

Keywords: bile duct ligation, chemokines, cholestasis, neutrophils
Abstract
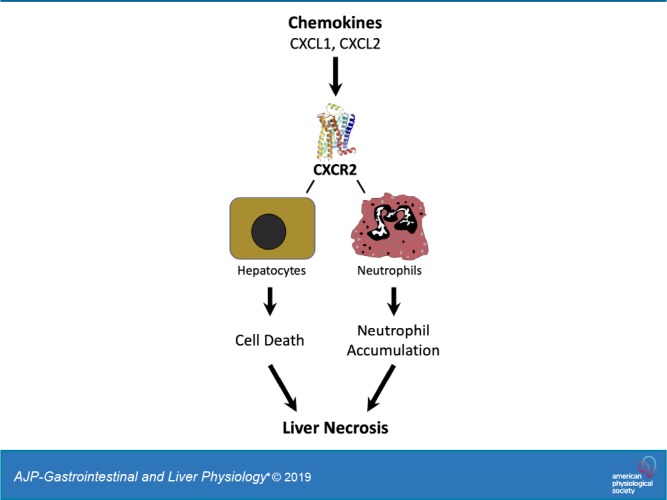
The CXC chemokine receptor 2 (CXCR2) is critical for neutrophil recruitment and hepatocellular viability but has not been studied in the context of cholestatic liver injury following bile duct ligation (BDL). The present study sought to elucidate the cell-specific roles of CXCR2 on acute liver injury after BDL. Wild-type and CXCR2−/− mice were subjected BDL. CXCR2 chimeric mice were created to assess the cell-specific role of CXCR2 on liver injury after BDL. SB225002, a selective CXCR2 antagonist, was administrated intraperitoneally after BDL to investigate the potential of pharmacological inhibition. CXCR2−/− mice had significantly less liver injury than wild-type mice at 3 and 14 days after BDL. There was no difference in biliary fibrosis among groups. The chemokines CXCL1 and CXCL2 were induced around areas of necrosis and biliary structures, respectively, both areas where neutrophils accumulated after BDL. CXCR2−/− mice showed significantly less neutrophil accumulation in those injured areas. CXCR2Liver+/Myeloid+ and CXCR2Liver−/Myeloid− mice recapitulated the wild-type and CXCR2-knockout phenotypes, respectively. CXCR2Liver+/Myeloid+ mice suffered higher liver injury than CXCR2Liver+/Myeloid− and CXCR2Liver−/Myeloid+; however, only those chimeras with knockout of myeloid CXCR2 (CXCR2Liver+/Myeloid− and CXCR2Liver−/Myeloid−) showed reduction of neutrophil accumulation around areas of necrosis. Daily administration of SB225002 starting after 3 days of BDL reduced established liver injury at 6 days. In conclusion, neutrophil CXCR2 guides the cell to the site of injury, while CXCR2 on liver cells affects liver damage independent of neutrophil accumulation. CXCR2 appears to be a viable therapeutic target for cholestatic liver injury.
NEW & NOTEWORTHY This study is the first to reveal cell-specific roles of the chemokine receptor CXCR2 in cholestatic liver injury caused by bile duct ligation. CXCR2 on neutrophils facilitates neutrophil recruitment to the liver, while CXCR2 on liver cells contributes to liver damage independent of neutrophils. CXCR2 may represent a viable therapeutic target for cholestatic liver injury.
INTRODUCTION
Cholestatic liver disease may be induced by a variety of different causes disrupting bile flow as well as autoimmune disease and drug toxicities (8). Hepatic inflammation and injury followed by liver dysfunction are the primary characteristics of cholestatic liver disease. Bile duct ligation (BDL) is one of the most common experimental models of obstructive cholestasis that triggers hepatocellular injury, cholangiocyte proliferation, and liver fibrosis (23). BDL prevents bile flow from liver to gut, which results in retention of bile acids in the liver. While the accumulation of bile acids can directly lead to hepatocellular apoptosis (15, 21), bile acids also induce innate immune response by upregulating proinflammatory cytokines from hepatocytes (2), and consequent neutrophil infiltration causes hepatocellular damage by releasing reactive oxidative stress (7). Previous reports have shown that the CXC chemokines CXC ligand 1 (CXCL1), and CXC ligand 2 (CXCL2) are upregulated in the liver after BDL (2, 16, 27); however, the role of these chemokines and their receptor, CXC chemokine receptor 2 (CXCR2), in liver injury following BDL has not been elucidated.
CXCR2 is a receptor that binds specifically to CXC chemokines that contain the amino terminus sequence ELR (Glu-Leu-Arg), such as CXCL1 and CXCL2 (1). Signaling through CXCR2 has been shown to contribute to liver injury in two distinct ways. First, insult-induced stimulation of chemokines, like CXCL1 and CXCL2, creates a chemotactic gradient, and the binding of these chemokines to CXCR2 expressed on neutrophils results in neutrophil recruitment to the site of injury. The blockade of CXC chemokines or CXCR2 has been shown to attenuate liver injury induced by ischemia/reperfusion by preventing neutrophil recruitment to the liver (5, 14). Second, CXC chemokine signaling through CXCR2 can directly affect hepatocyte viability depending on ligand concentration. In conditions in which CXCR2 chemokine expression is high, such as after ischemia/reperfusion, CXCR2 signaling in hepatocytes results in increased hepatocellular death (13). However, in models of hepatectomy, where there are more moderate increases in the liver expression of CXC chemokines, hepatocytes are induced to proliferate (18). We previously demonstrated that CXCR2 signaling in hepatocytes functions as a rheostat that regulates hepatocyte proliferation and cell death, depending on available ligand concentration (25). Therefore, CXCR2 may play divergent functions in the injury response depending on the cell type and ligand concentration. In this study, we sought to determine the role of CXCR2 on acute liver injury caused by BDL using CXCR2-knockout mice, bone marrow chimeric mice, and a pharmacological antagonist of CXCR2.
MATERIALS AND METHODS
Animals.
Male wild-type (Balb/c) and CXCR2−/− mice (on a Balb/c background) from Jackson Laboratory (Bar Harbor, ME) aged 8–12 wk were used in these experiments. To generate chimeric CXCR2 mice, bone marrow transplantation was performed. Mice received 8 Gy of gamma-radiation, given as a split dose of 4 and 4 Gy, separated by 3 h. Immediately thereafter, 10 × 106 cells of wild-type or CXCR2−/− bone marrow was injected into the tail vein. Mice underwent surgical procedure at 4 wk after bone marrow transplantaion. Analysis of blood leukocyte for CXCR2 expression was performed by flow cytometry as previously performed (24). This project was approved by the University of Cincinnati Animal Care and Use Committee a in compliance with the National Institutes of Health guidelines.
BDL was performed as described previously (23). Briefly, mice were anesthetized with pentobarbital sodium (60 mg/kg ip). A midline laparotomy was performed, and common bile duct was exposed and separated from portal vein and hepatic artery. The common bile duct was ligated with two surgical knots by 4–0 silk. Mice were euthanized after the indicated periods of operation, and blood and liver samples were taken for analysis. Sham mice underwent only laparatomy. Some mice were injected intraperitoneally with 4 mg/kg SB225002 (Calbiochem, San Diego, CA), a nonpeptide antagonist of CXCR2, or 3% DMSO + 0.3% Tween 80 solution as a vehicle at 24-h intervals starting after 3 days of surgery.
Blood analysis.
Blood was obtained by cardiac puncture. Measurement of serum alanine amino transferase (ALT) as an index of hepatocellular injury was made using a diagnosis kit by bioassay (Wiener Laboratories, Rosario, Argentina) according to the manufacturer’s instructions.
Histological analysis.
Liver tissues were fixed in 10% neutral-buffered formalin, processed, and then embedded in paraffin for light microscopy. Sections were stained with hematoxylin and eosin for histological examination and processed further for immunostaining. For immunohistochemical staining, sections had antigen retrieval and nonspecific binding and were blocked with hydrogen peroxide and avidin/biotin. Sections were then incubated with primary antibodies against CK7 (Abcam, Cambridge, UK), CXCL1 (Abcam), CXCL2 (Bio-Rad, Hercules, CA), F4/80 (Abcam), IRF5 (Abcam), CD163 (Abcam), and Ly-6G (Bio-Rad). Sections were washed and incubated with secondary antibodies followed by horseradish peroxidase. Immunoreactive proteins were detected by DAB. The number of neutrophils present around necrosis area and peribiliary area were counted in at least five high-power fields randomly. Sections were also stained with Sirius Red for determination of collagen deposition. Quantitative morphometric analysis of staining was performed with histologic sections at low power (×50) using ImageJ. Fibrosis area by Sirius Red staining area were expressed as percentage of total area.
RT-PCR analysis.
Total RNA was extracted from liver tissue using TRIzol (Invitrogen, Carlsbad, CA). cDNA was generated through the reverse transcription of 2 μg of RNA using High Capacity cDNA Reverse Transcription Kits (Applied Biosystems, Foster City, CA). Samples were incubated at 25°C for 10 min, 37°C for 120 min, and 85°C for 5 min to inactive the reverse transcriptase and then cooled at 5°C for 5 min. Four microliters of diluted cDNA samples were used for quantitative two-step PCR using SYBR Green PCR Master Mix (Applied Biosystems). Each sample was analyzed in duplicate. 18S was used as a housekeeping control gene. Threshold cycles were automatically calculated by the iCycler iQ Real-Time Detection System. Threshold cycle values were normalized to the housekeeping control (18S) to give relative genomic equivalence. Primers utilized were as follows: TNF-α: sense, TCGTAGCAAACCACCAAGTG and antisense, AGATAGCAAATCGGCTGACG; IL-1β: sense, GTGGCAGCTACCTGTGTCTT and antisense, CTCTGCTTGTGAGGTGCTGA; IL-6: sense, GCCTTCTTGGGACTGATGCT and antisense, GCCACTCCTTCTGTGACTCC; IL-12: sense, AAACCACCTCAGTTTGGCCA and antisense, CACCCTGTTGATGGTCACGA; α-smooth muscle actin (α-SMA): sense, GTCCCAGACATCAGGGAGTAA and antisense, TCGGATACTTCAGCGTCAGGA; collagen 1α1: sense, GAGCGGAGAGTACTGGATCG and antisense, GCTTCTTTTCCTTGGGGTTC; tissue inhibitor of metalloproteinase-1 (TIMP-1): sense, CCTTGCAAACTGGAGAGTGACA and antisense, AAGCAAAGTGACGGCTCTGGT; CXCL1: sense AATGAGCTGCGCTGTCAGT and antisense, ACTTGGGGACACCTTTTAGCA; CXCL2: sense, GCTCCTCAGTGCTGCACT and antisense, GCAAACTTTTTGACCGCCCT; and 18S: sense, AGTCCCTGCCCTTTGTACACA and antisense, GATCCGAGGGCCTCAAAC.
Hydroxyproline assay.
Liver tissues were homogenized, and tissue lysates were used for measuring hydroxyproline level in the liver by hydroxyproline assay kit (Abcam) according to the manufacturer’s instructions.
Statistical analysis.
All data are expressed as the means ± SE. Data were analyzed with a one-way ANOVA with subsequent Student t test. Differences were considered significant when P < 0.05.
RESULTS
CXCR2 contributes to liver injury but not fibrosis after BDL.
Because CXCR2 has been shown to regulate liver damage in other models of acute liver injury (9, 13, 18), we examined how CXCR2 signaling affects cholestatic liver injury induced by BDL. Wild-type and CXCR2−/− mice underwent BDL and liver injury and were assessed at 3 and 14 days after operation. As expected, wild-type mice suffered liver injury as evidenced by increased serum ALT levels at both time points (Fig. 1A). However, CXCR2−/− mice had significantly lower ALT levels than wild-type mice (Fig. 1A). We also confirmed the extent of liver injury histologically by measuring the percentage of necrosis area in the ligated liver. Livers from wild-type mice displayed significant necrotic area, while livers from CXCR2−/− mice had significantly less liver necrosis than wild-type mice at both 3 and 14 days after BDL (Fig. 1B). Then, we examined the level of inflammatory cytokines that provoked liver injury. There was no difference in IL-1β, IL-6, and IL-12 mRNA expression in the liver at any time point, while CXCR2−/− mice had significantly higher TNF-α mRNA expression at 14 days than wild-type mice (Fig. 1C). These data suggest that CXCR2 augments liver injury following BDL independent of inflammatory cytokine expression. Because BDL also results in liver fibrosis (23), we determined whether CXCR2 contributes to the development of liver fibrosis after BDL. While we observed that CXCR2−/− mice had lower mRNA expression of the profibrotic genes α-SMA, collagen 1α1, and TIMP-1 than wild-type (Fig. 2A), there was no difference in biliary fibrosis, as determined by Sirius Red staining, between CXCR2−/− and wild-type mice at either time point after BDL (Fig. 2B). We also did not see a significant difference in the liver hydroxyproline level between wild-type mice and CXCR2−/− mice after BDL (sham: wild-type 2.76 ± 0.30 μg/mg, CXCR2−/− 3.00 ± 0.24 μg/mg; 3 days: wild-type 4.00 ± 0.90 μg/mg, CXCR2−/− 3.74 ± 0.52 μg/mg; and 14 days: wild-type 3.63 ± 0.31 μg/mg, CXCR2−/− 4.90 ± 0.72 μg/mg). These data suggest that CXCR2 is not substantively involved in the development of biliary fibrosis after BDL. Furthermore, there was no difference in ductular reaction detected by CK7 immunohistochemical staining at 14 days after BDL between wild-type mice and CXCR2−/− mice (Fig. 2C).
Fig. 1.

Role of CXC chemokine receptor 2 (CXCR2) on liver injury after bile duct ligation (BDL). A: liver injury was measured by serum level of alanine aminotransferase (ALT). B: liver histology was assessed by staining with hematoxylin and eosin. Normal hepatic architecture was observed in sham-operated wild-type (WT) mice and CXCR2−/− [knockout (KO)] mice. After 3 and 14 days, livers from wild-type mice had large areas of necrosis, whereas livers from CXCR2−/− mice showed less evidence of necrosis. Original magnification: ×50. Liver injury was quantitated by percentage of necrosis area histologically. BDL caused an increase in ALT and necrosis area; however, CXCR2−/− mice showed significantly lower ALT and necrosis area than wild-type mice at 3 and 14 days after BDL. C: mRNA expression of inflammatory cytokines in the liver after BDL. There was no significant difference in the TNF-α, IL-1β, IL-6, and IL-12 expression after BDL between wild-type mice and CXCR2−/−mice except TNF-α expression at 14 days. Data are means ± SE with n = 3–9 per group. *P < 0.05, compared with sham. #P < 0.05, compared with wild type.
Fig. 2.

Role of CXC chemokine receptor 2 (CXCR2) on biliary fibrosis after bile duct ligation (BDL). A: mRNA expression of profibrotic genes in the liver after BDL. α-Smooth muscle actin (α-SMA), collagen 1α1, and tissue inhibitor of metalloproteinase-1 (TIMP-1) expression was significantly less in CXCR2−/− [knockout (KO)] mice than wild-type (WT) mice at 3 days. α-SMA expression was significantly less in CXCR2−/− mice than wild-type mice at 14 days. KO, knockout. B: Sirius Red staining. There was no difference in biliary fibrosis between wild-type mice and CXCR2−/− mice after BDL. Quantitative morphometric analysis of Sirius Red staining showed no significant difference in fibrosis between wild-type mice and CXCR2−/− mice. Original magnification: ×100. Data are means ± SE with n = 3–9 per group. Student’s t test was used for the statistical analysis. *P < 0.05, compared with wild type. C: CK7 staining. There was no difference in ductular reaction between wild-type mice and CXCR2−/− mice at 14 days after BDL.
Compartmentalized expression of CXCL1 and CXCL2 after BDL.
CXCL1 and CXCL2 are the primary ligands for CXCR2 in mice. After BDL, we found that liver expression of CXCL1 and CXCL2 mRNA was significantly increased (Fig. 3A). While these results were not altogether unexpected, we found striking differences between the pattern of CXCL1 and CXCL2 protein expression by immunohistochemistry. In livers of sham mice, only a few CXCL2 staining hepatocytes were randomly seen in the liver parenchyma. However, 3 and 14 days after BDL, hepatocytes surrounding necrosis area were preferably stained with CXCL2 (Fig. 3B). In contrast, the primary pattern of immunostaining for CXCL1 3 days and 14 days after BDL was confined around biliary structures (Fig. 3C).
Fig. 3.

CXC chemokine receptor 1 and 2 (CXCR1 and CXCL2) expression after bile duct ligation (BDL). A: liver mRNA expression of CXCL1 and CXCL2 after BDL. CXCL1 and CXCL2 expression in the liver was upregulated at 3 and 14 days after BDL. CXCR2−/− [knockout (KO)] mice showed significantly higher CXCL1 expression at 3 days and higher CXCL1 and CXCL2 expression at 14 days than wild-type (WT) mice. Data are means ± SE with n = 3–9 per group. Student’s t test was used for the statistical analysis. *P < 0.05, compared with sham. #P < 0.05, compared with wild type. B: CXCL2 immunohistochemical staining. CXCL2 protein was found to be expressed around areas of hepatocyte necrosis after BDL in wild-type mice. Original magnification: ×400. C: CXCL1 immunohistochemical staining. CXCL1 protein was found to be expressed surrounding biliary structures after BDL in wild-type mice. Original magnification: ×400.
CXCR2 does not affect macrophage accumulation in the liver.
Kupffer cells and macrophages contribute to both liver injury and liver repair (11). To investigate whether CXCR2 affects accumulation and function of macrophage after BDL, we assessed macrophage distribution and their polarization by immunohistochemically. F4/80 staining revealed that wild-type mice had infiltration of macrophages to the necrotic area and periportal area at 3 and 14 days after BDL (Fig. 4A). Regarding macrophage polarization in wild-type mice, IRF5-positive M1 macrophages preferably accumulated to the injured area, while CD163-positive M2 macrophages were found throughout the liver tissue (Fig. 4B). CXCR2−/− mice showed a similar pattern of macrophage accumulation and polarization after BDL (Fig. 4, A and B). These results suggest that CXCR2 does not affect macrophage accumulation or polarization after BDL.
Fig. 4.

Macrophage infiltration after bile duct ligation (BDL). A: F4/80 immunohistochemical staining. Macrophages accumulated to the necrotic area and periportal area in both wild-type (WT) mice and CXC chemokine receptor 2 knockout (CXCR2−/−; CXCR2KO) mice. Original magnification: ×200. B: macrophage polarization after BDL. IRF5 immunohistochemical staining showed that M1 macrophages preferably accumulated to the injured area in wild type. C: CD163 immunohistochemical staining showed that M2 macrophages existed in the sinusoid throughout liver tissue in wild type. CXCR2−/− mice also had similar pattern of the accumulation of M1 and M2 macrophages. Original magnification: ×200.
CXCR2 regulates neutrophil migration in the liver.
Neutrophils are one of the key cell types causing liver injury after BDL (7), and CXC chemokines have been reported to contribute to neutrophil recruitment after the insult (20). To investigate whether signaling via CXCR2 was responsible for neutrophil recruitment to the liver after BDL, we examined neutrophil recruitment by Ly-6G immunostaining, which allowed us to assess both the number of neutrophils and their localization in the liver. Wild-type mice showed significant increases in neutrophil numbers around areas of focal hepatocyte necrosis (Fig. 5A) and around biliary structures (Fig. 5B) at 3 and 14 days after BDL. CXCR2−/− mice had significantly less neutrophils compared with wild-type mice at both locations and time points (Fig. 5, A and B). Interestingly, 14 days after BDL, CXCR2−/− mice displayed intense aggregation of neutrophils around the periphery of portal triads (Fig. 5B). Ly-6G staining was confirmed by staining sections for myeloperoxidase, which showed similar staining patterns (data not shown). These data suggest that CXCR2 contributes directly to the recruitment and intrahepatic localization of neutrophils in the injured liver after BDL.
Fig. 5.

Neutrophil accumulation in the liver after bile duct ligation (BDL). Ly-6G immunohistochemical staining. A: neutrophils accumulated around focal necrosis after BDL. CXC chemokine receptor 2 knockout (CXCR2−/−; CXCR2KO) mice showed significant less neutrophil accumulation around focal necrosis than wild-type (WT) mice. HPF, high-power field. Original magnification: ×400. B: neutrophils accumulated in peribiliary areas after BDL. CXCR2−/− mice showed significant less neutrophil accumulation around biliary structures than wild-type mice. Data are means ± SE. Sham: wild-type n = 3, CXCR2−/− n = 3; 3 days: wild-type n = 7, CXCR2−/− n = 6; and 14 days: wild-type n = 9, CXCR2−/− n = 7. Student’s t test was used for the statistical analysis. *P < 0.05, compared with wild type. Original magnification: ×200.
CXCR2 on neutrophils and liver cells contribute to liver injury after BDL.
Our data so far suggest that CXCR2 guides neutrophils to the injured/necrotic site and amplifies liver injury after BDL. Since CXCR2 is expressed by cells other than neutrophils, including hepatocytes, endothelial cells, and hepatic stellate cells (3, 17, 22), we sought to clarify hepatic vs myeloid CXCR2 function after BDL. To address this issue, we created CXCR2 chimeric mice and assessed their response after BDL. Using bone marrow transplantation, we created mice expressing CXCR2 on both liver and myeloid cells (CXCR2Liver+/Myeloid+), only on liver cells (CXCR2Liver+/Myeloid−), only on myeloid cells (CXCR2Liver−/Myeloid+), or on neither liver or myeloid cells (CXCR2Liver−/Myeloid−). Flow cytometry analysis confirmed that mice receiving bone marrow from wild-type (CXCR2Liver+/Myeloid+ and CXCR2Liver−/Myeloid+) showed the expression of CXCR2 on neutrophils, whereas mice receiving bone marrow from CXCR2−/− (CXCR2Liver+/Myeloid− and CXCR2Liver−/Myeloid−) lacked neutrophil CXCR2 expression (Fig. 6A). Consistent with our previous data using wild-type mice and global CXCR2−/− mice, CXCR2Liver+/Myeloid+ mice had a significantly higher serum ALT level and higher percentage of necrotic area than CXCR2Liver−/Myeloid− mice after BDL (Fig. 6, B–D). CXCR2Liver+/Myeloid+ and CXCR2Liver−/Myeloid− mice recapitulated the wild-type and CXCR2-knockout phenotypes, respectively. CXCR2Liver+/Myeloid+ mice also had significant higher serum ALT level and significant larger areas of necrosis than CXCR2Liver+/Myeloid− mice and larger areas of necrosis than CXCR2Liver−/Myeloid+ mice (Fig. 6, B–D). These data suggest that both myeloid and liver CXCR2 contributes to liver injury after BDL. To examine the effects of myeloid and liver CXCR2 on recruitment of neutrophils to necrotic areas, we stained sections of the ligated liver with Ly-6G. Interestingly, only those chimeras with knockout of myeloid CXCR2 (CXCR2Liver+/Myeloid− and CXCR2Liver−/Myeloid−) showed reduction of neutrophil numbers and localization around necrotic areas (Fig. 6E). Ly-6G staining was confirmed by staining sections for myeloperoxidase, which showed similar staining patterns (data not shown). This suggests that liver CXCR2 does not contribute to neutrophil accumulation after BDL and that the decreased necrosis seen in mice lacking liver CXCR2 may be a result of direct effects, as has been shown in other liver injury models (13, 25).
Fig. 6.

Cell-specific role of CXC chemokine receptor 2 (CXCR2) on liver injury after bile duct ligation (BDL). Irradiated mice underwent bone marrow transplantation. After engraftment, BDL was performed. Samples were collected at 3 days after BDL A: CXCR2 expression on neutrophils in chimeric mice. Expression of CXCR2 on neutrophils in blood were analyzed using flow cytometry confirmed bone marrow transplantation. WT, wild type; KO, knockout. B: liver injury was measured by serum level of alanine aminotransferase (ALT) and hematoxylin and eosin staining. C and D: liver injury was also measured by percentage of necrosis area determined histologically. CXCR2Liver+/Myeloid+ mice had significantly higher serum ALT level and larger necrosis area than CXCR2Liver+/Myeloid− and CXCR2Liver−/Myeloid− mice after BDL. CXCR2Liver+/Myeloid+ mice also had significant larger necrosis area than CXCR2Liver−/Myeloid+ mice. Original magnification: ×50. E: neutrophil accumulation around areas of necrosis. Only chimeric mice lacking neutrophil CXCR2 had reduced neutrophil recruitment after BDL. HPF, high-power field. Original magnification: ×400. Data are means ± SE with n = 7–9 per group. Student’s t-test was used for the statistical analysis. *P < 0.05, compared with wild type.
Blockade of CXCR2 reduces established liver injury by bile duct ligation.
There have been few reports of pharmacological inhibitors that may have attenuating effects on cholestatic liver injury. Because we found that knockout of CXCR2 improved liver injury after BDL, we next examined if CXCR2 could be targeted therapeutically to treat cholestatic liver injury. Wild-type mice subjected to BDL were intraperitoneally injected with vehicle control or 4 mg/kg SB225002, a nonpeptide antagonist of CXCR2, 3 days after BDL and every 24 h thereafter (Fig. 7A). Mice were euthanized 6 days after BDL, and liver injury was assessed. Therapeutic administration of SB225002 significantly reduced liver injury, as measured by serum ALT and histological analysis of liver sections (Fig. 7, B and C). These results suggest that blockade of CXCR2 may be a relevant therapeutic target for the treatment of cholestatic liver injury.
Fig. 7.

The effect of CXC chemokine receptor 2 (CXCR2) antagonist on established liver injury after bile duct ligation (BDL). A: mice were injected intraperitoneally with vehicle or SB225002 every day beginning 3 days after BDL. Samples were collected at 6 days. B: liver injury was measured by serum level of alanine aminotransferase (ALT). C: liver injury was also measured by percentage of necrosis area determined histologically. SB225002 treatment significantly decreased serum ALT and necrosis area. Data are means ± SE with n = 7 per group. Student’s t test was used for the statistical analysis. *P < 0.05, compared with vehicle group. Original magnification: ×50.
DISCUSSION
CXC chemokine signaling via CXCR2 has been implicated as an important regulatory pathway in several hepatic pathologies (2, 6, 12, 18, 20). The current study provides new information regarding the compartmentalized expression of different CXC chemokines, the different functions of myeloid- versus liver-expressed CXCR2, and the viability of therapeutic treatment with CXCR2 antagonists after BDL.
Neutrophil recruitment is known to be an important component of BDL-induced liver injury. CD18-deficient mice had less neutrophil accumulation and neutrophil-derived oxidant stress in the liver after BDL, resulting in reduced acute cholestatic liver injury (7). CXC chemokines expressed in the injured liver bind to CXCR2 expressed on neutrophils to mediate the recruitment to the site(s) of injury. Our study demonstrates that different CXC ligands are expressed in different regions of the liver after BDL. We found that CXCL2 was expressed preferentially in hepatocytes surrounding areas of focal necrosis, while CXCL1 was expressed around biliary structures. While the mechanism of this differential expression of CXC chemokines remains to be determined, it is possibly due to the cellular milieu at these different sites. Bile acid leakage occurring around biliary structures could be one cause. Bile acids are known to induce various inflammatory mediators, including chemokines (2, 27), and could influence a differential expression of chemokines in these areas. Alternatively, it could be a cell-specific effect. In a murine model of biliary atresia induced by rotavirus, biliary epithelial cells were shown to express much higher levels of CXCL1 than CXCL2 (10). Interestingly, we found that CXCR2−/− mice had much higher liver expression of CXCL1 and CXCL2 than wild-type mice, which could be the result of the loss of a negative feedback loop with the deletion of CXCR2.
A previous study demonstrated that prophylactic blockade of the CXC chemokines CXCL1 and CXCL2, using antibodies reduced neutrophil accumulation and liver damage after BDL in an iNKT-cell-deficient mouse model (26). Our findings, using CXCR2 gene knockout mice in a conventional model of BDL, are consistent with that report. CXCR2−/− mice showed significant reductions in neutrophil recruitment and liver injury after BDL. However, in addition to being expressed on neutrophils, CXCR2 is also expressed on many liver cells, and in hepatocytes we and others have shown that CXCR2 signaling may directly affect cell viability (4, 13, 22). Therefore, we used bone marrow transplantation to create mice with chimeric expression of CXCR2 to determine any functional differences. First, we found that chimeric mice expressing CXCR2 on both myeloid and liver cells (CXCR2Liver+/Myeloid+) and chimeric mice lacking expression of CXCR2 on both these cell types (CXCR2Liver−/Myeloid−) recapitulated the wild-type and global CXCR2-knockout phenotypes, respectively. More interestingly, we found that only chimeric mice lacking neutrophil CXCR2 had reduced neutrophil recruitment after BDL, suggesting that CXCR2 expressed on liver cells played no role in this phenomenon. That is particularly important as chimeric mice that lacked liver cell expression of CXCR2 had attenuated liver injury, despite having unchanged recruitment of neutrophils. These data suggest that CXCR2 expressed on liver cells, such as hepatocytes, directly influenced liver cell injury. This is consistent with our findings in a model of hepatic ischemia/reperfusion injury (13).
Liver fibrosis is also an important consequence of cholestatic liver injury (23). In CXCR2−/− mice, we observed reduced profibrotic gene expression at 3 days and αSMA expression at 14 days. Despite these effects on gene expression, we saw no difference in biliary fibrosis after BDL between wild-type and CXCR2−/− mice. Neutrophil recruitment was greatly reduced in CXCR2 deficient mice, which suggests that neutrophil-mediated liver injury is not a prerequisite for fibrosis after BDL, as reported previously (19).
Finally, we assessed the effect of therapeutic pharmacological inhibition of CXCR2 on BDL-induced liver injury. We administered SB225002, a selective CXCR2 antagonist, 3 days after BDL, a time that would be consistent with clinical diagnosis/confirmation of cholestatic disease. Our results showed that inhibition of CXCR2 reduced established hepatocellular injury. These findings suggest that CXCR2 may represent a therapeutic target for cholestatic liver disease.
In conclusion, the current study demonstrates different functional roles for CXCR2 in liver injury induced by BDL. CXCR2 expressed on neutrophils is responsible for their recruitment to injured areas of liver, while CXCR2 expressed on both neutrophils and liver cells contributes to liver injury after BDL. Gene deletion of CXCR2 had no effect on the development of liver fibrosis. Finally, our findings demonstrate that pharmacologic antagonism may represent a viable treatment strategy for reducing hepatic damage in patients with cholestatic liver disease.
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-56029 (to A. B. Lentsch).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.K. and A.B.L. conceived and designed research; T.K., R.M.S., and H.S.G. performed experiments; T.K., R.M.S., H.S.G., and C.C.C. analyzed data; T.K., C.C.C., and A.B.L. interpreted results of experiments; T.K. prepared figures; T.K., R.M.S., and A.B.L. drafted manuscript; T.K., C.C.C. and A.B.L. edited and revised manuscript; R.M.S., H.S.G., C.C.C., and A.B.L. approved final version of manuscript;
ACKNOWLEDGMENTS
We thank Jeff Bailey in Cincinnati Children’s Comprehensive Mouse and Cancer Core for the technical support of bone marrow transplantation.
REFERENCES
- 1.Addison CL, Daniel TO, Burdick MD, Liu H, Ehlert JE, Xue YY, Buechi L, Walz A, Richmond A, Strieter RM. The CXC chemokine receptor 2, CXCR2, is the putative receptor for ELR+ CXC chemokine-induced angiogenic activity. J Immunol 165: 5269–5277, 2000. doi: 10.4049/jimmunol.165.9.5269. [DOI] [PubMed] [Google Scholar]
- 2.Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol 178: 175–186, 2011. doi: 10.1016/j.ajpath.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bone-Larson CL, Hogaboam CM, Evanhoff H, Strieter RM, Kunkel SL. IFN-gamma-inducible protein-10 (CXCL10) is hepatoprotective during acute liver injury through the induction of CXCR2 on hepatocytes. J Immunol 167: 7077–7083, 2001. doi: 10.4049/jimmunol.167.12.7077. [DOI] [PubMed] [Google Scholar]
- 4.Colletti LM, Green M, Burdick MD, Kunkel SL, Strieter RM. Proliferative effects of CXC chemokines in rat hepatocytes in vitro and in vivo. Shock 10: 248–257, 1998. doi: 10.1097/00024382-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Colletti LM, Kunkel SL, Walz A, Burdick MD, Kunkel RG, Wilke CA, Strieter RM. Chemokine expression during hepatic ischemia/reperfusion-induced lung injury in the rat. The role of epithelial neutrophil activating protein. J Clin Invest 95: 134–141, 1995. doi: 10.1172/JCI117630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorman RB, Gujral JS, Bajt ML, Farhood A, Jaeschke H. Generation and functional significance of CXC chemokines for neutrophil-induced liver injury during endotoxemia. Am J Physiol Gastrointest Liver Physiol 288: G880–G886, 2005. doi: 10.1152/ajpgi.00317.2004. [DOI] [PubMed] [Google Scholar]
- 7.Gujral JS, Farhood A, Bajt ML, Jaeschke H. Neutrophils aggravate acute liver injury during obstructive cholestasis in bile duct-ligated mice. Hepatology 38: 355–363, 2003. doi: 10.1053/jhep.2003.50341. [DOI] [PubMed] [Google Scholar]
- 8.Hirschfield GM, Heathcote EJ, Gershwin ME. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology 139: 1481–1496, 2010. doi: 10.1053/j.gastro.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 9.Hu B, Colletti LM. CXC receptor-2 knockout genotype increases X-linked inhibitor of apoptosis protein and protects mice from acetaminophen hepatotoxicity. Hepatology 52: 691–702, 2010. doi: 10.1002/hep.23715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jafri M, Donnelly B, Bondoc A, Allen S, Tiao G. Cholangiocyte secretion of chemokines in experimental biliary atresia. J Pediatr Surg 44: 500–507, 2009. doi: 10.1016/j.jpedsurg.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ju C, Tacke F. Hepatic macrophages in homeostasis and liver diseases: from pathogenesis to novel therapeutic strategies. Cell Mol Immunol 13: 316–327, 2016. doi: 10.1038/cmi.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konishi T, Lentsch AB. Hepatic ischemia/reperfusion: mechanisms of tissue injury, repair, and regeneration. Gene Expr 17: 277–287, 2017. doi: 10.3727/105221617X15042750874156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuboki S, Shin T, Huber N, Eismann T, Galloway E, Schuster R, Blanchard J, Edwards MJ, Lentsch AB. Hepatocyte signaling through CXC chemokine receptor-2 is detrimental to liver recovery after ischemia/reperfusion in mice. Hepatology 48: 1213–1223, 2008. doi: 10.1002/hep.22471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lentsch AB, Yoshidome H, Cheadle WG, Miller FN, Edwards MJ. Chemokine involvement in hepatic ischemia/reperfusion injury in mice: roles for macrophage inflammatory protein-2 and Kupffer cells. Hepatology 27: 507–512, 1998. doi: 10.1002/hep.510270226. [DOI] [PubMed] [Google Scholar]
- 15.Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev 90: 1165–1194, 2010. doi: 10.1152/physrev.00061.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morita Y, Yoshidome H, Kimura F, Shimizu H, Ohtsuka M, Takeuchi D, Mitsuhashi N, Iida A, Miyazaki M. Excessive inflammation but decreased immunological response renders liver susceptible to infection in bile duct ligated mice. J Surg Res 146: 262–270, 2008. doi: 10.1016/j.jss.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 17.Murdoch C, Monk PN, Finn A. Cxc chemokine receptor expression on human endothelial cells. Cytokine 11: 704–712, 1999. doi: 10.1006/cyto.1998.0465. [DOI] [PubMed] [Google Scholar]
- 18.Ren X, Carpenter A, Hogaboam C, Colletti L. Mitogenic properties of endogenous and pharmacological doses of macrophage inflammatory protein-2 after 70% hepatectomy in the mouse. Am J Pathol 163: 563–570, 2003. doi: 10.1016/S0002-9440(10)63684-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saito JM, Bostick MK, Campe CB, Xu J, Maher JJ. Infiltrating neutrophils in bile duct-ligated livers do not promote hepatic fibrosis. Hepatol Res 25: 180–191, 2003. doi: 10.1016/S1386-6346(02)00247-4. [DOI] [PubMed] [Google Scholar]
- 20.Saito JM, Maher JJ. Bile duct ligation in rats induces biliary expression of cytokine-induced neutrophil chemoattractant. Gastroenterology 118: 1157–1168, 2000. doi: 10.1016/S0016-5085(00)70369-6. [DOI] [PubMed] [Google Scholar]
- 21.Spivey JR, Bronk SF, Gores GJ. Glycochenodeoxycholate-induced lethal hepatocellular injury in rat hepatocytes. Role of ATP depletion and cytosolic free calcium. J Clin Invest 92: 17–24, 1993. doi: 10.1172/JCI116546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stefanovic L, Brenner DA, Stefanovic B. Direct hepatotoxic effect of KC chemokine in the liver without infiltration of neutrophils. Exp Biol Med (Maywood) 230: 573–586, 2005. doi: 10.1177/153537020523000809. [DOI] [PubMed] [Google Scholar]
- 23.Tag CG, Sauer-Lehnen S, Weiskirchen S, Borkham-Kamphorst E, Tolba RH, Tacke F, Weiskirchen R. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp (96): 2015. doi: 10.3791/52438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van Sweringen HL, Sakai N, Quillin RC, Bailey J, Schuster R, Blanchard J, Goetzman H, Caldwell CC, Edwards MJ, Lentsch AB. Roles of hepatocyte and myeloid CXC chemokine receptor-2 in liver recovery and regeneration after ischemia/reperfusion in mice. Hepatology 57: 331–338, 2013. doi: 10.1002/hep.26049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson GC, Kuboki S, Freeman CM, Nojima H, Schuster RM, Edwards MJ, Lentsch AB. CXC chemokines function as a rheostat for hepatocyte proliferation and liver regeneration. PLoS One 10: e0120092, 2015. doi: 10.1371/journal.pone.0120092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wintermeyer P, Cheng CW, Gehring S, Hoffman BL, Holub M, Brossay L, Gregory SH. Invariant natural killer T cells suppress the neutrophil inflammatory response in a mouse model of cholestatic liver damage. Gastroenterology 136: 1048–1059, 2009. doi: 10.1053/j.gastro.2008.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Hong JY, Rockwell CE, Copple BL, Jaeschke H, Klaassen CD. Effect of bile duct ligation on bile acid composition in mouse serum and liver. Liver Int 32: 58–69, 2012. doi: 10.1111/j.1478-3231.2011.02662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]