

Keywords: autoantibody, enteric nervous system, gastrointestinal motility, glia, macrophage
Abstract
Intestinal functions, including motility and secretion, are locally controlled by enteric neural networks housed within the wall of the gut. The fidelity of these functions depends on the precision of intercellular signaling among cellular elements, including enteric neurons, epithelial cells, immune cells, and glia, all of which are vulnerable to disruptive influences during inflammatory events. This review article describes current knowledge regarding inflammation-induced neuroplasticity along key elements of enteric neural circuits, what is known about the causes of these changes, and possible therapeutic targets for protecting and/or repairing the integrity of intrinsic enteric neurotransmission. Changes that have been detected in response to inflammation include increased epithelial serotonin availability, hyperexcitability of intrinsic primary afferent neurons, facilitation of synaptic activity among enteric neurons, and attenuated purinergic neuromuscular transmission. Dysfunctional propulsive motility has been detected in models of colitis, where causes include the changes described above, and in models of multiple sclerosis and other autoimmune conditions, where autoantibodies are thought to mediate dysmotility. Other cells implicated in inflammation-induced neuroplasticity include muscularis macrophages and enteric glia. Targeted treatments that are discussed include 5-hydroxytryptamine receptor 4 agonists, cyclooxygenase inhibitors, antioxidants, B cell depletion therapy, and activation of anti-inflammatory pathways.
INTRODUCTION
Dating back to the work of Bayliss and Starling (4), and of Trendelenberg (83), it has long been recognized that the nervous system of the intestines can generate local reflex responses. In fact, this knowledge led Langley (48) to define the enteric nervous system (ENS) as a distinct, third division of the autonomic nervous system, in addition to the parasympathetic and sympathetic divisions, in his book titled The Autonomic Nervous System. As gastrointestinal physiologists and pharmacologists gained an appreciation for the diversity of neurotransmitters and associated receptors that are found in the ENS, the intestine became the tissue of choice for studying neurotransmitter receptors and for testing pharmacological compounds. In turn, a variety of treatment strategies involving ENS-related targets have been exploited to alter gut functions in an effort to improve symptoms. These include drugs that target opioid, serotonergic, dopaminergic and cholinergic receptors, the serotonin transporter, as well as synthetic and degradative enzymes.
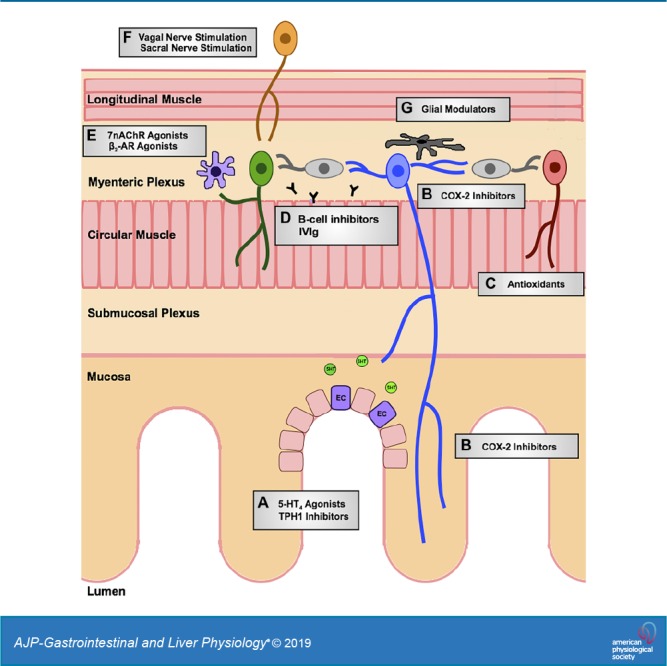
During the past two decades, as the focus has shifted from the ENS in healthy conditions to pathological models, neuroplastic changes have been identified that likely contribute to altered intestinal function and sensation in gastrointestinal disorders. Inflammation-related changes have been documented all along the enteric reflex pathways, including mucosal serotonin signaling, intrinsic afferent neurons, interneurons, and motor neurons. Roles for enteric glial cells and macrophages in inflammatory neuroplasticity have also been described. This review article is meant to highlight these changes in enteric neural circuitry in inflammatory conditions and to outline potential approaches to prevent or reverse them. Figure 1 is a schematic diagram that highlights potential therapeutic targets.
Fig. 1.

Schema of potential therapeutic targets to mediate inflammation-induced enteric neuroplasticity. Targets include A) 5-hydroxytryptamine receptor 4 (5-HT4) agonists and tryptophan hydroxylase-1 (TPH1) inhibitors at the mucosa, B) cyclooygenase-2 (COX-2) inhibitors acting at intrinsic primary afferent neurons, C) antioxidants to restore inhibitory junction potentials, D) B cell inhibitors and intravenous immunoglobulin (IVIg) at the myenteric plexus, E) α7-nicotinic acetylcholine receptor (α7nAChR) and β2-adrenoceptor (β2-AR) agonists of muscularis macrophages, F) vagal and sacral nerve stimulation of extrinsic efferents, and G) glial modulators within the myenteric plexus.
SEROTONIN SIGNALING
Changes in Serotonin Signaling in Inflammatory and Functional Gastrointestinal Disorders
Several lines of evidence support the concept that serotonin released from enterochromaffin (EC) cells in the epithelial lining is capable of stimulating motor, secretory, and vasodilatory reflexes within the gastrointestinal tract, and it can also activate spinal and vagal reflexes (78). Because of its role as a signaling molecule at the afferent terminal of intrinsic and extrinsic gut reflexes, changes in serotonin signaling could have wide-ranging impacts. This has led to a number of studies of the status of mucosal serotonin signaling in various functional and inflammatory disorders of the gastrointestinal tract.
In both inflammatory conditions and in functional gastrointestinal disorders, there is an increase in serotonin availability, and one of the major contributing factors is a decrease in expression of the serotonin reuptake transporter (SERT) by intestinal epithelial cells (3, 20, 49, 62, 64). Decreased SERT has been reported in a number of animal models of small intestinal and colonic inflammation and in rectal biopsy specimens from human subjects with ulcerative colitis, diverticulitis, and both diarrhea- and constipation-predominant irritable bowel syndrome (IBS).
The Caco-2 cell line, which is derived from a human adenocarcinoma and natively expresses SERT, has been used by a number of groups to investigate regulation of SERT expression and function. Using this approach, addition of interferon-γ (IFNγ) and tumor necrosis factor-α (TNFα) to the culture medium decreases SERT RNA and protein levels as well as serotonin uptake (20). These findings indicate that, during inflammation, IFNγ and TNFα could decrease epithelial SERT activity, leading to increased serotonin levels following EC cell release events. Work from the Barbara laboratory at the University of Bologna has linked inflammation with decreased SERT found in irritable bowel syndrome (IBS) (3). They demonstrated that biopsy samples from individuals with IBS contain elevated IFNγ levels. Furthermore, adding IBS biopsy supernatant to Caco-2 cultures causes a decrease in SERT that is partially reversed when IFNγ receptor expression is suppressed. Collectively, these results support the concept that the proinflammatory cytokines IFNγ and TNFα contribute to decreased SERT and altered serotonin signaling in intestinal inflammation and in IBS.
In addition to proinflammatory cytokines, Toll-like receptor (TLR) activation also decreases SERT expression in Caco-2 cells. Alcaide and colleagues have demonstrated that activation of TLR2 (49) or TLR3 (64) can lead to decreased SERT expression and function, with the cAMP/PKA and p38 MAPK pathways contributing to the TLR2 response and the p38 MAPK pathway mediating the TLR3 response. Consistent with the notion that TLRs can have a negative impact on SERT in the intestinal epithelium, mice lacking TLR2 have elevated SERT levels (49).
Given that decreased SERT expression is a consistent feature of intestinal inflammation and IBS, and that alterations in SERT activity likely lead to changes in serotonin signaling, manipulations that enhance SERT expression may normalize intestinal functions and sensitivity. Once again, using Caco-2 cells as an in vitro model, Gill and colleagues (24, 67) reported that both epidermal growth factor (EGF) and transforming growth factor-β1 (TGFβ1) can increase SERT function.
Protective Actions of 5-Hydroxytryptamine Receptor 4 Activation
5-Hydroxytryptamine receptor 4 (5-HT4 receptors) are expressed in the intestinal epithelium, and in the colon they are expressed by enterocytes, goblet cells, and EC cells (33). Treatment of mice by enema with a 5-HT4 agonist at the onset of dextran sulfate sodium (DSS) or 2,4,6-trinitrobenzenesulfonic acid (TNBS) colitis decreases the extent of colitis, and treatment of mice with active DSS or TNBS colitis speeds up the recovery from colitis (77). Although it is not clear whether 5-HT4 agonists have an anti-inflammatory effect through an immunological mechanism, a number of epithelial actions have been established from these studies that could aid in the maintenance and restoration of the epithelium in instances of colitis. These include mucus secretion from goblet cells (33), resistance to oxidative stress (77), and epithelial cell proliferation and migration (68, 77).
The protective actions of 5-HT4 agonist treatment in inflammatory models are accompanied by improvements in motility and neuromuscular transmission (77). As described below, colitis is associated with disrupted propulsive motility and an attenuation of the purinergic component of the inhibitory junction potentials (IJPs). In guinea pigs with TNBS colitis, fecal pellet propulsive motility is significantly improved in isolated distal colon preparations, and in mice with DSS colitis, colonic motility measured in vivo is significantly improved in animals that received 5-HT4 agonist treatment by enema. Furthermore, IJPs are of normal amplitude in guinea pigs with TNBS colitis that receive 5-HT4 agonist treatment.
In addition to these protective actions in models of colitis, 5-HT4 agonist treatment has also been shown to have a beneficial effect in postoperative ileus (80). The 5-HT4 agonist prucalopride reduces intestinal inflammation and prevents postoperative ileus in mice when administered before, but not after, abdominal surgery. Furthermore, in humans undergoing a Whipple procedure, prucalopride improves clinical recovery (80). These actions are thought to involve increased release of acetylcholine from enteric cholinergic neurons and, in turn, a decrease in activation of muscularis macrophages. The site of action of 5-HT4 agonists is highlighted in Fig. 1A.
It appears that 5-HT4 receptors also have an impact on the enteric neuronal population during development and in adulthood. For example, 5-HT4 knockout (KO) mice (54) and mice with an overactive form of SERT exhibit fewer enteric neurons, which is reversed by 5-HT4 agonist treatment (58), and in DSS colitis, 5-HT promotes neurogenesis by stimulating 5-HT4 receptors (7). 5-HT4 receptor stimulation also causes a transdifferentiation of glial cells to neurons (7, 54). In addition to promoting formation of neurons, 5-HT4 receptor stimulation promotes repair of neural circuits in the ENS, as demonstrated by Takaki et al. (60), who showed improved recovery of the rectoanal reflex when guinea pigs that underwent rectal resection and anastomosis were treated with the 5-HT4 agonist mosapride citrate.
Proinflammatory Actions of Serotonin
In addition to the protective actions that serotonin can mediate through the activation of 5-HT4 receptors, as described above, serotonin can also act as a proinflammatory molecule. Experimental colitis is worsened in mice lacking SERT (8), which leads to increased serotonin levels. Colitis is less severe in mice with lower serotonin levels due to inhibition of tryptophan hydroxylase (TPH), the rate-limiting enzyme of serotonin synthesis, by deletion of the TPH1 gene or treatment with a TPH inhibitor (23, 43) (Fig. 1A). The proinflammatory actions of serotonin may involve activation of 5-HT7 receptors on dendritic cells (42, 50); however, it must be noted that activation of 5-HT7 receptors has also been reported to have an anti-inflammatory effect (37). A “sword and shield” hypothesis for the actions of serotonin has been proposed by Dr. Michael Gershon, of Columbia University, in which the proinflammatory actions of serotonin serve as a sword by activating an immune reaction to protect the gut from invasion (22).
INFLAMMATION-INDUCED ENTERIC NEUROPLASTICITY
Intestinal inflammation is associated with gastrointestinal dysmotility that often persists upon resolution of the inflammation, detracting from the patient’s quality of life and contributing to disease morbidity (11). One therapeutic target that may prevent hyperexcitability of after-hyperpolarization (AH) neurons associated with gastrointestinal dysmotility in inflammatory conditions is through inhibition of cyclooxygenase-2 (COX-2) (Fig. 1B).
Studies of inflammation-induced plasticity in a guinea pig model of TNBS colitis demonstrate altered electrophysiological and synaptic characteristics of both AH neurons (including intrinsic primary afferent neurons and interneurons), and synaptic (S) neurons (reflecting motor neurons and ascending interneurons) of the myenteric (53) and submucosal plexus (55). TNBS colitis results in increased number of action potentials following a depolarizing current pulse and more spontaneous action potentials in addition to reduced amplitude of after-hyperpolarization in AH neurons of both plexuses (53, 55).
In the myenteric plexus, this synaptic facilitation is attributed to an increase in the hyperpolarization-dependent cation current (IH) (53). In the submucosal plexus, synaptic facilitation is associated with the shorter duration of action potentials in AH neurons, reducing the influx of Ca2+ from internal and external stores, thus decreasing the Ca2+-activated K+ current. Subsequently, some aspects of TNBS colitis, including myenteric AH neuron hyperexcitability and decreased propulsive motility, are found to be remediated by inhibition of COX-2 when the inhibitor DFU [5,5-dimethyl-3-(3-fluorophenyl)-4(4-methyl-sulfonyl)- phenyl-2-(5H)-furanone] is administered during the acute phase of the disease (52). Synthesis of prostaglandins and thromboxane at the level of the mucosa are found to be mediated by COX-1, as selective inhibition of COX-1 by SC-560 [5-(4-chlorophenyl)-1-(4-methoxyphenyl)-3-trifluoromethyl-pyrazole] administration returns them to control levels (52). Other disease signatures, such as increased mucosal 5-HT availability, mucosal architecture, and inflammation, are not altered by COX-1 or COX-2 inhibition. COX-2 and the related prostaglandin PGE2 are necessary for the onset of hyperexcitability induced by TNBS colitis but do not play a major role in its maintenance (45). TNBS colitis results in chronic disrupted propulsive motility persisting at 56 days, and COX-2 inhibition during the chronic phase does not improve motility (45). Instead, it is believed that persistent electrophysiological changes in neuronal properties that occur during active inflammation contribute to the prolonged dysmotility.
These findings support the concept that early COX-2 inhibition in gastrointestinal inflammation could prevent the onset of hyperexcitability that contributes to maintained dysmotility in these conditions but would be of little benefit to normalize motility and synaptic facilitation after the resolution of inflammation (45, 52). Inhibition of IH, which improves motility of inflamed colons ex vivo (32), would not be a viable treatment strategy because of the importance of this current in repolarizing the cardiac muscle action potential.
PURINERGIC NEUROMUSCULAR TRANSMISSION
To identify the specific mechanisms underlying neuromuscular deficits in colitis-induced inflammation, we and others studied the electrophysiological properties associated with dysmotility of intestinal smooth muscle. Propulsive motility is significantly disrupted at sites of colonic lesions in a guinea pig model of TNBS colitis and is associated with a decreased amplitude and frequency of IJPs (72, 82), as well as the AH neuron hyperexcitability described above (32). Further analysis of the IJP pharmacology revealed that the purinergic, but not the nitrergic, component is significantly reduced in inflamed regions of the colon. The role of purinergic contributions of the IJP on intestinal motility was validated by the disruption of propulsive motility in control colons following application of a P2Y1 antagonist (82) and in mice deficient in P2Y1 (35). In animals with TNBS colitis, IJP attenuation is recovered by day 56, although dysmotility persists, suggesting that purinergic signaling is implicated in the onset of dysmotility but does not directly mediate its persistence (82).
A likely mechanism leading to disrupted purinergic signaling in inflammation is free radical oxidative damage of mitochondria and reduction of purine synthesis. In TNBS colitis, the release of purines from nerve terminals in the muscularis as measured by HPLC is decreased (72). Similarly, inhibition of ATP synthesis by oligomycin and/or dicyclohexylcarbodiimide in healthy colon preparations results in a decreased amplitude of smooth muscle IJPs, suggesting that reduced purine synthesis is a major contributor to altered contractility in colitis. This effect can also be demonstrated by acute application of H2O2. When the antioxidant Tempol is added to the drinking water of TNBS-treated guinea pigs and DSS-treated mice, the IJP amplitude is normalized, but it is not clear whether this is a protective or restorative effect. Tempol also improves motility in guinea pig colitis. In addition to protecting and/or recovering the purinergic IJP in guinea pig TNBS colitis, these studies demonstrate the significance of mitochondrial oxidative damage on gastrointestinal neuromuscular function and highlight the therapeutic potential of antioxidant free radical scavengers on inflammation-induced dysmotility in colitis (Fig. 1C).
ANTIBODY-MEDIATED GASTROINTESTINAL DYSMOTILITY
Autoimmune gastrointestinal dysmotility is a recognized clinical condition (19). Gastrointestinal disorders are reported in a wide variety of systemic autoimmune diseases, including lupus erythematosus, systemic sclerosis, scleroderma, and rheumatoid arthritis (15). Furthermore, circulating autoantibodies targeting the ENS and leading to disrupted motility have been demonstrated in paraneoplastic syndromes (31), Lambert Eaton syndrome (79), Chagas disease (17, 25), diabetes (38, 79), and achalasia (63, 65). In some of these disorders, it has been established that molecular mimicry results in antibodies targeting proteins that are important for neuromuscular transmission, such as Ca2+ channels (Lambert Eaton syndrome) or muscarinic receptors (Chagas disease). In other conditions, such as achalasia, the autoimmune disruption in motor function involves a loss of neurons.
Autoantibodies, which are often not detectable by traditional immunoblot assays, may interact with cells critical to motility and secretion and contribute to altered functional disorders. Jackson et al. (38) were among the first to report that functional autoantibodies in type 1 diabetes (T1D) contribute to enteric neuropathy, resulting in altered intestinal motility. Both in vitro application and passive transfer of purified IgG from patients with diabetes alters migrating myoelectrical complexes (MMCs) in an L-type calcium channel-dependent manner in mouse colon preparations (38). Administration of intravenous immunoglobulin (IVIg) into mice before passive transfer of T1D IgG is sufficient to reduce the disruption of IgG on colonic motor contractility (84).
Similar approaches have been used to detect the presence in the blood of narcolepsy patients of functional autoantibodies that amplify cholinergic activity, first demonstrated in murine bladder (74) and subsequently in colon (39). These approaches led to the discovery that autoantibodies in narcolepsy act by inhibiting voltage-gated sodium channels (40).
Gastrointestinal dysmotility occurs in experimental autoimmune encephalomyelitis (EAE), a murine model of multiple sclerosis (MS) (76, 85). Delayed colonic motility is exhibited in wild-type (WT) mice induced with EAE but is absent in B cell-deficient muMt− mice, suggesting that the observed dysmotility is antibody mediated (76). Importantly, there is no difference in somatic motor symptoms between the two strains of mice, and while a significant linear relationship exists between colonic motility and somatic disease score in WT mice, this relationship is abolished in muMt− mice. Serum from humans with MS and from mice with EAE exhibit immunoreactivity against neurons and/or glia in the myenteric plexus (76, 85). Wunsch et al. (85) demonstrated that immune cell infiltration in the intestinal muscularis of EAE mice immunized against the fusion protein MP4 is observable before mice exhibit somatic motor symptoms and continues through disease progression. MP4-mediated EAE is associated with delayed gastrointestinal motility and myenteric axonal injury, the latter of which is also observed in resected colon samples from human subjects with MS. Antibodies generated in EAE mice target enteric neurons and glia, and several potential antigens, including apolipoprotein A-I, are present in clinical serum samples.
These studies bring to light the potential role of modulating antibody production in treatment of gastrointestinal involvement in autoimmune diseases. Such targets have proved effective against symptoms in other causes of autoimmune gastrointestinal dysmotility. For example, IVIg has been shown to reduce bowel and bladder symptoms in patients with Sjögren’s syndrome (75) and scleroderma (70). Additionally, IVIg and the anti-CD20 antibody Rituxan have been used to protect against gastrointestinal symptoms associated with antibodies in paraneoplastic syndrome (16, 27). Enhanced understanding of antibody mechanisms underlying gastrointestinal complications in T1D and MS could increase the range of therapeutic options for these symptoms (Fig. 1D).
THERAPEUTIC TARGETS INVOLVING MUSCULARIS MACROPHAGES
Another cell population that contributes to the maintenance of ENS homeostasis is muscularis macrophages (MMs). MMs predominantly display the tissue protective, “M2” phenotype, and are involved in the resolution of inflammation and dysmotility in postoperative ileus (18) but are known to shift toward a proinflammatory state during some conditions, including aging (5). These cells are phenotypically and functionally distinct from lamina propria macrophages (LPMs) and are intimately associated with ENS neurons. MM secretion of bone morphogenic protein-2 (BMP2) stimulates neurons to release colony-stimulating factor 1 (CSF1); thus, MMs and neurons provide reciprocal support in healthy states (66). MM deficiency induced by administration of anti-CSF1R monoclonal antibody or through the use of Csf1op/op transgenic mice results in uncoordinated motility, disorganization of myenteric neurons, slower colonic motility in vivo, and hyperreactive colonic stretch reflexes ex vivo (66). An additional role of MMs in homeostasis is through the phagocytosis of apoptotic neurons and cellular debris in the muscularis, a process that occurs regularly in the healthy intestine (46).
The intestinal microbiota is an important contributor to normal intestinal motility and is thought to be a regulator of macrophage-neuronal communication in the muscularis. For example, germ-free mice or mice lacking functional innate immune receptors exhibit significant intestinal dysmotility that is thought to occur by disruption of communication between MMs and neurons (66). This is supported by the finding that antibiotic treatment causes a decrease in MM Bmp2 and loss of MMs. The microbiota may interact directly with enteric neurons as well, as antibiotic treatment results in decreased Csf1, whereas treatment of enteric neurons with LPS results in increased Csf1. Fecal microbial transplant is able to restore motility and homeostatic MM-neuron interactions in mice treated with antibiotic administration (66) and in models of aging (6). Several microbial factors may be involved in maintenance of MM-neuronal homeostasis, including LPS signaling (66) and bile acid metabolism (6).
Cholinergic Anti-Inflammatory Pathway
The cholinergic anti-inflammatory pathway (CAIP) describes stimulation of α7-nicotinic acetylcholine receptor (α7nAChR) receptors through activation of the vagus nerve, subsequently depressing TNFα and NF-κB activity in immune cells, including macrophages, dendritic cells, and splenocytes (Fig. 1, E and F). The CAIP reduces inflammatory cytokines in the spleen in models of sepsis (34) and is efficacious in modulating inflammation in the intestinal muscularis. Kimura et al. (44) recently demonstrated the multidimensional targets of CAIP, with α7nAChR mediating macrophage trafficking to the gut, and muscarinic acetylcholine receptor M2 receptor mediating the neutrophil infiltration. Vagal nerve stimulation (VNS) results in activity of extrinsic parasympathetic innervation to the gut that synapses with enteric cholinergic neurons (14, 61). Matteoli et al. (61) demonstrated that the anti-inflammatory effect of VNS and normalization of gastrointestinal motility in a mouse model of postoperative ileus is dependent on vagal innervation of cholinergic neurons in the gut, stimulating α7nAChR signaling on MMs and effectively reducing macrophage activation. One limitation of VNS is the fact that the vagal nerve does not innervate the entire length of the colon, as the hindgut receives parasympathetic preganglionic input from the sacral spinal cord. Moreover, sacral nerve stimulation (SNS) reduces mucosal inflammation and improves epithelial barrier integrity, and it has demonstrated efficacy in the treatment of ulcerative proctitis (10) and experimentally in a porcine model of TNBS colitis (9). An additional pathway that activates the CAIP is through anti-inflammatory effects of serotonergic receptors, including the 5-HT4 receptor (44). As described previously, stimulation of this pathway by the 5-HT4 agonist Prucalopride before intestinal manipulation prevents the onset of postoperative ileus through stimulation of cholinergic enteric neurons and α7nAChR signaling in MMs (80). Other methods of stimulating the cholinergic anti-inflammatory pathway include high-fat enteral feeding (56, 57) and α7nAChR agonists, the latter of which have not been examined in the gut in depth. The anti-inflammatory effects of enteric cholinergic stimulation in colitis, postoperative ileus, and pancreatitis have been widely tested in animal models and have been summarized succinctly by Goverse et al. (26). Little research has been done to assess the anti-inflammatory role of neuronal α7nAChR in the ENS during colitis, but Lakhan et al. (47) report an upregulation of α7nAChR mRNA in the muscularis of mice induced with DSS-colitis, suggesting its involvement in inflammation induced plasticity. Vagal nerve stimulation is already used clinically, and future studies could uncover additional therapeutics to target the CAIP (Fig. 1, E and F).
Macrophage β2-Adrenergic Receptors
Another way in which MMs are distinct from LPMs is through their high expression of Adrb2, which codes for the β2-adrenergic receptor (β2-AR) (21). The genes downstream of Adrb2 are largely involved in tissue-protective and wound-healing effects, including arginase-1 (Arg1) and mannose receptor C type 1 (Mrc1). Mucida and colleagues demonstrated the role of macrophage β2-AR on inflammation and neuronal loss associated with enteric infection. MMs deficient in Adrb2 exhibit decreased expression of Arg1 compared with mice heterozygous for Adrb2 upon infection with the mutant strain of Salmonella typhimurium (SpiB), suggesting that β2-AR is critical for maintenance of the tissue-protective macrophage phenotype. Mice treated with a β2-AR agonist during SpiB infection exhibit a MM-dependent protective effect of enteric neuron cell death. This protective effect is diminished in LysMCre;β2ARfl/fl mice, in which Cre recombinase is expressed under the Lyz2 lysozyme promoter, resulting in β2-AR deficiency in cells of the myeloid origin, including macrophages (59). Protection against enteric neuron cell death following SpiB exposure is also exhibited in WT mice treated with β2-AR antibodies (59). These studies support the role of muscularis β2-AR for its therapeutic potential to mediate enteric neuroplasticity (Fig. 1E).
THERAPEUTIC TARGETS INVOLVING ENTERIC GLIA
Over the past decade or so, enteric glia have become recognized as important players in the regulation of intestinal motility and secretion, and barrier function (12, 28, 69, 71) (Fig. 1G). Through studies that used tamoxifen-inducible knockout of glial fibrillary acidic protein (GFAP)-expressing enteric glia, these cells were proposed to significantly contribute to intestinal barrier function (73). It was subsequently found that enteric glia represent broad transcriptional heterogeneity, with GFAP expression limited to small subsets compared with proteolipid protein (PLP) (71). The deletion of PLP1-expressing enteric glia did not result in significant intestinal barrier permeability (71).
Glia are involved in intestinal motility, where a loss of PLP1-expressing glia results in slower whole gastrointestinal transit, limited to a change in colonic motility without affecting gastric or small intestinal motility (71). Interestingly, the role of enteric glia on motility is sexually dimorphic, with female mice requiring glia for normal motility, whereas the glia are dispensable in male mice (71). This finding contrasts with a previous study that reported no change in gastrointestinal motility in mice that lacked the GFAP-expressing subpopulation of glia (73).
Enteric glia also play an active role in the response to inflammation, where enteric glia have been reported to contribute both to neuronal loss and to provide protection. For example, enteric glia are involved in neurodegeneration through a sequence of events that includes ATP release from glial cells via connexin-43 channels, which leads to stimulation of neuronal P2X purinoceptor 7 (P2X7) receptors and activation of pannexin-1 channels (29). Furthermore, the neurotoxic connexin-43 activity is driven by nitric oxide produced by inducible nitric oxide synthase, which is upregulated in glia during inflammation (12). It has been proposed that modulation of connexin-43, P2X7 receptors, and pannexin-1 channels could be beneficial in intestinal inflammatory conditions (30). There is a significant relationship between many purinergic genes and inflammatory genes in human reactive enteric glia, supporting the role of purinergic therapies (51). Glia can also exert neuroprotective actions through the production of antioxidant glutathione (1, 13), and epithelial barrier maintenance and suppression of activated T cells through the release of glial-derived neurotrophic factor (GDNF) (36, 41). In response to GDNF release, neuropeptide Y autocrine signaling in enteric neurons plays an important role in maintenance of neuronal integrity (2). An additional target is inhibition of interleukin-1 (IL-1); treatment with either an IL-1 receptor antagonist or antibodies that target IL-1, results in reduced severity of postoperative ileus in mice (81). Together, these mechanisms represent many promising therapeutic targets to protect against inflammation induced neuroplasticity through glial modulation.
SUMMARY AND CONCLUDING REMARKS
This review has provided a summary of the key sites along the enteric neural circuitry that controls motor activity where inflammation-induced changes have been detected. These include enhanced mucosal serotonin signaling, hyperexcitable intrinsic primary afferent neurons, amplified interneuronal synaptic communication, and attenuated inhibitory neuromuscular transmission. These changes are brought about by a number of proinflammatory factors including cytokines, prostaglandins, oxidative stress, and autoantibodies, and some of these changes persist beyond recovery from inflammation. Potential treatment strategies, that have been shown to have benefit in animal models, include cyclooxygenase inhibitors, free radical scavengers, and B cell depletion, as well as CAIP stimulation and 5-HT4 agonist treatment.
GRANTS
This work was supported by grants from the National Institute of Diabetes and Digestive and Kidney Diseases (F31 DK-108540 to E. T. Spear and R01 DK-113800 to G. M. Mawe), and from the National Multiple Sclerosis Society (PP2248 to G. M. Mawe).
DISCLOSURES
Dr. Mawe receives research funding from Takeda Pharmaceuticals and is on the Scientific Advisory Board of Dignify Therapeutics.
AUTHOR CONTRIBUTIONS
E.T.S. and G.M.M. prepared figures; E.T.S. and G.M.M. drafted manuscript; E.T.S. and G.M.M. edited and revised manuscript; E.T.S. and G.M.M. approved final version of manuscript.
REFERENCES
- 1.Abdo H, Derkinderen P, Gomes P, Chevalier J, Aubert P, Masson D, Galmiche JP, Vanden Berghe P, Neunlist M, Lardeux B. Enteric glial cells protect neurons from oxidative stress in part via reduced glutathione. FASEB J 24: 1082–1094, 2010. doi: 10.1096/fj.09-139519. [DOI] [PubMed] [Google Scholar]
- 2.Anitha M, Chandrasekharan B, Salgado JR, Grouzmann E, Mwangi S, Sitaraman SV, Srinivasan S. Glial-derived neurotrophic factor modulates enteric neuronal survival and proliferation through neuropeptide Y. Gastroenterology 131: 1164–1178, 2006. doi: 10.1053/j.gastro.2006.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbaro MR, Di Sabatino A, Cremon C, Giuffrida P, Fiorentino M, Altimari A, Bellacosa L, Stanghellini V, Barbara G. Interferon-γ is increased in the gut of patients with irritable bowel syndrome and modulates serotonin metabolism. Am J Physiol Gastrointest Liver Physiol 310: G439–G447, 2016. doi: 10.1152/ajpgi.00368.2015. [DOI] [PubMed] [Google Scholar]
- 4.Bayliss WM, Starling EH. The movements and innervation of the small intestine. J Physiol 24: 99–143, 1899. doi: 10.1113/jphysiol.1899.sp000752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker L, Nguyen L, Gill J, Kulkarni S, Pasricha PJ, Habtezion A. Age-dependent shift in macrophage polarisation causes inflammation-mediated degeneration of enteric nervous system. Gut 67: 827–836, 2018. doi: 10.1136/gutjnl-2016-312940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becker L, Spear ET, Sinha SR, Haileselassie Y, Habtezion A. Age-related changes in gut microbiota alter phenotype of muscularis macrophages and disrupt gastrointestinal motility. Cell Mol Gastroenterol Hepatol 7: 243–245.e2, 2019. doi: 10.1016/j.jcmgh.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belkind-Gerson J, Hotta R, Nagy N, Thomas AR, Graham H, Cheng L, Solorzano J, Nguyen D, Kamionek M, Dietrich J, Cherayil BJ, Goldstein AM. Colitis induces enteric neurogenesis through a 5-HT4-dependent mechanism. Inflamm Bowel Dis 21: 870–878, 2015. doi: 10.1097/MIB.0000000000000326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bischoff SC, Mailer R, Pabst O, Weier G, Sedlik W, Li Z, Chen JJ, Murphy DL, Gershon MD. Role of serotonin in intestinal inflammation: knockout of serotonin reuptake transporter exacerbates 2,4,6-trinitrobenzene sulfonic acid colitis in mice. Am J Physiol Gastrointest Liver Physiol 296: G685–G695, 2009. doi: 10.1152/ajpgi.90685.2008. [DOI] [PubMed] [Google Scholar]
- 9.Brégeon J, Coron E, Da Silva AC, Jaulin J, Aubert P, Chevalier J, Vergnolle N, Meurette G, Neunlist M. Sacral nerve stimulation enhances early intestinal mucosal repair following mucosal injury in a pig model. J Physiol 594: 4309–4323, 2016. doi: 10.1113/JP271783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brégeon J, Neunlist M, Bossard C, Biraud M, Coron E, Bourreille A, Meurette G. Improvement of refractory ulcerative proctitis with sacral nerve stimulation. J Clin Gastroenterol 49: 853–857, 2015. doi: 10.1097/MCG.0000000000000331. [DOI] [PubMed] [Google Scholar]
- 11.Brierley SM, Linden DR. Neuroplasticity and dysfunction after gastrointestinal inflammation. Nat Rev Gastroenterol Hepatol 11: 611–627, 2014. doi: 10.1038/nrgastro.2014.103. [DOI] [PubMed] [Google Scholar]
- 12.Brown IA, McClain JL, Watson RE, Patel BA, Gulbransen BD. Enteric glia mediate neuron death in colitis through purinergic pathways that require connexin-43 and nitric oxide. Cell Mol Gastroenterol Hepatol 2: 77–91, 2016. doi: 10.1016/j.jcmgh.2015.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown IAM, Gulbransen BD. The antioxidant glutathione protects against enteric neuron death in situ, but its depletion is protective during colitis. Am J Physiol Gastrointest Liver Physiol 314: G39–G52, 2018. doi: 10.1152/ajpgi.00165.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cailotto C, Costes LM, van der Vliet J, van Bree SH, van Heerikhuize JJ, Buijs RM, Boeckxstaens GE. Neuroanatomical evidence demonstrating the existence of the vagal anti-inflammatory reflex in the intestine. Neurogastroenterol Motil 24: 191–e93, 2012. doi: 10.1111/j.1365-2982.2011.01824.x. [DOI] [PubMed] [Google Scholar]
- 15.Cojocaru M, Cojocaru IM, Silosi I, Vrabie CD. Gastrointestinal manifestations in systemic autoimmune diseases. Maedica (Buchar) 6: 45–51, 2011. [PMC free article] [PubMed] [Google Scholar]
- 16.Coret F, Bosca I, Fratalia L, Perez-Griera J, Pascual A, Casanova B. Long-lasting remission after rituximab treatment in a case of anti-Hu-associated sensory neuronopathy and gastric pseudoobstruction. J Neurooncol 93: 421–423, 2009. doi: 10.1007/s11060-008-9787-y. [DOI] [PubMed] [Google Scholar]
- 17.De Giorgio R, Guerrini S, Barbara G, Stanghellini V, De Ponti F, Corinaldesi R, Moses PL, Sharkey KA, Mawe GM. Inflammatory neuropathies of the enteric nervous system. Gastroenterology 126: 1872–1883, 2004. doi: 10.1053/j.gastro.2004.02.024. [DOI] [PubMed] [Google Scholar]
- 18.Farro G, Stakenborg M, Gomez-Pinilla PJ, Labeeuw E, Goverse G, Di Giovangiulio M, Stakenborg N, Meroni E, D’Errico F, Elkrim Y, Laoui D, Lisowski ZM, Sauter KA, Hume DA, Van Ginderachter JA, Boeckxstaens GE, Matteoli G. CCR2-dependent monocyte-derived macrophages resolve inflammation and restore gut motility in postoperative ileus. Gut 66: 2098–2109, 2017. doi: 10.1136/gutjnl-2016-313144. [DOI] [PubMed] [Google Scholar]
- 19.Flanagan EP, Saito YA, Lennon VA, McKeon A, Fealey RD, Szarka LA, Murray JA, Foxx-Orenstein AE, Fox JC, Pittock SJ. Immunotherapy trial as diagnostic test in evaluating patients with presumed autoimmune gastrointestinal dysmotility. Neurogastroenterol Motil 26: 1285–1297, 2014. doi: 10.1111/nmo.12391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foley KF, Pantano C, Ciolino A, Mawe GM. IFN-γ and TNF-α decrease serotonin transporter function and expression in Caco2 cells. Am J Physiol Gastrointest Liver Physiol 292: G779–G784, 2007. doi: 10.1152/ajpgi.00470.2006. [DOI] [PubMed] [Google Scholar]
- 21.Gabanyi I, Muller PA, Feighery L, Oliveira TY, Costa-Pinto FA, Mucida D. Neuro-immune interactions drive tissue programming in intestinal macrophages. Cell 164: 378–391, 2016. doi: 10.1016/j.cell.2015.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gershon MD. Serotonin is a sword and a shield of the bowel: serotonin plays offense and defense. Trans Am Clin Climatol Assoc 123: 268–280, 2012. [PMC free article] [PubMed] [Google Scholar]
- 23.Ghia JE, Li N, Wang H, Collins M, Deng Y, El-Sharkawy RT, Côté F, Mallet J, Khan WI. Serotonin has a key role in pathogenesis of experimental colitis. Gastroenterology 137: 1649–1660, 2009. doi: 10.1053/j.gastro.2009.08.041. [DOI] [PubMed] [Google Scholar]
- 24.Gill RK, Anbazhagan AN, Esmaili A, Kumar A, Nazir S, Malakooti J, Alrefai WA, Saksena S. Epidermal growth factor upregulates serotonin transporter in human intestinal epithelial cells via transcriptional mechanisms. Am J Physiol Gastrointest Liver Physiol 300: G627–G636, 2011. doi: 10.1152/ajpgi.00563.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goin JC, Sterin-Borda L, Bilder CR, Varrica LM, Iantorno G, Ríos MC, Borda E. Functional implications of circulating muscarinic cholinergic receptor autoantibodies in chagasic patients with achalasia. Gastroenterology 117: 798–805, 1999. doi: 10.1016/S0016-5085(99)70337-9. [DOI] [PubMed] [Google Scholar]
- 26.Goverse G, Stakenborg M, Matteoli G. The intestinal cholinergic anti-inflammatory pathway. J Physiol 594: 5771–5780, 2016. doi: 10.1113/JP271537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grover M, Camilleri M. Treatment with methylnaltrexone and IVIG for paraneoplastic gastrointestinal dysmotility. Gastroenterol Hepatol (N Y) 9: 51–53, 2013. [PMC free article] [PubMed] [Google Scholar]
- 28.Grubišić V, Gulbransen BD. Enteric glial activity regulates secretomotor function in the mouse colon but does not acutely affect gut permeability. J Physiol 595: 3409–3424, 2017. doi: 10.1113/JP273492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gulbransen BD, Bashashati M, Hirota SA, Gui X, Roberts JA, MacDonald JA, Muruve DA, McKay DM, Beck PL, Mawe GM, Thompson RJ, Sharkey KA. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med 18: 600–604, 2012. doi: 10.1038/nm.2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gulbransen BD, Christofi FL. Are we close to targeting enteric glia in gastrointestinal diseases and motility disorders? Gastroenterology 155: 245–251, 2018. doi: 10.1053/j.gastro.2018.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hirano I, Pandolfino J. Chronic intestinal pseudo-obstruction. Dig Dis 18: 83–92, 2000. doi: 10.1159/000016969. [DOI] [PubMed] [Google Scholar]
- 32.Hoffman JM, McKnight ND, Sharkey KA, Mawe GM. The relationship between inflammation-induced neuronal excitability and disrupted motor activity in the guinea pig distal colon. Neurogastroenterol Motil 23: 673–e279, 2011. doi: 10.1111/j.1365-2982.2011.01702.x. [DOI] [PubMed] [Google Scholar]
- 33.Hoffman JM, Tyler K, MacEachern SJ, Balemba OB, Johnson AC, Brooks EM, Zhao H, Swain GM, Moses PL, Galligan JJ, Sharkey KA, Greenwood-Van Meerveld B, Mawe GM. Activation of colonic mucosal 5-HT(4) receptors accelerates propulsive motility and inhibits visceral hypersensitivity. Gastroenterology 142: 844–854.e4, 2012. doi: 10.1053/j.gastro.2011.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huston JM, Wang H, Ochani M, Ochani K, Rosas-Ballina M, Gallowitsch-Puerta M, Ashok M, Yang L, Tracey KJ, Yang H. Splenectomy protects against sepsis lethality and reduces serum HMGB1 levels. J Immunol 181: 3535–3539, 2008. doi: 10.4049/jimmunol.181.5.3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang SJ, Blair PJ, Durnin L, Mutafova-Yambolieva V, Sanders KM, Ward SM. P2Y1 purinoreceptors are fundamental to inhibitory motor control of murine colonic excitability and transit. J Physiol 590: 1957–1972, 2012. doi: 10.1113/jphysiol.2011.224634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ibiza S, García-Cassani B, Ribeiro H, Carvalho T, Almeida L, Marques R, Misic AM, Bartow-McKenney C, Larson DM, Pavan WJ, Eberl G, Grice EA, Veiga-Fernandes H. Glial-cell-derived neuroregulators control type 3 innate lymphoid cells and gut defence. Nature 535: 440–443, 2016. doi: 10.1038/nature18644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Idzko M, Panther E, Stratz C, Müller T, Bayer H, Zissel G, Dürk T, Sorichter S, Di Virgilio F, Geissler M, Fiebich B, Herouy Y, Elsner P, Norgauer J, Ferrari D. The serotoninergic receptors of human dendritic cells: identification and coupling to cytokine release. J Immunol 172: 6011–6019, 2004. doi: 10.4049/jimmunol.172.10.6011. [DOI] [PubMed] [Google Scholar]
- 38.Jackson MW, Gordon TP, Waterman SA. Disruption of intestinal motility by a calcium channel-stimulating autoantibody in type 1 diabetes. Gastroenterology 126: 819–828, 2004. doi: 10.1053/j.gastro.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 39.Jackson MW, Reed JH, Smith AJ, Gordon TP. An autoantibody in narcolepsy disrupts colonic migrating motor complexes. J Neurosci 28: 13303–13309, 2008. doi: 10.1523/JNEUROSCI.4489-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson MW, Spencer NJ, Reed JH, Smith AJ, Gordon TP. Potentiation of a functional autoantibody in narcolepsy by a cholinesterase inhibitor. Lab Invest 89: 1332–1339, 2009. doi: 10.1038/labinvest.2009.108. [DOI] [PubMed] [Google Scholar]
- 41.Kermarrec L, Durand T, Neunlist M, Naveilhan P, Neveu I. Enteric glial cells have specific immunosuppressive properties. J Neuroimmunol 295-296: 79–83, 2016. doi: 10.1016/j.jneuroim.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 42.Kim JJ, Bridle BW, Ghia JE, Wang H, Syed SN, Manocha MM, Rengasamy P, Shajib MS, Wan Y, Hedlund PB, Khan WI. Targeted inhibition of serotonin type 7 (5-HT7) receptor function modulates immune responses and reduces the severity of intestinal inflammation. J Immunol 190: 4795–4804, 2013. doi: 10.4049/jimmunol.1201887. [DOI] [PubMed] [Google Scholar]
- 43.Kim JJ, Wang H, Terc JD, Zambrowicz B, Yang QM, Khan WI. Blocking peripheral serotonin synthesis by telotristat etiprate (LX1032/LX1606) reduces severity of both chemical- and infection-induced intestinal inflammation. Am J Physiol Gastrointest Liver Physiol 309: G455–G465, 2015. doi: 10.1152/ajpgi.00299.2014. [DOI] [PubMed] [Google Scholar]
- 44.Kimura H, Imura YK, Tomiyasu H, Mihara T, Kaji N, Ohno K, Unno T, Tanahashi Y, Jan TR, Tsubone H, Ozaki H, Hori M. Neural anti-inflammatory action mediated by two types of acetylcholine receptors in the small intestine. Sci Rep 9: 5887, 2019. doi: 10.1038/s41598-019-41698-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krauter EM, Strong DS, Brooks EM, Linden DR, Sharkey KA, Mawe GM. Changes in colonic motility and the electrophysiological properties of myenteric neurons persist following recovery from trinitrobenzene sulfonic acid colitis in the guinea pig. Neurogastroenterol Motil 19: 990–1000, 2007. doi: 10.1111/j.1365-2982.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- 46.Kulkarni S, Micci MA, Leser J, Shin C, Tang SC, Fu YY, Liu L, Li Q, Saha M, Li C, Enikolopov G, Becker L, Rakhilin N, Anderson M, Shen X, Dong X, Butte MJ, Song H, Southard-Smith EM, Kapur RP, Bogunovic M, Pasricha PJ. Adult enteric nervous system in health is maintained by a dynamic balance between neuronal apoptosis and neurogenesis. Proc Natl Acad Sci USA 114: E3709–E3718, 2017. doi: 10.1073/pnas.1619406114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lakhan SE, Kirchgessner A. Anti-inflammatory effects of nicotine in obesity and ulcerative colitis. J Transl Med 9: 129, 2011. doi: 10.1186/1479-5876-9-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langley JN. The Autonomic Nervous System. Cambridge, UK: Cambridge, Heffer; 1921. [Google Scholar]
- 49.Latorre E, Layunta E, Grasa L, Castro M, Pardo J, Gomollón F, Alcalde AI, Mesonero JE. Intestinal serotonin transporter inhibition by Toll-like receptor 2 activation. A feedback modulation. PLoS One 11: e0169303, 2016. doi: 10.1371/journal.pone.0169303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li N, Ghia JE, Wang H, McClemens J, Cote F, Suehiro Y, Mallet J, Khan WI. Serotonin activates dendritic cell function in the context of gut inflammation. Am J Pathol 178: 662–671, 2011. doi: 10.1016/j.ajpath.2010.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liñán-Rico A, Turco F, Ochoa-Cortes F, Harzman A, Needleman BJ, Arsenescu R, Abdel-Rasoul M, Fadda P, Grants I, Whitaker E, Cuomo R, Christofi FL. Molecular signaling and dysfunction of the human reactive enteric glial cell phenotype: implications for GI Infection, IBD, POI, neurological, motility, and GI disorders. Inflamm Bowel Dis 22: 1812–1834, 2016. doi: 10.1097/MIB.0000000000000854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linden DR, Sharkey KA, Ho W, Mawe GM. Cyclooxygenase-2 contributes to dysmotility and enhanced excitability of myenteric AH neurones in the inflamed guinea pig distal colon. J Physiol 557: 191–205, 2004. doi: 10.1113/jphysiol.2004.062174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Linden DR, Sharkey KA, Mawe GM. Enhanced excitability of myenteric AH neurones in the inflamed guinea-pig distal colon. J Physiol 547: 589–601, 2003. doi: 10.1113/jphysiol.2002.035147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu MT, Kuan YH, Wang J, Hen R, Gershon MD. 5-HT4 receptor-mediated neuroprotection and neurogenesis in the enteric nervous system of adult mice. J Neurosci 29: 9683–9699, 2009. doi: 10.1523/JNEUROSCI.1145-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lomax AE, Mawe GM, Sharkey KA. Synaptic facilitation and enhanced neuronal excitability in the submucosal plexus during experimental colitis in guinea-pig. J Physiol 564: 863–875, 2005. doi: 10.1113/jphysiol.2005.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lubbers T, Luyer MD, de Haan JJ, Hadfoune M, Buurman WA, Greve JW. Lipid-rich enteral nutrition reduces postoperative ileus in rats via activation of cholecystokinin-receptors. Ann Surg 249: 481–487, 2009. doi: 10.1097/SLA.0b013e318194d187. [DOI] [PubMed] [Google Scholar]
- 57.Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med 202: 1023–1029, 2005. doi: 10.1084/jem.20042397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Margolis KG, Li Z, Stevanovic K, Saurman V, Israelyan N, Anderson GM, Snyder I, Veenstra-VanderWeele J, Blakely RD, Gershon MD. Serotonin transporter variant drives preventable gastrointestinal abnormalities in development and function. J Clin Invest 126: 2221–2235, 2016. doi: 10.1172/JCI84877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matheis F, Muller P, Graves C, Gabanyi I, Kerner ZJ, Costa-Borges D, Mucida D. Adrenergic signaling in muscularis macrophages limits neuronal death following enteric infection (Preprint). bioXRV 556340, 2019. doi: 10.1101/556340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsuyoshi H, Kuniyasu H, Okumura M, Misawa H, Katsui R, Zhang GX, Obata K, Takaki M. A 5-HT(4)-receptor activation-induced neural plasticity enhances in vivo reconstructs of enteric nerve circuit insult. Neurogastroenterol Motil 22: 806–e226, 2010. doi: 10.1111/j.1365-2982.2010.01474.x. [DOI] [PubMed] [Google Scholar]
- 61.Matteoli G, Gomez-Pinilla PJ, Nemethova A, Di Giovangiulio M, Cailotto C, van Bree SH, Michel K, Tracey KJ, Schemann M, Boesmans W, Vanden Berghe P, Boeckxstaens GE. A distinct vagal anti-inflammatory pathway modulates intestinal muscularis resident macrophages independent of the spleen. Gut 63: 938–948, 2014. doi: 10.1136/gutjnl-2013-304676. [DOI] [PubMed] [Google Scholar]
- 62.Mawe GM, Hoffman JM. Serotonin signalling in the gut–functions, dysfunctions and therapeutic targets. Nat Rev Gastroenterol Hepatol 10: 473–486, 2013. doi: 10.1038/nrgastro.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McMillan HJ, Srinivasan J. Achalasia, chronic sensory neuropathy, and N-type calcium channel autoantibodies: beneficial response to IVIG. Clin J Gastroenterol 3: 78–82, 2010. doi: 10.1007/s12328-010-0140-6. [DOI] [PubMed] [Google Scholar]
- 64.Mendoza C, Matheus N, Latorre E, Castro M, Mesonero JE, Alcalde AI. Toll-like receptor 3 activation affects serotonin transporter activity and expression in human enterocyte-like Caco-2 cells. Cell Physiol Biochem 30: 187–198, 2012. doi: 10.1159/000339057. [DOI] [PubMed] [Google Scholar]
- 65.Moses PL, Ellis LM, Anees MR, Ho W, Rothstein RI, Meddings JB, Sharkey KA, Mawe GM. Antineuronal antibodies in idiopathic achalasia and gastro-oesophageal reflux disease. Gut 52: 629–636, 2003. doi: 10.1136/gut.52.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Muller PA, Koscsó B, Rajani GM, Stevanovic K, Berres ML, Hashimoto D, Mortha A, Leboeuf M, Li XM, Mucida D, Stanley ER, Dahan S, Margolis KG, Gershon MD, Merad M, Bogunovic M. Crosstalk between muscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 158: 1210, 2014. doi: 10.1016/j.cell.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 67.Nazir S, Kumar A, Chatterjee I, Anbazhagan AN, Gujral T, Priyamvada S, Saksena S, Alrefai WA, Dudeja PK, Gill RK. Mechanisms of intestinal serotonin transporter (SERT) upregulation by TGF-β1 induced non-Smad pathways. PLoS One 10: e0120447, 2015. doi: 10.1371/journal.pone.0120447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Park CJ, Armenia SJ, Zhang L, Cowles RA. The 5-HT4 receptor agonist prucalopride stimulates mucosal growth and enhances carbohydrate absorption in the ileum of the mouse. J Gastrointest Surg 23: 1198–1205, 2019. doi: 10.1007/s11605-018-3907-6. [DOI] [PubMed] [Google Scholar]
- 69.Pochard C, Coquenlorge S, Jaulin J, Cenac N, Vergnolle N, Meurette G, Freyssinet M, Neunlist M, Rolli-Derkinderen M. Defects in 15-HETE production and control of epithelial permeability by human enteric glial cells from patients with Crohn’s disease. Gastroenterology 150: 168–180, 2016. doi: 10.1053/j.gastro.2015.09.038. [DOI] [PubMed] [Google Scholar]
- 70.Raja J, Nihtyanova SI, Murray CD, Denton CP, Ong VH. Sustained benefit from intravenous immunoglobulin therapy for gastrointestinal involvement in systemic sclerosis. Rheumatology (Oxford) 55: 115–119, 2016. doi: 10.1093/rheumatology/kev318. [DOI] [PubMed] [Google Scholar]
- 71.Rao M, Rastelli D, Dong L, Chiu S, Setlik W, Gershon MD, Corfas G. Enteric glia regulate gastrointestinal motility but are not required for maintenance of the epithelium in mice. Gastroenterology 153: 1068–1081.e7, 2017. doi: 10.1053/j.gastro.2017.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Roberts JA, Durnin L, Sharkey KA, Mutafova-Yambolieva VN, Mawe GM. Oxidative stress disrupts purinergic neuromuscular transmission in the inflamed colon. J Physiol 591: 3725–3737, 2013. doi: 10.1113/jphysiol.2013.254136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Savidge TC, Newman P, Pothoulakis C, Ruhl A, Neunlist M, Bourreille A, Hurst R, Sofroniew MV. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology 132: 1344–1358, 2007. doi: 10.1053/j.gastro.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 74.Smith AJ, Jackson MW, Neufing P, McEvoy RD, Gordon TP. A functional autoantibody in narcolepsy. Lancet 364: 2122–2124, 2004. doi: 10.1016/S0140-6736(04)17553-3. [DOI] [PubMed] [Google Scholar]
- 75.Smith AJ, Jackson MW, Wang F, Cavill D, Rischmueller M, Gordon TP. Neutralization of muscarinic receptor autoantibodies by intravenous immunoglobulin in Sjögren syndrome. Hum Immunol 66: 411–416, 2005. doi: 10.1016/j.humimm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 76.Spear ET, Holt EA, Joyce EJ, Haag MM, Mawe SM, Hennig GW, Lavoie B, Applebee AM, Teuscher C, Mawe GM. Altered gastrointestinal motility involving autoantibodies in the experimental autoimmune encephalomyelitis model of multiple sclerosis. Neurogastroenterol Motil 30: e13349, 2018. doi: 10.1111/nmo.13349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Spohn SN, Bianco F, Scott RB, Keenan CM, Linton AA, O’Neill CH, Bonora E, Dicay M, Lavoie B, Wilcox RL, MacNaughton WK, De Giorgio R, Sharkey KA, Mawe GM. Protective actions of epithelial 5-hydroxytryptamine 4 receptors in normal and inflamed colon. Gastroenterology 151: 933–944.e3, 2016. doi: 10.1053/j.gastro.2016.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Spohn SN, Mawe GM. Non-conventional features of peripheral serotonin signalling - the gut and beyond. Nat Rev Gastroenterol Hepatol 14: 412–420, 2017. doi: 10.1038/nrgastro.2017.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Srinivasan S, Wiley JW. New insights into neural injury, repair, and adaptation in visceral afferents and the enteric nervous system. Curr Opin Gastroenterol 16: 78–82, 2000. doi: 10.1097/00001574-200001000-00014. [DOI] [PubMed] [Google Scholar]
- 80.Stakenborg N, Labeeuw E, Gomez-Pinilla PJ, De Schepper S, Aerts R, Goverse G, Farro G, Appeltans I, Meroni E, Stakenborg M, Viola MF, Gonzalez-Dominguez E, Bosmans G, Alpizar YA, Wolthuis A, D’Hoore A, Van Beek K, Verheijden S, Verhaegen M, Derua R, Waelkens E, Moretti M, Gotti C, Augustijns P, Talavera K, Vanden Berghe P, Matteoli G, Boeckxstaens GE. Preoperative administration of the 5-HT4 receptor agonist prucalopride reduces intestinal inflammation and shortens postoperative ileus via cholinergic enteric neurons. Gut 68: 1406–1416, 2019. doi: 10.1136/gutjnl-2018-317263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stoffels B, Hupa KJ, Snoek SA, van Bree S, Stein K, Schwandt T, Vilz TO, Lysson M, Veer CV, Kummer MP, Hornung V, Kalff JC, de Jonge WJ, Wehner S. Postoperative ileus involves interleukin-1 receptor signaling in enteric glia. Gastroenterology 146: 176–87.e1, 2014. doi: 10.1053/j.gastro.2013.09.030. [DOI] [PubMed] [Google Scholar]
- 82.Strong DS, Cornbrooks CF, Roberts JA, Hoffman JM, Sharkey KA, Mawe GM. Purinergic neuromuscular transmission is selectively attenuated in ulcerated regions of inflamed guinea pig distal colon. J Physiol 588: 847–859, 2010. doi: 10.1113/jphysiol.2009.185082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trendelenburg P. Physiological and pharmacological investigations of small intestinal peristalsis. [Translation “Physiologische und pharmakologische Versuche über die Dünndarmperistaltik”. Arch Exp Pathol Pharmakol 81: 55–129, 1917]. Naunyn Schmiedebergs Arch Pharmacol 373: 101–133, 2006. doi: 10.1007/s00210-006-0052-7. [DOI] [PubMed] [Google Scholar]
- 84.Wan EC, Gordon TP, Jackson MW. Autoantibodies to calcium channels in type 1 diabetes mediate autonomic dysfunction by different mechanisms in colon and bladder and are neutralized by antiidiotypic antibodies. J Autoimmun 31: 66–72, 2008. doi: 10.1016/j.jaut.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 85.Wunsch M, Jabari S, Voussen B, Enders M, Srinivasan S, Cossais F, Wedel T, Boettner M, Schwarz A, Weyer L, Göcer O, Schroeter M, Maeurer M, Woenckhaus M, Pollok K, Radbruch H, Klotz L, Scholz CJ, Nickel J, Friebe A, Addicks K, Ergün S, Lehmann PV, Kuerten S. The enteric nervous system is a potential autoimmune target in multiple sclerosis. Acta Neuropathol 134: 281–295, 2017. doi: 10.1007/s00401-017-1742-6. [DOI] [PubMed] [Google Scholar]