

Keywords: clostridium, metformin, microbiome, obesity, pancreatic ductal adenocarcinoma, type 2 diabetes mellitus
Abstract
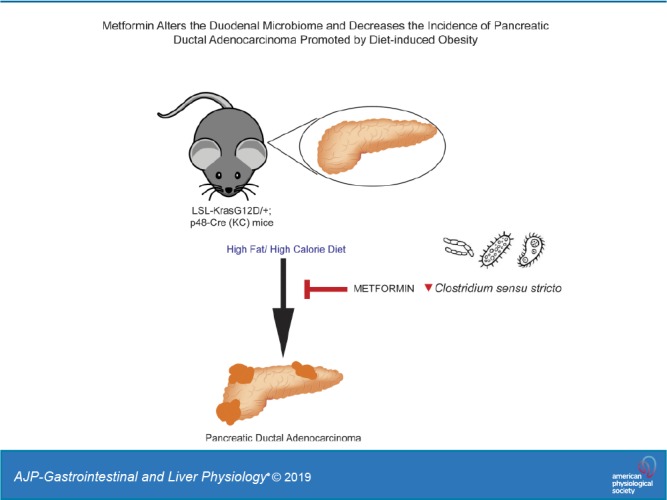
Pancreatic ductal adenocarcinoma (PDAC)’s growing incidence has been linked to the rise in obesity and type 2 diabetes mellitus. In previous work, we have shown that metformin can prevent the increased incidence of PDAC in a KrasG12D mouse model subjected to a diet high in fat and calories (HFCD). One potential way that metformin can affect the host is through alterations in the gut microbiome. Therefore, we investigated microbial associations with PDAC development and metformin use in the same mouse model. Lox-Stop-Lox Kras G12D/+ (LSL-Kras G12D/+); p48-Cre (KC) mice were given control diet, HFCD, or HFCD with 5 mg/mL metformin in drinking water for 3 mo. At the end of the 3 mo, 16S rRNA sequencing was performed to characterize microbiome composition of duodenal mucosal, duodenal luminal, and cecal luminal samples. KC mice on an HFCD demonstrated depletion of intact acini and formation of advanced pancreatic intraepithelial neoplasia. This effect was completely abrogated by metformin treatment. HFCD was associated with significant changes in microbial composition and diversity in the duodenal mucosa and lumen, much of which was prevented by metformin. In particular, Clostridium sensu stricto was negatively correlated with percent intact acini and seemed to be inhibited by the addition of metformin while on an HFCD. Administration of metformin eliminated PDAC formation in KC mice. This change was associated with significant microbial changes in both the mucosal and luminal microbiome of the duodenum. This suggests that the microbiome may be a potential mediator of the chemopreventive effects of metformin.
NEW & NOTEWORTHY Pancreatic ductal adenocarcinoma (PDAC)’s growing incidence has been linked to the rise in obesity and type 2 diabetes mellitus. Administration of metformin eliminated PDAC formation in KC mice with diet-induced obesity. This change was associated with significant microbial changes in both the mucosal and luminal microbiome of the duodenum. This suggests that the microbiome may be a potential mediator of the chemopreventive effects of metformin.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is currently the third leading cause of cancer-related deaths in the United States. In 2019, 56,770 new cases are expected in the United States (1). With one of the lowest overall 5-yr survival rates of cancers at 8%, PDAC is an extremely aggressive and fatal cancer. Unfortunately, because of the lack of early diagnostic tools and therapies, by 2030, it is estimated that PDAC will become the second leading cause of cancer-related mortality (41). Therefore, there is a pressing need to increase research efforts to identify novel targets and potential preventative measures.
Studies have demonstrated a link between PDAC risk and chronic conditions, including obesity and type 2 diabetes mellitus (T2DM) (5, 13, 24). Current research on animal models of PDAC showed that diets high in fat and obesity have tumor-promoting effects (12, 14, 57). Today, as the prevalence of obesity, metabolic syndromes, and T2DM increases, mechanistic insight into the relationship between obesity-related conditions and PDAC is of considerable interest to identify potential biomarkers and develop novel treatment options.
Metformin (1,1-dimethylbiguanide hydrochloride) is one of the most commonly prescribed medications for T2DM. Systemically, it lowers the level of circulating glucose by reducing liver gluconeogenesis and improving the sensitivity of peripheral tissues to insulin, thereby increasing glucose disposal in skeletal muscle and adipose tissue. Patients with diabetes who have been prescribed metformin have displayed a reduction in incidence recurrence and mortality of cancer, although a therapeutic effect has not been shown universally (17, 26, 29, 51, 55). A meta-analysis suggested that the cancer-reducing effects of metformin depended on tumor stage, with survival benefits in patients with local PDAC (31, 54, 59). Overall, current epidemiological evidence points to a potentially beneficial role of metformin in the preventative/interceptive rather than therapeutic setting in PDAC, though these studies have limitations (5a).
The mechanisms of the cancer-reducing effects of metformin are still not completely understood. Previously, we demonstrated that metformin inhibited PDAC growth in vitro and disrupted crosstalk between G protein-coupled receptor and insulin/IGF receptor signaling pathways (27). Metformin inhibited signaling via the mammalian target of rapamycin 1 and extracellular signal-regulated kinase (ERK) pathways in PDAC cells, with metformin-induced AMP-activated protein kinase activation amplified in PDAC cells in the presence of physiological levels of glucose (34, 45, 47). Metformin also inhibited the growth of PDAC cells in vivo when administered to nude mice with subcutaneous or orthotopic xenografts (28).
Over the last 10 years, a rapidly growing body of evidence in animal models and human studies has implicated the gut microbiome in obesity and obesity-related conditions (16, 33, 52). Reported mechanisms by which microbes can cause host metabolic changes include shifts in energy extraction, changes in short-chain fatty acid production, alterations of gut hormones, and changes in Toll-like receptor signaling (38). Importantly, several studies have found that metformin affects the gut microbiome, potentially contributing to its antiobesity effect. In animal models, metformin has been reported to induce Akkermansia, which by itself attenuated metabolic phenotypes in mice fed a high-fat diet (44). A second study found that metformin shifted the proximal small intestinal microbiome in such a way as to confer increased small intestinal glucose sensing by sodium-glucose transport protein 1 that was transferrable via the microbiome (3). In humans, both a large observational study of 784 individuals and a randomized controlled study of 40 individuals have identified a robust microbiome signature of metformin in feces (18, 56). In the latter study, Wu et al. (56) further demonstrated that the beneficial effects of metformin can be transferred to germ-free mice through fecal transplantation, providing causal evidence that the alteration of the gut microbiome mediates some of metformin’s antidiabetic effects.
We hypothesized that modulation of the intestinal microbiome by metformin could contribute to reduced PDAC incidence. To investigate the interplay between obesity, metformin, PDAC, and the microbiome, we employed an animal model we previously described in which PDAC development and progression was promoted by diet-induced obesity (DIO) (12). Mice expressing oncogenic Kras in the pancreas were exposed to a diet containing high amounts of fats and calories (HFCD). The HFCD-fed mice gained more weight and had metabolic disturbances such as hyperglycemia, hyperinsulinemia, and hyperleptinemia in comparison with lean control diet mice. Obese KC mice developed pancreatic inflammation and displayed increased PDAC incidence. Using the same animal model, we further showed that oral administration of metformin attenuated or even prevented metabolic disturbances and the DIO-associated increase in PDAC incidence, suggesting that metformin could be a preventative measure for obesity-associated PDAC (11). The aim of the current study was to characterize the effects of metformin on the proximal small intestinal and colonic microbiome of mice with DIO-associated PDAC and examine how these microbial differences correlated with disease outcomes.
METHODS
Experimental animals.
Offspring of LSL-KrasG12D/+ and p48-Cre+/− (KC) mice (21) were randomly allocated to a control diet (CD), HFCD, or HFCD with metformin in the drinking water (5 mg/mL) at 1 mo of age. There were at least eight mice in each group, with an equal number of female and male mice. All mice had free access to the diet. Mice fed the HFCD had free access to either regular drinking water or drinking water (reverse osmosis) supplemented with metformin (5 mg/mL). Water supplemented with metformin was made fresh and replenished twice weekly. Body weights were measured weekly, and the general health and behavior of animals were assessed daily. At 3 mo of age, cohorts of mice (male and female) were euthanized and tissues harvested. All studies involving animals were reviewed and approved by the Chancellor’s Animal Research Committee of the University of California, Los Angeles, in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (protocol number: 2011–118).
Genotyping analysis.
LSL-KRASG12D and Cre alleles were detected before randomizing to the experimental diets by PCR analysis of genomic DNA, as described (4). Mutant (KC) mice expressed both LSL-KRASG12D and Cre alleles and animals carrying neither allele were labeled as wild type.
Experimental diets.
Diets were prepared by Dyets (Bethlehem, PA). At 1 mo of age, mice were randomized to receive either the CD, HFCD, or HFCD with 5 mg/mL metformin (Sigma-Aldrich, St. Louis, MO) in the drinking water. A detailed composition of the diets was described previously (14). The CD contained 12% calories from fat, whereas the HFCD contained 40% of calories from fat (corn oil based). All diets were stored at −20°C (long-term) or 4°C (short-term) in sealed containers and prepared under low-light conditions. The diets were replaced on a weekly basis, and the metformin-containing water was refreshed twice a week. The concentration of metformin was determined to be a biologically effective dose as previously published (11).
Pancreas histology.
Hematoxylin and eosin-stained tissue sections of the pancreas, fixed in formalin and embedded in paraffin, were assessed by gastrointestinal pathologists blinded to the experimental groups. The presence and stage of murine pancreatic intraepithelial neoplasia (PanINs) and PDACs were analyzed according to previously published criteria (22, 23). For each animal, ~100 pancreatic ducts (tail of the pancreas) were quantified, and the proportion of murine PanIN-3 to the overall number of pancreatic ducts was recorded.
Microbiome characterization: 16S ribosomal RNA sequencing.
Because we postulate that local microbiome shifts would exert a greater effect on PDAC development, we examined the luminal and mucosal microbiome of the duodenum as well as the tissue-associated microbiome of the pancreas. We also examined the ileal and cecal luminal microbiome as a comparison to assess whether any microbiome changes with metformin were conserved across the gastrointestinal tract. Duodenal, ileal, and cecal luminal content were released by flushing with distilled deionized water, and then the mucosa-associated bacteria in the duodenum were released by DTT treatment according to our published protocol (25). Genomic DNA was extracted from samples using the Qiagen Powersoil kit as per the manufacturer’s instructions. The V4 region of 16S ribosomal RNA genes was amplified and underwent paired end sequencing on an Illumina MiSeq as previously described (50). The 253 base pair reads were processed to amplicon sequence variants using the DADA2 package in R (version 3.5.2) as previously described (7). A phylogenetic tree was constructed using the DECIPHER package in R for multiple alignments and the phangorn package in R for phylogenetic construction, as previously published (8). Amplicon sequence variants were removed if they were present in <10% of samples. α-Diversity (i.e., diversity within a sample) and β-diversity (differences in composition across samples) were calculated in QIIME (9) using genus-level data rarefied to the sample with the lowest sequence at 63,697 sequences [mean: 88,144 ± 9,162 (SD)].
Quantitative 16S PCR.
The total number of copies of the bacterial 16S rRNA gene was determined by a TaqMan-based quantitative PCR (qPCR) approach. Previously published universal primers designed to detect total bacterial 16S rRNA were used (35). The primer set included the forward primer, 5′-TCCTACGGGAGGCAGCAGT-3′ (T m, 59.4°C), the reverse primer, 5′-GGACTACCAGGGTATCTAATCCTGTT-3′ (T m, 58.1°C), and the probe, (6-carboxyfluorescein)-5′-CGTATTACCGCGGCTGCTGGCAC-3′-(carboxytetramethylrhodamine) (T m, 69.9°C). Using 5 μL of an iTaq Universal Probes Supermix (Bio-Rad), 0.2 μM of forward and reverse primers, 0.25 µM of probe, 1 μL of sample DNA, and PCR-grade water, a total volume of 10 μL was used for each qPCR reaction. All samples were run in triplicates, and a stock Escherichia coli sample of a known quantity (1.7 × 1010 colony-forming units) was used to create a standard dilution curve. The PCR reaction was performed using a Bio-Rad CFX384 Touch Real-Time PCR Detection System (Bio-Rad). The reaction conditions for amplification of DNA were 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 15 s and 60°C for 1 min. A linear regression model was fitted to threshold cycle values of the dilution curve and then used to determine the number of copies of the 16S rRNA gene present in each sample. Data are presented as the number of copies of the 16S rRNA gene per gram of sample before DNA extraction.
Statistical analyses.
Microbiome data were analyzed for alpha diversity, beta diversity, and association of taxa abundance with treatment group. Alpha diversity refers to metrics of diversity within a community (i.e., mouse sample), which pertain to the total number of species (richness), the total length along a phylogenetic tree covered by a community (phylogenetic diversity), or how evenly distributed the members of a community are among the species present (evenness) (32). For this study, we employed Faith’s phylogenetic diversity, Chao1 (a metric of richness), and Shannon index (a combined metric of richness/evenness). The significance of differences in alpha diversity was calculated by a two-tailed t test. Beta diversity refers to comparison of microbial composition across communities (i.e., mouse samples) based upon which species are present/absent or their relative abundances (20). In this study, beta diversity was calculated using the distance matrix based on the square root of the Jensen-Shannon divergence and visualized by principal coordinates analysis (39). Adonis, a permutational analysis of variance, was performed using 100,000 permutations to test for differences in distances across the treatment groups controlling for sex (2). Bacterial absolute abundances were calculated by multiplying relative abundance data from 16S rRNA sequencing with total number of 16S copies determined by qPCR. Differential absolute abundance was determined using the package limma in R, which employs an empirical Bayesian linear model, after removing low prevalent genera as defined as having a relative abundance of <0.1% (46). P values for differential abundance were converted to q values to correct for multiple hypothesis testing (<0.05 for significance) (49). Adonis and limma models included sex as a covariate.
RESULTS
Beta diversity and alpha diversity.
KC mice were randomly allocated to a CD, HFCD, or HFCD with metformin in the drinking water (5 mg/mL) at 1 mo of age. Overall, there was a significant difference in microbial composition across the three groups. The microbiome of mice on an HFCD (obese KC mice) are statistically different (P value < 0.0001) from either those that were on a control diet or on metformin (Fig. 1). This difference was consistent across the duodenum (lumen and mucosa) and the cecum. There was also a statistical difference between the groups in the ileum (Fig. 1D), but there was no difference in overall bacterial composition in the pancreas (Supplemental Fig. 1A; https://doi.org/10.6084/m9.figshare.8298155.v2).
Fig. 1.

Microbiome composition throughout the gastrointestinal tract is altered by a high-fat, high-calorie diet (HFCD) and metformin. Overall microbial composition is represented in principal coordinate analysis plots derived from the square root of the Jensen-Shannon divergence distances. Each symbol represents one mouse, with the color representing the treatment groups across various sites: duodenum (lumen) (A), duodenum (mucosa) (B), ileum (lumen) (C), and cecum (lumen) (D). P value for differences across groups is shown adjusting for sex. CD, control diet; PC, principal coordinate.
In the luminal duodenum, the HFCD caused a significant decrease in all α-diversity metrics (P value < 0.05), including phylogenetic diversity, Chao1 index (a metric for species richness), and Shannon index (a metric reflecting species evenness and richness) (Fig. 2). The addition of metformin tended to increase these metrics as compared with HFCD alone, but it did not reach significance. There was no difference in alpha diversity metrics between the control diet and HFCD with addition of metformin. A similar trend was seen in the cecum. The HFCD trended toward a decrease in all α-diversity metrics, but none reached statistical significance. However, in the cecum, mice on the HFCD with the addition of metformin had statistically lower phylogenetic diversity, Chao1, and Shannon index as compared with control (P value < 0.05). There was no statistically significant difference in any α-diversity metrics in the mucosa-associated microbiome of the duodenum, ileum, or pancreas across the different groups.
Fig. 2.

α-Diversity metrics are altered by a high-fat, high-calorie diet (HFCD) and metformin. Microbial diversity was assessed by Chao1 index (richness), Faith’s phylogenetic diversity, and Shannon index (evenness/richness) across different regions of the gastrointestinal tract. *Significance between control diet (CD) and HFCD; #significance between CD and HFCD + metformin.
Total bacterial quantity.
Bacterial load did not differ by the various groups across the different regions of the gastrointestinal tract except for in the cecum (Fig. 3). The duodenal mucosa-associated microbiome had the lowest bacterial quantity overall, and the cecum had the highest bacterial quantity. In the cecum, the addition of metformin significantly reduced the bacterial load as compared with a CD or HFCD alone. There was no difference in bacterial load in the cecum between CD and HFCD.
Fig. 3.

Overall bacterial quantity by different regions and different treatment groups: duodenum-mucosa (A), duodenum-lumen (B), ileum (C), and cecum (D). Overall bacterial load as determined by quantitative PCR of the different regions of the gastrointestinal tract. #Significance between control diet (CD) and high-fat, high-calorie diet (HFCD) with metformin; +significance between HFCD and HFCD with metformin.
Taxonomic composition between different treatments.
Microbial composition differed greatly across sites along the gastrointestinal tract as well as among the different treatments (Fig. 4). On a phylum level, the mucosa-associated microbiome of the duodenum had a higher relative abundance of Bacteroidetes and a lower relative abundance of Firmicutes than the luminal microbiome of the duodenum. In the lumen of the duodenum, the HFCD decreased the genera Dialister, Rikenellaceae RC9 gut group, and Acetatifactor while increasing Clostridium sensu stricto (Figs. 4D and 5B). The addition of metformin to the HFCD was associated with reversal of some of these differences, including, most strikingly, a reduction in Clostridium sensu stricto to levels lower than mice fed on a control diet. The addition of metformin to the HFCD also increased the abundance of Akkermansia compared with both the HFCD alone and control diet, though it did not reach statistical significance. The mucosa-associated microbiome of the duodenum had less drastic changes compared with the lumen, but some of the same trends held true for the mucosa-associated microbiome as it did for the lumen (Fig. 4, A and B). Similar to the lumen of the duodenum, the mucosa-associated microbiome of the duodenum showed a trend of increasing Clostridium sensu stricto on the HFCD, which was prevented by the addition of metformin. Metformin also increased the abundance of Akkermansia, similarly to its effects on the microbiome of the duodenal lumen. The changes seen in the lumen of the duodenum were similar to the changes seen in the lumen of the ileum (Supplemental Fig. 1, B and C). Similar to the findings in the duodenum, the HFCD increased the abundance of Muribaculum. There were no significant differences seen in the pancreas. The major genus present in the pancreas was Prevotella, and this genus did not differ among treatment groups (Supplemental Fig. 2).
Fig. 4.

Taxonomic composition varies by site and is altered by a high-fat, high-calorie diet (HFCD) and metformin. Taxonomic composition by phylum and genus of the duodenum (mucosa) (A and B), duodenum (lumen) (C and D), and cecum (lumen) (E and F). Each color within the bar graph represents a phylum or genus, with the area of the color proportional to absolute abundance. Only phyla or genera with >1% relative abundance are shown. *Genera that were differentially different between control diet (CD) and HFCD; #genera that were differentially different between HFCD and HFCD + metformin; +genera differences between CD and HFCD + metformin.
Fig. 5.

DESeq2 analysis showing genera that are differentially abundant between groups in the duodenal mucosa and lumen. Log2 fold changes (Log2FC) are shown for genera with differential abundance between different diets and treatment for mucosal duodenal samples (A) and luminal duodenal samples (B–D) in limma models at q value < 0.05. Dot size is proportional to the average absolute abundance of the genus, and color corresponds to phylum. CD, control diet; HFCD, high-fat, high-calorie diet.
To better understand the intestinal microbiome differences that are occurring regionally near the pancreas, differential abundance testing was performed at the genus level to determine the statistical difference between the treatment arms on the luminal microbiome (Fig. 5, B–D) and the mucosa-associated microbiome of the duodenum (Fig. 5A). The HFCD compared with control diet altered four genera in the lumen of the duodenum. They were Rikenellaceae (78-fold decrease), Acetatifactor (58-fold decrease), Dialister (47-fold decrease), and Clostridium sensu stricto (39-fold increase). The addition of metformin as compared with the HFCD alone altered three different genera. They were Romboutsia, Turicibacter, and Clostridium sensu stricto. This finding was also similar when comparing the HFCD with metformin to a control diet. The luminal changes of Clostridium sensu stricto and Romboustia in the duodenum continued through to the lumen of the ileum (Supplemental Fig. 1, D–F).
In the mucosa-associated microbiome of the duodenum, only one genus was differentially abundant between the various groups. Romboutsia was significantly decreased in the HFCD group with metformin as compared with the CD group. However, the trend of Clostridium sensu stricto was similar to those seen in the lumen of the duodenum, although it did not reach statistical significance.
Microbial shifts are linearly related to PDAC development.
Similar to our previous published work, we show that the HFCD in KC mice was able to promote PanIN development and progression. Figure 6A shows representative histological changes seen in the control mice, mice fed the HFCD, and mice fed the HFCD with metformin. The percentage of intact acini was greatly reduced in the HFCD group and returned to control levels with the addition of metformin (Fig. 6B). This reduction in intact acini was associated with a respective increase in the percent of pancreatic intraepithelial neoplasia 3 (PanIN3) (Fig. 6C). Through linear regression, we show that the Clostridium sensu stricto count in the lumen of the duodenum negatively correlated with percentage intact acini (P value = 0.001), suggesting a positive correlation to PDAC development. No other genera in the duodenum were as strongly associated with PDAC development. Contrastingly, no genera in the ileum or cecum were linearly associated with PDAC development.
Fig. 6.

Metformin decreases pancreatic ductal adenocarcinoma (PDAC) incidence induced by a high-fat, high-calorie diet (HFCD) in KC mice by altering the expression of Clostridium sensu stricto. A: representative hematoxylin and eosin staining of the pancreas of KC mice fed on a control diet (left), HFCD (middle), or HFCD with metformin (right). B: percentage (%) of intact acini in KC mice by treatment groups. C: percentage (%) of pancreatic intraepithelial neoplasia (PanIN)-3 lesions in KC mice by treatment group. D: linear regression of Clostridium sensu stricto from luminal duodenal samples by percent intact acini. *P < 0.05. CD, control diet.
DISCUSSION
Currently, the high death rate associated with PDAC along with the very limited treatment options available underscore the need for novel strategies to treat and prevent this lethal disease. Given the rise in obesity and its connection to the microbiome and cancer, understanding the interplay between obesity and the microbiome in PDAC development could lead to novel strategies for the prevention/treatment of PDAC. Potentially relevant microbial communities include the duodenum (in immediate proximity to the pancreas), more distal regions of the gut (acting through systemic effects on immunity and metabolism), and the pancreas itself. The normal pancreas was previously considered not to have a microbiome of its own. However, the intestinal flora can migrate into the pancreas and potentially influence the microenvironment (40). In the present study, we show how orally administered metformin significantly alters the regional microbiome of the duodenum and inhibits the development of PanIN lesions in a diet-induced obesity model of pancreatic cancer.
In our previous study, we showed that the HFCD led to a significant increase in weight gain that was also associated with a rise in circulating insulin and leptin, similar to many patients who are overweight or obese and have some degree of insulin and leptin resistance (11). In this study, we show that an HFCD can increase the percentage of advanced PanIN lesions, recognized precursors to PDAC, in the KC mouse model of pancreatic cancer and that this change was associated with a significant shift in the microbial composition and diversity of the duodenum. The oral administration of metformin was able to prevent the changes seen in the pancreas as well as in the microbiome of the duodenum. Specifically, within the duodenum, a reduction in alpha diversity was shown in the HFCD group as compared with the control group based on a decrease in species richness, species diversity, and species evenness. The reduction in alpha diversity is a sign of dysbiosis or microbial imbalance, which has been associated with the progression of various cancers (10). Therefore, the increase in α-diversity shown in the group with metformin suggests that metformin may play a role in resolving the microbial dysbiosis in the duodenum associated with the HFCD. However, the analysis of the mucosa-associated microbiome showed no significant changes in α-diversity, suggesting that the HFCD may not play as critical a role in the diversity of the mucosal-associated microbiome. Similar to the findings in the duodenum, an HFCD also decreases the α-diversity of the cecum. But unlike the duodenum, the addition of metformin further decreased the alpha diversity as compared with both the control and HFCD group. This is consistent with previously published works that showed fecal or colonic α-diversity decreasing with the addition of metformin (30). This difference highlights the possibility that metformin may have varying effects on the microbiome based on location.
When analyzing the taxonomic profiles of the three groups, there were distinct differences between the control diet, HFCD, and HFCD with metformin. In small case studies, patients with PDAC were found to have altered fecal and pancreatic tissue microbial signatures by 16S rRNA sequencing compared with healthy controls, including increased Proteobacteria and Actinobacteria (19, 40). Our study showed similar findings in the HFCD group at the site of the duodenum. Specifically, within the duodenum at a phylum level, an HFCD led to an increase in Actinobacteria. At the genus level, Clostridium sensu stricto increased dramatically in the HFCD group, and the addition of metformin was able to prevent this change in the duodenal microbiome. These data corroborate a recent publication by Pushalkar et al. (40). They showed that KC germ-free mice were protected against PDAC development and that the introduction of microbes from patients with PDAC, which are abundant in Actinobacteria and Bifidobacterium, accelerated cancer development in a Toll-like receptor-dependent manner (40).
Finally, our study reveals that Clostridium sensu stricto is potentially related to pancreatic cancer. Clostridium sensu stricto encompasses a subset of closely genetically related species within Clostridium, including pathogens such as Clostridium tetani, Clostridium botulinum, and Clostridium perfringens, and has previously been associated with necrotizing enterocolitis (6, 42, 53). The HFCD induced the growth of Clostridium sensu stricto in the duodenum, whereas the addition of metformin inhibited it. This bacterium was negatively correlated to the percentage of intact acini in the pancreas. Although no previous research has linked these bacteria to pancreatic cancer, Clostridium has been linked to liver cancer. A previous study proposed that Clostridium species may increase the production of deoxycholic acid, which is known to cause DNA damage and potentially initiate oncogenesis (58) and/or enhance EGF receptor signaling (36). Therefore, it is possible that Clostridium sensu stricto acts via similar mechanisms to promote PDAC development. Of note, there was no correlation of dysplasia to any genus in the ileum or cecum. This corroborates the idea that if the microbiome is involved in PDAC development, it is likely due to local regional signaling and translocation.
Some have postulated that the way the microbiome may mitigate cancer development is by altering the local immune response. Microbial ablation led to a reduction in immunosuppressive CD206+ M2-like tumor-associated macrophages with a concomitant increase in tumor-protective M1-like tumor-associated macrophages in the orthotopic Pdx1Cre; LSL-KrasG12D; Trp53R172H (KPC) model, changes that reversed with the repopulation of the microbiome (40). Similarly, another study using wild-type C57BL/6J, recombination activating 1 knockout (a model that lacks mature T and B cells), KPC, and phosphatase and tensin homolog floxed mice showed that gut microbial depletion with antibiotics resulted in an increase in T helper type 1 (interferon γ+CD4+CD3+) and Type 1 T cells (interferon γ+CD8+CD3+) cells in the tumor microenvironment and a reduction in pancreatic tumor burden (43).
Although this study does show a novel and possible mechanistic interplay between obesity, the microbiome, metformin, and PDAC, the study has several limitations. First, 16S sequencing is limited to only compositional analysis of the microbiome. There may be shifts in microbial function (e.g., gene expression and metabolites) on the HFCD and with metformin that are relevant to PDAC development but not detectable by this strategy. Second, although we would have also liked to see major shifts in the microbiome of the pancreas, the relatively low abundance of bacterial DNA that is present in the pancreas may have limited our ability to resolve differences. Future studies should involve shotgun sequencing and germ-free KC mice models to determine if the changes in the microbiome that are seen in this study are sufficient in inducing PDAC development.
A major strength of our research is the incorporation of quantitative PCR data to determine absolute bacterial abundances instead of relative abundances. The majority of papers that have examined the microbiome and PDAC development have depended on measurements of relative abundance. By doing so, they may falsely report differences in differential abundance testing by not accounting for the effect of treatment on overall bacterial quantity. Although there were no statistical differences between any of the groups in the duodenum or ileum, we found that the addition of metformin greatly reduces the overall bacterial quantity in the cecum. This is important for future studies that examine the effect of metformin on the colonic and fecal microbiome of mice and humans.
In conclusion, our results support a connection between obesity and the microbiome in diet-associated PDAC. In addition, our data suggest that metformin’s chemopreventive effects on PDAC, like its antidiabetic effects, could be potentially mediated through changes in the local regional microbiome.
GRANTS
National Institutes of Health (NIH) Grant P01-CA-163200 supported the work of the laboratory of G. Eibl. The work in the laboratory of E. Rozengur was supported by NIH Grants R01-DK-100405, P30-DK-41301, and P01-CA-163200 and by the Department of Veterans Affair Merit Award 1I01BX001473. Additional funding for E. Rozengur and G. Eibl’s laboratories came from the Hirshberg Foundation for Pancreatic Cancer Research. T. S. Dong was supported by NIH/National Institute of Diabetes and Digestive and Kidney Diseases T32-DK-07180, and J.P.J. was supported by VA Career Development Award IK2CX001717.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.R. and G.E. conceived and designed research; H.-H.C., M.H., V.L., and W.K. performed experiments; T.S.D., E.R., and J.P.J. analyzed data; T.S.D., W.K., E.R., J.P.J., and G.E. interpreted results of experiments; T.S.D. and H.-H.C. prepared figures; T.S.D. and H.-H.C. drafted manuscript; T.S.D., H.-H.C., M.H., V.L., W.K., E.R., J.P.J., and G.E. edited and revised manuscript; T.S.D., H.-H.C., M.H., V.L., W.K., E.R., J.P.J., and G.E. approved final version of manuscript.
REFERENCES
- 1.American Cancer Society Cancer Facts and Figures 2019 (Online). https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2019/cancer-facts-and-figures-2019.pdf.
- 2.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol 26: 32–46, 2001. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- 3.Bauer PV, Duca FA, Waise TMZ, Rasmussen BA, Abraham MA, Dranse HJ, Puri A, O’Brien CA, Lam TKT. Metformin alters upper small intestinal microbiota that impact a glucose-SGLT1-sensing glucoregulatory pathway. Cell Metab 27: 101–117.e5, 2018. doi: 10.1016/j.cmet.2017.09.019. [DOI] [PubMed] [Google Scholar]
- 4.Biankin AV, Kench JG, Biankin SA, Lee CS, Morey AL, Dijkman FP, Coleman MJ, Sutherland RL, Henshall SM. Pancreatic intraepithelial neoplasia in association with intraductal papillary mucinous neoplasms of the pancreas: implications for disease progression and recurrence. Am J Surg Pathol 28: 1184–1192, 2004. doi: 10.1097/01.pas.0000131556.22382.3c. [DOI] [PubMed] [Google Scholar]
- 5.Bracci PM. Obesity and pancreatic cancer: overview of epidemiologic evidence and biologic mechanisms. Mol Carcinog 51: 53–63, 2012. doi: 10.1002/mc.20778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5a.Broadhurst PJ, Hart AR. Metformin as an adjunctive therapy for pancreatic cancer: a review of the literature on its potential therapeutic use. Dig Dis Sci 63: 2840–2852, 2018. doi: 10.1007/s10620-018-5233-y. [DOI] [PubMed] [Google Scholar]
- 6.Call L, Stoll B, Oosterloo B, Ajami N, Sheikh F, Wittke A, Waworuntu R, Berg B, Petrosino J, Olutoye O, Burrin D. Metabolomic signatures distinguish the impact of formula carbohydrates on disease outcome in a preterm piglet model of NEC. Microbiome 6: 111, 2018. doi: 10.1186/s40168-018-0498-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods 13: 581–583, 2016. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Callahan BJ, Sankaran K, Fukuyama JA, McMurdie PJ, Holmes SP. Bioconductor workflow for microbiome data analysis: from raw reads to community analyses. F1000 Res 5: 1492, 2016. doi: 10.12688/f1000research.8986.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7: 335–336, 2010. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carding S, Verbeke K, Vipond DT, Corfe BM, Owen LJ. Dysbiosis of the gut microbiota in disease. Microb Ecol Health Dis 26: v.26, 2015, doi: 10.3402/mehd.v26.26191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang HH, Moro A, Chou CEN, Dawson DW, French S, Schmidt AI, Sinnett-Smith J, Hao F, Hines OJ, Eibl G, Rozengurt E. Metformin decreases the incidence of pancreatic ductal adenocarcinoma promoted by diet-induced obesity in the conditional KrasG12D mouse model. Sci Rep 8: 5899, 2018. doi: 10.1038/s41598-018-24337-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang H-H, Moro A, Takakura K, Su HY, Mo A, Nakanishi M, Waldron RT, French SW, Dawson DW, Hines OJ, Li G, Go VLW, Sinnett-Smith J, Pandol SJ, Lugea A, Gukovskaya AS, Duff MO, Rosenberg DW, Rozengurt E, Eibl G. Incidence of pancreatic cancer is dramatically increased by a high fat, high calorie diet in KrasG12D mice. PLoS One 12: e0184455, 2017. doi: 10.1371/journal.pone.0184455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chari ST, Leibson CL, Rabe KG, Ransom J, de Andrade M, Petersen GM. Probability of pancreatic cancer following diabetes: a population-based study. Gastroenterology 129: 504–511, 2005. doi: 10.1016/j.gastro.2005.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dawson DW, Hertzer K, Moro A, Donald G, Chang HH, Go VL, Pandol SJ, Lugea A, Gukovskaya AS, Li G, Hines OJ, Rozengurt E, Eibl G. High-fat, high-calorie diet promotes early pancreatic neoplasia in the conditional KrasG12D mouse model. Cancer Prev Res (Phila) 6: 1064–1073, 2013. doi: 10.1158/1940-6207.CAPR-13-0065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong TS, Gupta A. Influence of early life, diet, and the environment on the microbiome. Clin Gastroenterol Hepatol 17: 231–242, 2019. doi: 10.1016/j.cgh.2018.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ 330: 1304–1305, 2005. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Pedersen HK, Arumugam M, Kristiansen K, Voigt AY, Vestergaard H, Hercog R, Costea PI, Kultima JR, Li J, Jørgensen T, Levenez F, Dore J, Nielsen HB, Brunak S, Raes J, Hansen T, Wang J, Ehrlich SD, Bork P, Pedersen O; MetaHIT consortium . Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528: 262–266, 2015. doi: 10.1038/nature15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geller LT, Barzily-Rokni M, Danino T, Jonas OH, Shental N, Nejman D, Gavert N, Zwang Y, Cooper ZA, Shee K, Thaiss CA, Reuben A, Livny J, Avraham R, Frederick DT, Ligorio M, Chatman K, Johnston SE, Mosher CM, Brandis A, Fuks G, Gurbatri C, Gopalakrishnan V, Kim M, Hurd MW, Katz M, Fleming J, Maitra A, Smith DA, Skalak M, Bu J, Michaud M, Trauger SA, Barshack I, Golan T, Sandbank J, Flaherty KT, Mandinova A, Garrett WS, Thayer SP, Ferrone CR, Huttenhower C, Bhatia SN, Gevers D, Wargo JA, Golub TR, Straussman R. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357: 1156–1160, 2017. doi: 10.1126/science.aah5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, Knight R, Ley RE. Conducting a microbiome study. Cell 158: 250–262, 2014. doi: 10.1016/j.cell.2014.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hingorani SR, Petricoin EF III, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4: 437–450, 2003. doi: 10.1016/S1535-6108(03)00309-X. [DOI] [PubMed] [Google Scholar]
- 22.Hruban RH, Adsay NV, Albores-Saavedra J, Compton C, Garrett ES, Goodman SN, Kern SE, Klimstra DS, Klöppel G, Longnecker DS, Lüttges J, Offerhaus GJ. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol 25: 579–586, 2001. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 23.Hruban RH, Fukushima N. Pancreatic adenocarcinoma: update on the surgical pathology of carcinomas of ductal origin and PanINs. Mod Pathol 20, Suppl 1: S61–S70, 2007. doi: 10.1038/modpathol.3800685. [DOI] [PubMed] [Google Scholar]
- 24.Huxley R, Ansary-Moghaddam A, Berrington de González A, Barzi F, Woodward M. Type-II diabetes and pancreatic cancer: a meta-analysis of 36 studies. Br J Cancer 92: 2076–2083, 2005. doi: 10.1038/sj.bjc.6602619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobs JP, Lin L, Goudarzi M, Ruegger P, McGovern DPB, Fornace AJ Jr, Borneman J, Xia L, Braun J. Microbial, metabolomic, and immunologic dynamics in a relapsing genetic mouse model of colitis induced by T-synthase deficiency. Gut Microbes 8: 1–16, 2017. doi: 10.1080/19490976.2016.1257469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HJ, Lee S, Chun KH, Jeon JY, Han SJ, Kim DJ, Kim YS, Woo JT, Nam MS, Baik SH, Ahn KJ, Lee KW. Metformin reduces the risk of cancer in patients with type 2 diabetes: An analysis based on the Korean National Diabetes Program Cohort. Medicine (Baltimore) 97: e0036, 2018. doi: 10.1097/MD.0000000000010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res 69: 6539–6545, 2009. doi: 10.1158/0008-5472.CAN-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kisfalvi K, Moro A, Sinnett-Smith J, Eibl G, Rozengurt E. Metformin inhibits the growth of human pancreatic cancer xenografts. Pancreas 42: 781–785, 2013. doi: 10.1097/MPA.0b013e31827aec40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee DY, Yu JH, Park S, Han K, Kim NH, Yoo HJ, Choi KM, Baik SH, Kim NH, Seo JA. The influence of diabetes and antidiabetic medications on the risk of pancreatic cancer: a nationwide population-based study in Korea. Sci Rep 8: 9719, 2018. doi: 10.1038/s41598-018-27965-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee H, Ko G. Effect of metformin on metabolic improvement and gut microbiota. Appl Environ Microbiol 80: 5935–5943, 2014. doi: 10.1128/AEM.01357-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X, Li T, Liu Z, Gou S, Wang C. The effect of metformin on survival of patients with pancreatic cancer: a meta-analysis. Sci Rep 7: 5825, 2017. doi: 10.1038/s41598-017-06207-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lozupone CA, Knight R. Species divergence and the measurement of microbial diversity. FEMS Microbiol Rev 32: 557–578, 2008. doi: 10.1111/j.1574-6976.2008.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maruvada P, Leone V, Kaplan LM, Chang EB. The human microbiome and obesity: moving beyond associations. Cell Host Microbe 22: 589–599, 2017. doi: 10.1016/j.chom.2017.10.005. [DOI] [PubMed] [Google Scholar]
- 34.Ming M, Sinnett-Smith J, Wang J, Soares HP, Young SH, Eibl G, Rozengurt E. Dose-dependent AMPK-dependent and independent mechanisms of berberine and metformin inhibition of mTORC1, ERK, DNA synthesis and proliferation in pancreatic cancer cells. PLoS One 9: e114573, 2014. doi: 10.1371/journal.pone.0114573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nadkarni MA, Martin FE, Jacques NA, Hunter N. Determination of bacterial load by real-time PCR using a broad-range (universal) probe and primers set. Microbiology 148: 257–266, 2002. doi: 10.1099/00221287-148-1-257. [DOI] [PubMed] [Google Scholar]
- 36.Nagathihalli NS, Beesetty Y, Lee W, Washington MK, Chen X, Lockhart AC, Merchant NB. Novel mechanistic insights into ectodomain shedding of EGFR Ligands Amphiregulin and TGF-α: impact on gastrointestinal cancers driven by secondary bile acids. Cancer Res 74: 2062–2072, 2014. doi: 10.1158/0008-5472.CAN-13-2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Parekh PJ, Balart LA, Johnson DA. The influence of the gut microbiome on obesity, metabolic syndrome and gastrointestinal disease. Clin Transl Gastroenterol 6: e91, 2015. doi: 10.1038/ctg.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Preheim SP, Perrotta AR, Friedman J, Smilie C, Brito I, Smith MB, Alm E. Computational methods for high-throughput comparative analyses of natural microbial communities. Methods Enzymol 531: 353–370, 2013. doi: 10.1016/B978-0-12-407863-5.00018-6. [DOI] [PubMed] [Google Scholar]
- 40.Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, Mohan N, Aykut B, Usyk M, Torres LE, Werba G, Zhang K, Guo Y, Li Q, Akkad N, Lall S, Wadowski B, Gutierrez J, Kochen Rossi JA, Herzog JW, Diskin B, Torres-Hernandez A, Leinwand J, Wang W, Taunk PS, Savadkar S, Janal M, Saxena A, Li X, Cohen D, Sartor RB, Saxena D, Miller G. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov 8: 403–416, 2018. doi: 10.1158/2159-8290.CD-17-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 74: 2913–2921, 2014. doi: 10.1158/0008-5472.CAN-14-0155. [DOI] [PubMed] [Google Scholar]
- 42.Romano-Keeler J, Shilts MH, Tovchigrechko A, Wang C, Brucker RM, Moore DJ, Fonnesbeck C, Meng S, Correa H, Lovvorn HN III, Tang YW, Hooper L, Bordenstein SR, Das SR, Weitkamp JH. Distinct mucosal microbial communities in infants with surgical necrotizing enterocolitis correlate with age and antibiotic exposure. PLoS One 13: e0206366, 2018. doi: 10.1371/journal.pone.0206366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sethi V, Kurtom S, Tarique M, Lavania S, Malchiodi Z, Hellmund L, Zhang L, Sharma U, Giri B, Garg B, Ferrantella A, Vickers SM, Banerjee S, Dawra R, Roy S, Ramakrishnan S, Saluja A, Dudeja V. Gut microbiota promotes tumor growth in mice by modulating immune response. Gastroenterology 155: 33–37.e6, 2018. doi: 10.1053/j.gastro.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, Lee MS, Bae JW. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63: 727–735, 2014. doi: 10.1136/gutjnl-2012-303839. [DOI] [PubMed] [Google Scholar]
- 45.Sinnett-Smith J, Kisfalvi K, Kui R, Rozengurt E. Metformin inhibition of mTORC1 activation, DNA synthesis and proliferation in pancreatic cancer cells: dependence on glucose concentration and role of AMPK. Biochem Biophys Res Commun 430: 352–357, 2013. doi: 10.1016/j.bbrc.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3, 2004. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 47.Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different patterns of Akt and ERK feedback activation in response to rapamycin, active-site mTOR inhibitors and metformin in pancreatic cancer cells. PLoS One 8: e57289, 2013. doi: 10.1371/journal.pone.0057289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA 100: 9440–9445, 2003. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tong M, Jacobs JP, McHardy IH, Braun J, Angeles L. Sampling of intestinal microbiota and targeted amplification of bacterial 16S rRNA genes for microbial ecologic analysis. Curr Protoc Immunol 107: 1–11, 2014. doi: 10.1002/0471142735.im0741s107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tseng CH. Metformin and pancreatic cancer risk in patients with type 2 diabetes. Pancreas 47: e57–e59, 2018. doi: 10.1097/MPA.0000000000001130. [DOI] [PubMed] [Google Scholar]
- 52.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444: 1027–1031, 2006. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 53.Udaondo Z, Duque E, Ramos J-L. The pangenome of the genus Clostridium. Environ Microbiol 19: 2588–2603, 2017. doi: 10.1111/1462-2920.13732. [DOI] [PubMed] [Google Scholar]
- 54.Wan G, Sun X, Li F, Wang X, Li C, Li H, Yu X, Cao F. Survival benefit of metformin adjuvant treatment for pancreatic cancer patients: a systematic review and meta-analysis. Cell Physiol Biochem 49: 837–847, 2018. doi: 10.1159/000493214. [DOI] [PubMed] [Google Scholar]
- 55.Wang Z, Lai ST, Xie L, Zhao JD, Ma NY, Zhu J, Ren ZG, Jiang GL. Metformin is associated with reduced risk of pancreatic cancer in patients with type 2 diabetes mellitus: a systematic review and meta-analysis. Diabetes Res Clin Pract 106: 19–26, 2014. doi: 10.1016/j.diabres.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 56.Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, Ståhlman M, Olsson LM, Serino M, Planas-Fèlix M, Xifra G, Mercader JM, Torrents D, Burcelin R, Ricart W, Perkins R, Fernàndez-Real JM, Bäckhed F. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 23: 850–858, 2017. doi: 10.1038/nm.4345. [DOI] [PubMed] [Google Scholar]
- 57.Xue L, Yang K, Newmark H, Leung D, Lipkin M. Epithelial cell hyperproliferation induced in the exocrine pancreas of mice by a western-style diet. J Natl Cancer Inst 88: 1586–1590, 1996. doi: 10.1093/jnci/88.21.1586. [DOI] [PubMed] [Google Scholar]
- 58.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499: 97–101, 2013. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 59.Zhou PT, Li B, Liu FR, Zhang MC, Wang Q, Li YY, Xu C, Liu YH, Yao Y, Li D. Metformin is associated with survival benefit in pancreatic cancer patients with diabetes: a systematic review and meta-analysis. Oncotarget 8: 25242–25250, 2017. doi: 10.18632/oncotarget.15692. [DOI] [PMC free article] [PubMed] [Google Scholar]