-VAD-FMK, and necrosulfonamide significantly reduced cell death in both vector and BAD cells, confirming one mechanism of cell death might be activated when the other is compromised. Therefore, BAD stimulates docetaxel-induced cell death through necroptosis.
Figure 5.
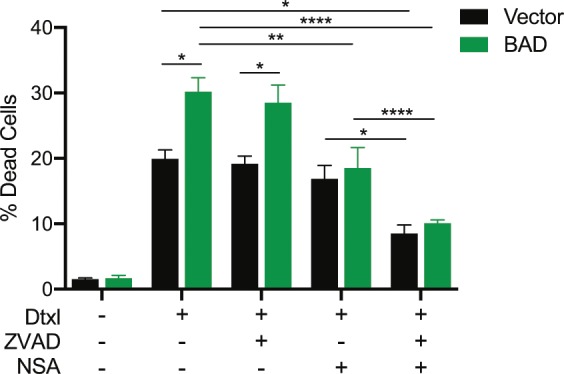
Docetaxel is killing the cells through a necroptotic mechanism. MDA-MB-231 cells stably expressing vector or BAD were treated with 125 nM docetaxel (Dtxl), 20 μM Z-VAD-FMK (ZVAD), or 5 μM necrosulfonamide (NSA) for 5 days. Cells were stained with Annexin V-647 and PI and analyzed via flow cytometry. The Annexin V+/PI+ population is represented in a bar graph. Two-way ANOVA with Tukey’s post-hoc test; n = 3.
Oxidative phosphorylation regulates docetaxel cell death
We have previously shown that BAD increases mitochondrial metabolism to promote cell survival7. AMP-activated protein kinase (AMPK), a cell energy sensor, boosts mitochondrial respiration during low energy status in mitosis and inhibits ROS-mediated necroptosis36,37. We therefore measured whether BAD expression altered oxygen consumption rate and AMPK activation upon docetaxel treatment. As expected, BAD expression increased oxygen consumption7 and this was unaffected by docetaxel treatment (Fig. 6a). Correspondingly, levels of active phosphorylated AMPK did not change in response to docetaxel. On the other hand, in vector control cells, docetaxel elevated the oxygen flux to reach similar levels to BAD-expressing cells (Fig. 6a). In line with this, AMPK activation also increased in response to docetaxel, suggesting that in vector cells, AMPK could attenuate necroptosis (Fig. 6b). Therefore, these data suggest BAD maintains adequate energy levels during mitotic arrest to circumvent AMPK activation and facilitate increased necroptosis.
Figure 6.

BAD-mediated mitotic arrest is downstream of AMPK. (a) MDA-MB-231 cells stably expressing vector or BAD were treated with 125 nM docetaxel for 48 hours prior to high-resolution respirometry. One-way ANOVA with Dunnett’s post-hoc test; n = 3. (b) Cells were treated with 125 nM docetaxel for 16, 24, or 48 hours. DMSO control treatment was for 48 hours. Right: Quantification of pAMPK-Thr172 levels over AMPK total protein were normalized to tubulin. One-way ANOVA with Dunnett’s post-hoc test; n = 3.
BAD requires reactive oxygen species for docetaxel induced cell death
BAD promotes cell survival and mitochondrial metabolism by stimulating complex I activity of the electron transport chain7, which is a main site of cellular ROS production38. Reactive oxygen species (ROS) are metabolic by-products of mitochondrial respiration and are required for maintaining redox-homeostasis of cancer cells39. Additionally, ROS has been shown to facilitate necroptosis40,41. BAD-expressing cells significantly increased ROS levels in response to docetaxel relative to vector control cells (Fig. 7a). To determine if BAD-enhanced necroptosis was dependent on ROS, we measured cell death in the presence of the ROS scavenger N-acetyl cysteine (NAC). Cell death was significantly attenuated only in docetaxel-treated BAD cells with NAC (Fig. 7b). We further examined whether ROS-dependent cell death in BAD cells was dependent on complex I. Inhibition of complex I by rotenone did not attenuate docetaxel cell death in BAD cells (Fig. 7c). Thus, these data suggest BAD confers docetaxel cell death that is dependent on ROS but does not require complex I activity. In conclusion, BAD prolongs docetaxel-mediated mitotic arrest to promote ROS-dependent necroptotic cell death (Fig. 7d).
Figure 7.

BAD requires ROS for docetaxel cell death. (a) MDA-MB-231 cells stably expressing vector or BAD were treated with 125 nM docetaxel or DMSO control for 48 hours prior to staining with CM-H2DCFDA to measure reactive oxygen species (ROS). Mean fluorescence intensity (MFI) was measured via flow cytometry. Fold increase of docetaxel/control is graphed. Student’s t-test; n = 3. (b) MDA-MB-231 cells stably expressing vector or BAD were treated with 125 nM docetaxel or 10 mM N-acetyl cysteine (NAC) for 5 days. Annexin V+/PI+ staining flow cytometry analysis of cells is graphed. Two-way ANOVA with Tukey’s post-hoc test; n = 3. (c) MDA-MB-231 cells expressing BAD were treated with 125 nM docetaxel or 50 nM rotenone for 5 days. Annexin V+/PI+ staining flow cytometry analysis of cells is graphed. Student’s t-test; n = 3. (d) Schematic representation of docetaxel-mediated cell death. BAD increases length in mitotic arrest by inhibiting cyclin B1 degradation, leading to a ROS-dependent necroptotic cell death.
Discussion
In the current study, we show that BAD sensitizes cells to docetaxel treatment in vitro and in vivo. Docetaxel treatment did not significantly trigger apoptotic cell death and instead displayed necrotic cell death morphologies. Inhibition of the necroptosis executioner, MLKL, attenuated cell death, indicating that BAD enhanced docetaxel toxicity via necroptosis. BAD enhanced death in mitosis in association with increased length in mitotic arrest and elevated cyclin B1. Thus, BAD enhances docetaxel sensitivity by facilitating longer mitotic arrest and activating necroptotic cell death in mitosis.
Our previous results reveal high BAD protein levels are associated with a 3.7-fold increased probability of overall survival of primary breast cancer patients treated with taxane5. BAD stimulates cell cycle progression leading to increased breast cancer cell number and tumor growth7. Similarly, ectopic BAD expression increased prostate cancer cell number and tumor growth42. The present data validated BAD-dependent tumor growth in a mouse model and demonstrated that this increased tumor growth is sensitive to docetaxel. Docetaxel is an anti-mitotic drug that targets actively proliferating cells by stabilizing microtubules and inducing cell death43. Thus, docetaxel may target BAD cells more effectively due to their increased proliferation.
Necrosis is an unregulated form of cell death, while necroptosis is programmed necrosis characterized by the activation of the RIP3-dependent pathway44. Necroptosis is activated in response to many anticancer drugs and contributes to their cytotoxicity45. TNF was the first documented inducer of the necroptotic pathway46. In response to TNF, overactivation of PARP1 by ROS-mediated DNA damage was shown to cause necrosis47. More recently, TNF-induced necroptosis and PARP-1-mediated necrosis have been established as two distinct pathways to programmed necrosis48. Our results showed no difference in PARP cleavage between vector and BAD cells in response to docetaxel, suggesting that BAD did not specifically stimulate PARP-mediated necroptosis. Inhibiting apoptotic caspases with zVAD-FMK alone did not inhibit docetaxel-induced cell death, similar to previous reports by others with MDA-MB-231 cells49. Therefore, apoptosis is not a major cell death pathway for docetaxel in these triple-negative breast carcinoma cells. Recently, necroptosis activation has been shown to overcome chemotherapy resistance50. Aldehyde dehydrogenase inhibitors kill ovarian cancer stem cells by activating necroptosis, in part, by the induction of mitochondrial uncoupling proteins and reduction in mitochondrial oxidative phosphorylation51. In line with this, another group showed that docetaxel-resistant prostate cancer cells induced a shift from glycolysis to oxidative phosphorylation to confer a survival advantage52. In our model system, we saw a similar increase in mitochondrial metabolism in vector cells exposed to docetaxel, suggesting this was associated with a higher resistance to cell death than BAD cells.
BAD-enhanced docetaxel-mediated cell death was dependent on ROS. Impaired oxidative phosphorylation can lead to loss of inner transmembrane potential, reduction of ATP, and production of mitochondrial ROS53. However, oxidative phosphorylation was not impaired in docetaxel-treated BAD cells. Additionally, ROS levels peak in G2 and mitosis and can cause oxidative damage54. BAD increased length in docetaxel-mediated mitotic arrest, suggesting that increased ROS were a result of extended mitosis. Chemotherapeutic agents generate ROS in cancer cells to push ROS levels over a threshold to induce cell death39. Additionally, ROS can activate necroptosis in certain cell types55. Therefore, we hypothesize BAD-mediated prolonged mitotic arrest alters redox homeostasis during taxane treatment to cause cell death.
Docetaxel has been a standard chemotherapeutic regimen both alone and in combination for many different cancers56. Although docetaxel prolongs overall and disease-free survival, a significant number of patients eventually acquire chemoresistance and succumb to their disease. Polyploidization is a key factor in resistance and relapse of docetaxel therapy, and can arise following docetaxel-mediated mitotic arrest57. Polyploid cells are large, multinucleated cells that aberrantly exited mitosis without undergoing cytokinesis, through a process known as ‘mitotic slippage’12. Polyploid cells are characterized by excessive centrosomes, with unstable chromosomes, and are highly resistant to chemotherapy13. Slipped polyploid cells can continue to cycle, or emerge from chemotherapy-induced senescence with stem cell-like features and display a more aggressive phenotype58,59. In support of this, prostate cancer cells treated with docetaxel generated a population of ‘slipped’ cells, of which a small percentage survived and gave rise to a chemoresistant and cancer stem cell positive population60. We observed that BAD promotes death in mitosis over mitotic slippage. BAD cells had a significantly longer mitotic arrest phase and underwent significantly more mitotic necroptosis. These data suggest that this longer mitotic arrest allowed the cell death signal to accrue leading to a cell fate of death over cyclin B1 degradation-mediated slippage23. Thus, in response to taxane chemotherapy, BAD high patient tumors may undergo greater cell death with less mitotic slippage, and decreased capacity to evolve chemoresistance and cancer stem-like properties, in line with the increased overall survival of these patients.
In summary, BAD increases cell death in response to docetaxel treatment. We have shown BAD expression increases length in mitotic arrest, with more death in mitosis and less mitotic slippage and polyploidy. Additionally, this cell death is necroptotic and dependent on reactive oxygen species. Understanding the mechanism of docetaxel cell death will aid in understanding BAD’s function as a prognostic biomarker in breast cancer. This may guide future studies examining whether BAD predicts patient response to taxane therapy and may suggest non-taxane chemotherapy for breast cancer patients with low BAD levels.
Materials and Methods
Cell culture and reagents
MDA-MB-231 cells were from ATCC. Cells were cultured in RPMI 1640 medium (Life Technologies) with 10% FBS as previously described5. Ectopic BAD expression and stable cell line generation was as before7. Cell lines were routinely tested for mycoplasma contamination and passaged a maximum of 25 times. Z-VAD-FMK was from R&D systems, and necrosulfonamide was from Calbiochem.
Flow cytometry analysis of cell death and cell cycle
Cells were incubated with 125 nM docetaxel (Sigma-Aldrich) or 2.5 μM staurosporine (Sigma-Aldrich) for the indicated times. Cells were harvested and washed twice with 1 X PBS prior to incubation with Annexin-V 647 (Invitrogen) in 1 X Annexin V binding buffer (1:20 dilution) for 15 minutes at room temperature in the dark, according to the manufacturer’s instructions. Cells were spun down to remove Annexin-V prior to addition of 20 μg/ml propidium iodide for 5 minutes (Life Technologies). Fluorescence was measured on the FL-4 and FL-2 channels with the BD Accuri™ C6 flow cytometer. To measure cell cycle, cells were fixed overnight in 70% ethanol and permeabilized with 0.25% Triton X-100 (Sigma-Aldrich) following addition of 10 μg/ml RNase A (Sigma-Aldrich) and 20 μg/ml PI.
Mouse studies
Animal procedures were performed in accordance with the guidelines and regulations set forth by the Canadian Council on Animal Care and approved by the University of Alberta Health Sciences 2 Animal Care and Use Committee (Protocol # AUP00000386). 3 × 106 cells were injected into the left mammary gland (#4) of nude mice (CrTac:NCr-Foxn1nu, Taconic) in 100 μl of a 1:1 matrigel/media mix. RPMI 1640 medium with no supplemental serum or antibiotic and Corning® Matrigel® Basement Membrane Matrix was used. Docetaxel (10 mg/kg) was administered via intraperitoneal injection on days as indicated. Animals were monitored weekly and sacrificed when tumors reached 20 mm in diameter. Tumors were collected and tumor volume (mm3) was calculated with the formula of (length × width × height)/2. Hematoxylin and eosin staining was performed as previously described7.
Protein analysis
Protocols for immunoblotting and co-immunoprecipitation (IP) were as previously described5. Antibodies were from the following sources: Cyclin B1, CDK1, phospho-histone H3 Ser10, pAMPK-Thr172, AMPK, Mcl-1, Bcl-2, Bcl-XL, pBcl-XL-Ser62 and cleaved PARP were from Cell Signaling Technologies. BAD, tubulin and BAX conformational antibody clone 6A7 were from Sigma-Aldrich. BAX was from Santa Cruz Biotechnology, vimentin was from Abcam, and cleaved caspase-3 was from Enzo Life Sciences.
Live-cell imaging
Cells were plated on Nunc™ Lab-Tek™ chambered coverglass prior to 125 nM docetaxel addition. A layer of mineral oil was layered over the medium to prevent evaporation. Cells were enclosed in a live-cell chamber with regulated temperature, humidity, and pH. Images were taken every 12 minutes up to 72 hours using a Zeiss AxioObserver Z1 Microscope, 20x objective lens.
Immunofluorescence
Immunofluorescence was performed as previously described7.
Reactive oxygen species
Cells were grown in 125 nM docetaxel or DMSO control for 48 hours prior to wash in Hank’s Balanced Salt Solution (ThermoFisher) followed by staining in 1.5 μM chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) (Invitrogen) for 45 minutes at 37 °C in the dark. Mean fluorescence intensity (MFI) was measured in the FL-1 channel using flow cytometry.
High-resolution respirometry
High-resolution respirometry was performed as previously described61. Briefly, respiration of intact cells (1 × 106 cells/mL) was measured in RPMI 1640 culture medium (10% fetal calf serum) under cellular routine conditions (ROUTINE). After inhibition of ATP-synthase with 2 μg/mL oligomycin, respiration declined to the resting or leak-compensating state (LEAK state). Uncoupling with stepwise titration to an optimal concentration of the protonophore carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone, FCCP, induced maximal noncoupled respiration as a measure of electron transfer system capacity (ET state).
Statistical analysis
GraphPad Prism Software was used for all statistical tests. A Student’s t-test was used for comparisons between two groups. For comparisons between greater than two groups with one variable, one-way ANOVA followed by a Dunnett’s (compared to one control group) or Tukey’s (comparing all groups to each other) multiple comparisons tests were performed. A two-way ANOVA was used when two variables were present. All data are presented as standard error of the mean (SEM). Experimental replicates are indicated and were performed at least three times. Statistical significance: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Supplementary information
Acknowledgements
We would like to thank the Women and Children’s Health Research Institute, Canadian Breast Cancer Foundation and Alberta Cancer Foundation for funding this research.
Author contributions
J.M. and I.S.G. conceived and planned the experiments. J.M. performed all experiments and wrote the manuscript with edits by I.S.G. R.M. and R.K. helped perform the mouse experiments. NY helped perform the respirometry experiment with interpretation and analysis from H.L.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-57282-1.
References
- 1.Yu K.-D., Zhu R., Zhan M., Rodriguez A. A., Yang W., Wong S., Makris A., Lehmann B. D., Chen X., Mayer I., Pietenpol J. A., Shao Z.-M., Symmans W. F., Chang J. C. Identification of Prognosis-Relevant Subgroups in Patients with Chemoresistant Triple-Negative Breast Cancer. Clinical Cancer Research. 2013;19(10):2723–2733. doi: 10.1158/1078-0432.CCR-12-2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonnefoi Hervé, Piccart Martine, Bogaerts Jan, Mauriac Louis, Fumoleau Pierre, Brain Etienne, Petit Thierry, Rouanet Philippe, Jassem Jacek, Blot Emmanuel, Zaman Khalil, Cufer Tanja, Lortholary Alain, Lidbrink Elisabet, André Sylvie, Litière Saskia, Lago Lissandra Dal, Becette Véronique, Cameron David A, Bergh Jonas, Iggo Richard. TP53 status for prediction of sensitivity to taxane versus non-taxane neoadjuvant chemotherapy in breast cancer (EORTC 10994/BIG 1-00): a randomised phase 3 trial. The Lancet Oncology. 2011;12(6):527–539. doi: 10.1016/S1470-2045(11)70094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murray S., Briasoulis E., Linardou H., Bafaloukos D., Papadimitriou C. Taxane resistance in breast cancer: Mechanisms, predictive biomarkers and circumvention strategies. Cancer Treatment Reviews. 2012;38(7):890–903. doi: 10.1016/j.ctrv.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Harris Lyndsay N., Ismaila Nofisat, McShane Lisa M., Andre Fabrice, Collyar Deborah E., Gonzalez-Angulo Ana M., Hammond Elizabeth H., Kuderer Nicole M., Liu Minetta C., Mennel Robert G., Van Poznak Catherine, Bast Robert C., Hayes Daniel F. Use of Biomarkers to Guide Decisions on Adjuvant Systemic Therapy for Women With Early-Stage Invasive Breast Cancer: American Society of Clinical Oncology Clinical Practice Guideline. Journal of Clinical Oncology. 2016;34(10):1134–1150. doi: 10.1200/JCO.2015.65.2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craik A C, Veldhoen R A, Czernick M, Buckland T W, Kyselytzia K, Ghosh S, Lai R, Damaraju S, Underhill D A, Mackey J R, Goping I S. The BH3-only protein Bad confers breast cancer taxane sensitivity through a nonapoptotic mechanism. Oncogene. 2010;29(39):5381–5391. doi: 10.1038/onc.2010.272. [DOI] [PubMed] [Google Scholar]
- 6.Janumyan YM, et al. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. Embo J. 2003;22:5459–5470. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mann Jasdeep, Githaka John Maringa, Buckland Timothy W., Yang Ning, Montpetit Rachel, Patel Namrata, Li Lei, Baksh Shairaz, Godbout Roseline, Lemieux Hélène, Goping Ing Swie. Non-canonical BAD activity regulates breast cancer cell and tumor growth via 14-3-3 binding and mitochondrial metabolism. Oncogene. 2019;38(18):3325–3339. doi: 10.1038/s41388-018-0673-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Musacchio Andrea. The Molecular Biology of Spindle Assembly Checkpoint Signaling Dynamics. Current Biology. 2015;25(20):R1002–R1018. doi: 10.1016/j.cub.2015.08.051. [DOI] [PubMed] [Google Scholar]
- 9.Gascoigne K. E., Taylor S. S. How do anti-mitotic drugs kill cancer cells? Journal of Cell Science. 2009;122(15):2579–2585. doi: 10.1242/jcs.039719. [DOI] [PubMed] [Google Scholar]
- 10.Silva Patrícia M.A., Ribeiro Nilza, Lima Raquel T., Andrade Cláudia, Diogo Vânia, Teixeira Joana, Florindo Cláudia, Tavares Álvaro, Vasconcelos M. Helena, Bousbaa Hassan. Suppression of spindly delays mitotic exit and exacerbates cell death response of cancer cells treated with low doses of paclitaxel. Cancer Letters. 2017;394:33–42. doi: 10.1016/j.canlet.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 11.Tsuda, Y. et al. Mitotic slippage and the subsequent cell fates after inhibition of Aurora B during tubulin-binding agent-induced mitotic arrest, Sci. Rep., 10.1038/s41598-017-17002-z (2017). [DOI] [PMC free article] [PubMed]
- 12.Cheng Bing, Crasta Karen. Consequences of mitotic slippage for antimicrotubule drug therapy. Endocrine-Related Cancer. 2017;24(9):T97–T106. doi: 10.1530/ERC-17-0147. [DOI] [PubMed] [Google Scholar]
- 13.Ogden Angela, Rida Padmashree C.G., Knudsen Beatrice S., Kucuk Omer, Aneja Ritu. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Letters. 2015;367(2):89–92. doi: 10.1016/j.canlet.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fulda Simone. The mechanism of necroptosis in normal and cancer cells. Cancer Biology & Therapy. 2013;14(11):999–1004. doi: 10.4161/cbt.26428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu Xinfang, Deng Qipan, Bode Ann M, Dong Zigang, Cao Ya. The role of necroptosis, an alternative form of cell death, in cancer therapy. Expert Review of Anticancer Therapy. 2013;13(7):883–893. doi: 10.1586/14737140.2013.811180. [DOI] [PubMed] [Google Scholar]
- 16.Degterev Alexei, Hitomi Junichi, Germscheid Megan, Ch'en Irene L, Korkina Olga, Teng Xin, Abbott Derek, Cuny Gregory D, Yuan Chengye, Wagner Gerhard, Hedrick Stephen M, Gerber Scott A, Lugovskoy Alexey, Yuan Junying. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nature Chemical Biology. 2008;4(5):313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho Young Sik, Park Hey Li. Exploitation of necroptosis for treatment of caspase-compromised cancers. Oncology Letters. 2017;14(2):1207–1214. doi: 10.3892/ol.2017.6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diao Yan, Ma Xiaobin, Min WeiLi, Lin Shuai, Kang HuaFeng, Dai ZhiJun, Wang Xijing, Zhao Yang. Dasatinib promotes paclitaxel-induced necroptosis in lung adenocarcinoma with phosphorylated caspase-8 by c-Src. Cancer Letters. 2016;379(1):12–23. doi: 10.1016/j.canlet.2016.05.003. [DOI] [PubMed] [Google Scholar]
- 19.Munkarah Adnan, Chuang Linus, Lotzová Eva, Cook Kenton, Morris Mitchell, Wharton J.Taylor. Comparative Studies of Taxol and Taxotere on Tumor Growth and Lymphocyte Functions. Gynecologic Oncology. 1994;55(2):211–216. doi: 10.1006/gyno.1994.1279. [DOI] [PubMed] [Google Scholar]
- 20.Rieder Conly L., Maiato Helder. Stuck in Division or Passing through. Developmental Cell. 2004;7(5):637–651. doi: 10.1016/j.devcel.2004.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Panvichian, R. et al. Paclitaxel-associated multimininucleation is permitted by the inhibition of caspase activation: A potential early step in drug resistance, Cancer Res. (1998). [PubMed]
- 22.Sinha Debottam, Duijf Pascal H.G., Khanna Kum Kum. Mitotic slippage: an old tale with a new twist. Cell Cycle. 2019;18(1):7–15. doi: 10.1080/15384101.2018.1559557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gascoigne Karen E., Taylor Stephen S. Cancer Cells Display Profound Intra- and Interline Variation following Prolonged Exposure to Antimitotic Drugs. Cancer Cell. 2008;14(2):111–122. doi: 10.1016/j.ccr.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22:R966–80. doi: 10.1016/j.cub.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 25.Haschka, M. D. et al. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest, Nat. Commun., 10.1038/ncomms7891 (2015). [DOI] [PMC free article] [PubMed]
- 26.Sakurikar Nandini, Eichhorn Joshua M., Chambers Timothy C. Cyclin-dependent Kinase-1 (Cdk1)/Cyclin B1 Dictates Cell Fate after Mitotic Arrest via Phosphoregulation of Antiapoptotic Bcl-2 Proteins. Journal of Biological Chemistry. 2012;287(46):39193–39204. doi: 10.1074/jbc.M112.391854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bah N, Maillet L, Ryan J, Dubreil S, Gautier F, Letai A, Juin P, Barillé-Nion S. Bcl-xL controls a switch between cell death modes during mitotic arrest. Cell Death & Disease. 2014;5(6):e1291–e1291. doi: 10.1038/cddis.2014.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Terrano D. T., Upreti M., Chambers T. C. Cyclin-Dependent Kinase 1-Mediated Bcl-xL/Bcl-2 Phosphorylation Acts as a Functional Link Coupling Mitotic Arrest and Apoptosis. Molecular and Cellular Biology. 2009;30(3):640–656. doi: 10.1128/MCB.00882-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang Elizabeth, Zha Jiping, Jockel Jennifer, Boise Lawrence H, Thompson Craig B, Korsmeyer Stanley J. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces bax and promotes cell death. Cell. 1995;80(2):285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 30.Adachi M, Imai K. The proapoptotic BH3-only protein BAD transduces cell death signals independently of its interaction with Bcl-2. Cell Death & Differentiation. 2002;9(11):1240–1247. doi: 10.1038/sj.cdd.4401097. [DOI] [PubMed] [Google Scholar]
- 31.Bertrand Richard, Solary Eric, O'Connor Patrick, Kohn Kurt W., Pommier Yves. Induction of a Common Pathway of Apoptosis by Staurosporine. Experimental Cell Research. 1994;211(2):314–321. doi: 10.1006/excr.1994.1093. [DOI] [PubMed] [Google Scholar]
- 32.Galluzzi Lorenzo, Kroemer Guido. Necroptosis: A Specialized Pathway of Programmed Necrosis. Cell. 2008;135(7):1161–1163. doi: 10.1016/j.cell.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 33.Sun Liming, Wang Huayi, Wang Zhigao, He Sudan, Chen She, Liao Daohong, Wang Lai, Yan Jiacong, Liu Weilong, Lei Xiaoguang, Wang Xiaodong. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell. 2012;148(1-2):213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 34.De Iuliis Francesca, Salerno Gerardo, Giuffrida Anna, Milana Bernardina, Taglieri Ludovica, Rubinacci Giovanna, Giantulli Sabrina, Terella Federica, Silvestri Ida, Scarpa Susanna. Breast cancer cells respond differently to docetaxel depending on their phenotype and on survivin upregulation. Tumor Biology. 2015;37(2):2603–2611. doi: 10.1007/s13277-015-4075-x. [DOI] [PubMed] [Google Scholar]
- 35.Liao Daohong, Sun Liming, Liu Weilong, He Sudan, Wang Xiaodong, Lei Xiaoguang. Necrosulfonamide inhibits necroptosis by selectively targeting the mixed lineage kinase domain-like protein. MedChemComm. 2014;5(3):333. doi: 10.1039/c3md00278k. [DOI] [Google Scholar]
- 36.Zhao Haixin, Li Teng, Wang Kai, Zhao Fei, Chen Jiayi, Xu Guang, Zhao Jie, Li Ting, Chen Liang, Li Lin, Xia Qing, Zhou Tao, Li Hui-Yan, Li Ai-Ling, Finkel Toren, Zhang Xue-Min, Pan Xin. AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nature Cell Biology. 2019;21(4):476–486. doi: 10.1038/s41556-019-0296-3. [DOI] [PubMed] [Google Scholar]
- 37.Wang Yi-Shu, Yu Peng, Wang Yong, Zhang Jing, Hang Wei, Yin Zhi-Xian, Liu Gang, Chen Jianfeng, Werle Kaitlin D., Quan Cheng-Shi, Gao Hang, Zeng Qinghua, Cui Rutao, Liang Jiyong, Ding Qiang, Li Yu-Lin, Xu Zhi-Xiang. AMP-activated protein kinase protects against necroptosis via regulation of Keap1-PGAM5 complex. International Journal of Cardiology. 2018;259:153–162. doi: 10.1016/j.ijcard.2018.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orr Adam L., Ashok Deepthi, Sarantos Melissa R., Shi Tong, Hughes Robert E., Brand Martin D. Inhibitors of ROS production by the ubiquinone-binding site of mitochondrial complex I identified by chemical screening. Free Radical Biology and Medicine. 2013;65:1047–1059. doi: 10.1016/j.freeradbiomed.2013.08.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dharmaraja Allimuthu T. Role of Reactive Oxygen Species (ROS) in Therapeutics and Drug Resistance in Cancer and Bacteria. Journal of Medicinal Chemistry. 2017;60(8):3221–3240. doi: 10.1021/acs.jmedchem.6b01243. [DOI] [PubMed] [Google Scholar]
- 40.Kim You-Sun, Morgan Michael J., Choksi Swati, Liu Zheng-gang. TNF-Induced Activation of the Nox1 NADPH Oxidase and Its Role in the Induction of Necrotic Cell Death. Molecular Cell. 2007;26(5):675–687. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 41.Zhang D.-W., Shao J., Lin J., Zhang N., Lu B.-J., Lin S.-C., Dong M.-Q., Han J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science. 2009;325(5938):332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 42.Smith Adrienne J., Karpova Yelena, D'Agostino Ralph, Willingham Mark, Kulik George. Expression of the Bcl-2 Protein BAD Promotes Prostate Cancer Growth. PLoS ONE. 2009;4(7):e6224. doi: 10.1371/journal.pone.0006224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia Patrick, Braguer Diane, Carles Gérard, Khyari Said El, Barra Yves, de Ines Concepcion, Barasoain Isabel, Briand Claudette. Comparative effects of taxol and Taxotere on two different human carcinoma cell lines. Cancer Chemotherapy and Pharmacology. 1994;34(4):335–343. doi: 10.1007/BF00686042. [DOI] [PubMed] [Google Scholar]
- 44.Wu Wei, Liu Peng, Li Jianyong. Necroptosis: An emerging form of programmed cell death. Critical Reviews in Oncology/Hematology. 2012;82(3):249–258. doi: 10.1016/j.critrevonc.2011.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Su Z, Yang Z, Xie L, DeWitt J P, Chen Y. Cancer therapy in the necroptosis era. Cell Death & Differentiation. 2016;23(5):748–756. doi: 10.1038/cdd.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Degterev Alexei, Huang Zhihong, Boyce Michael, Li Yaqiao, Jagtap Prakash, Mizushima Noboru, Cuny Gregory D, Mitchison Timothy J, Moskowitz Michael A, Yuan Junying. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nature Chemical Biology. 2005;1(2):112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 47.Los Marek, Mozoluk Malgorzata, Ferrari Davide, Stepczynska Anna, Stroh Christopher, Renz Andrea, Herceg Zdenko, Wang Zhao-Qi, Schulze-Osthoff Klaus. Activation and Caspase-mediated Inhibition of PARP: A Molecular Switch between Fibroblast Necrosis and Apoptosis in Death Receptor Signaling. Molecular Biology of the Cell. 2002;13(3):978–988. doi: 10.1091/mbc.01-05-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sosna Justyna, Voigt Susann, Mathieu Sabine, Lange Arne, Thon Lutz, Davarnia Parvin, Herdegen Thomas, Linkermann Andreas, Rittger Andrea, Chan Francis Ka-Ming, Kabelitz Dieter, Schütze Stefan, Adam Dieter. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cellular and Molecular Life Sciences. 2013;71(2):331–348. doi: 10.1007/s00018-013-1381-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hwang Eunjoo, Hwang Seong-Hye, Kim Jongjin, Park Jin Hyun, Oh Sohee, Kim Young A, Hwang Ki-Tae. ABT-737 ameliorates docetaxel resistance in triple negative breast cancer cell line. Annals of Surgical Treatment and Research. 2018;95(5):240. doi: 10.4174/astr.2018.95.5.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang, X. et al. Bypassing drug resistance by triggering necroptosis: Recent advances in mechanisms and its therapeutic exploitation in leukemia, J. Exp. Clin. Cancer Res., 10.1186/s13046-018-0976-z (2018). [DOI] [PMC free article] [PubMed]
- 51.Chefetz Ilana, Grimley Edward, Yang Kun, Hong Linda, Vinogradova Ekaterina V., Suciu Radu, Kovalenko Ilya, Karnak David, Morgan Cynthia A., Chtcherbinine Mikhail, Buchman Cameron, Huddle Brandt, Barraza Scott, Morgan Meredith, Bernstein Kara A., Yoon Euisik, Lombard David B., Bild Andrea, Mehta Geeta, Romero Iris, Chiang Chun-Yi, Landen Charles, Cravatt Benjamin, Hurley Thomas D., Larsen Scott D., Buckanovich Ronald J. A Pan-ALDH1A Inhibitor Induces Necroptosis in Ovarian Cancer Stem-like Cells. Cell Reports. 2019;26(11):3061-3075.e6. doi: 10.1016/j.celrep.2019.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ippolito, L. et al. Metabolic shift toward oxidative phosphorylation in docetaxel resistant prostate cancer cells, Oncotarget., 10.18632/oncotarget.11301 (2016). [DOI] [PMC free article] [PubMed]
- 53.Koo Michael Jakun, Rooney Kristen T., Choi Mary E., Ryter Stefan W., Choi Augustine M.K., Moon Jong-Seok. Impaired oxidative phosphorylation regulates necroptosis in human lung epithelial cells. Biochemical and Biophysical Research Communications. 2015;464(3):875–880. doi: 10.1016/j.bbrc.2015.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patterson Jesse C., Joughin Brian A., van de Kooij Bert, Lim Daniel C., Lauffenburger Douglas A., Yaffe Michael B. ROS and Oxidative Stress Are Elevated in Mitosis during Asynchronous Cell Cycle Progression and Are Exacerbated by Mitotic Arrest. Cell Systems. 2019;8(2):163-167.e2. doi: 10.1016/j.cels.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, Y. et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome, Nat. Commun., 10.1038/ncomms14329 (2017). [DOI] [PMC free article] [PubMed]
- 56.Kang Byung, Kwon Oh-Kyoung, Chung Ho, Yu Wansik, Kim Jong. Taxanes in the Treatment of Advanced Gastric Cancer. Molecules. 2016;21(5):651. doi: 10.3390/molecules21050651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin Ke-Chih, Torga Gonzalo, Sun Yusha, Axelrod Robert, Pienta Kenneth J., Sturm James C., Austin Robert H. The role of heterogeneous environment and docetaxel gradient in the emergence of polyploid, mesenchymal and resistant prostate cancer cells. Clinical & Experimental Metastasis. 2019;36(2):97–108. doi: 10.1007/s10585-019-09958-1. [DOI] [PubMed] [Google Scholar]
- 58.Wang Qin, Wu Peter C., Dong David Z., Ivanova Iana, Chu Elizabeth, Zeliadt Steven, Vesselle Hubert, Wu Daniel Y. Polyploidy road to therapy-induced cellular senescence and escape. International Journal of Cancer. 2012;132(7):1505–1515. doi: 10.1002/ijc.27810. [DOI] [PubMed] [Google Scholar]
- 59.Achuthan Santhi, Santhoshkumar Thankayyan R., Prabhakar Jem, Nair S. Asha, Pillai M. Radhakrishna. Drug-induced Senescence Generates Chemoresistant Stemlike Cells with Low Reactive Oxygen Species. Journal of Biological Chemistry. 2011;286(43):37813–37829. doi: 10.1074/jbc.M110.200675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mittal, K. et al. Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer, Br. J. Cancer., 10.1038/bjc.2017.78 (2017). [DOI] [PMC free article] [PubMed]
- 61.Yang Ning, Weinfeld Michael, Lemieux Hélène, Montpetit Ben, Goping Ing Swie. Photo-activation of the delocalized lipophilic cation D112 potentiates cancer selective ROS production and apoptosis. Cell Death & Disease. 2017;8(2):e2587–e2587. doi: 10.1038/cddis.2017.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.