

Abstract
Angiotensin II (ANG II) raises blood pressure partly by stimulating tubular Na+ reabsorption. The effects of ANG II on tubular Na+ transporters (i.e., channels, pumps, cotransporters, and exchangers) vary between short-term and long-term exposure. To better understand the physiological impact, we used a computational model of transport along the rat nephron to predict the effects of short- and long-term ANG II-induced transporter activation on Na+ and K+ reabsorption/secretion, and to compare measured and calculated excretion rates. Three days of ANG II infusion at 200 ng·kg−1·min−1 is nonpressor, yet stimulates transporter accumulation. The increase in abundance of Na+/H+ exchanger 3 (NHE3) or activated Na+-K+-2Cl− cotransporter-2 (NKCC2-P) predicted significant reductions in urinary Na+ excretion, yet there was no observed change in urine Na+. The lack of antinatriuresis, despite Na+ transporter accumulation, was supported by Li+ and creatinine clearance measurements, leading to the conclusion that 3-day nonpressor ANG II increases transporter abundance without proportional activation. Fourteen days of ANG II infusion at 400 ng·kg−1·min−1 raises blood pressure and increases Na+ transporter abundance along the distal nephron; proximal tubule and medullary loop transporters are decreased and urine Na+ and volume output are increased, evidence for pressure natriuresis. Simulations indicate that decreases in NHE3 and NKCC2-P contribute significantly to reducing Na+ reabsorption along the nephron and to pressure natriuresis. Our results also suggest that differential regulation of medullary (decrease) and cortical (increase) NKCC2-P is important to preserve K+ while minimizing Na+ retention during ANG II infusion. Lastly, our model indicates that accumulation of active Na+-Cl− cotransporter counteracts epithelial Na+ channel-induced urinary K+ loss.
Keywords: hypertension, kidney, mathematical model, pressure natriuresis
INTRODUCTION
Angiotensin II (ANG II), infused at a dose that does not acutely provoke a rise in blood pressure, eventually leads to hypertension. The chronic ANG II infusion model is commonly used to investigate essential hypertension in rodents, as it reproduces many aspects of human hypertension, such as renal injury (20). ANG II acts to elevate blood pressure in part by stimulating Na+ reabsorption in multiple nephron segments (4), but recent studies from our group and others have shown that the effects of ANG II on tubular Na+ transporters (i.e., channels, pumps, cotransporters, and exchangers) vary between short-term nonpressor ANG II and long-term exposure with accompanying hypertension.
An acute (20 min) infusion of ANG II elicits the redistribution of Na+ transporters in the proximal tubule (PT) and distal convoluted tubule (DCT), without changing overall expression (37, 39). A 3-day infusion of ANG II at nonpressor doses increases the abundance of both proximal and distal Na+ transporters (29). A 14-day infusion increases Na+ transport abundance and activity in the distal nephron [from the cortical thick ascending limb (cTAL) to the medullary collecting duct (CD)], but the resulting hypertension drives a pressure-natriuresis response in the PT and medullary loop (30), thereby overriding the effects of ANG II and lowering Na+ reabsorption, so as to match Na+ output to input. In other words, ANG II stimulates, whereas hypertension inhibits, tubular Na+ transport, and circulating fluid volume is maintained or restored owing to these counterbalancing effects. More specifically, we found that a 14-day infusion of ANG II in male rats increased the abundance of Na+-K+-2Cl− cotransporter-2 (NKCC2, and its activated form NKCC2-P) in the cTAL, Na+-Cl− cotransporter (NCC, and its activated form NCC-P) in the DCT, and epithelial Na+ channel (ENaC, and its activated cleaved forms), which is expressed in the convoluted tubule and throughout the CD. Conversely, the abundance of Na+/H+ exchanger 3 (NHE3) in the PT and that of medullary NKCC2 (mNKCC2) in the medullary thick ascending limb (mTAL) were decreased (30).
Changes in transporter abundance, however, do not necessarily imply parallel changes in fluxes, since transporter activity also depends on 1) the fraction of transporters in the plasma membrane versus intracellular membranes, 2) covalent modification of transporters, e.g., by phosphorylation or cleavage, and 3) load, that is, flow rates. In the present study, we used a computational model of transport along the rat nephron to predict the effects of short- and long-term ANG II-induced transporter activation to better understand the physiological impact of ANG II with and without hypertension. We assessed the specific contributions of individual transporters affected by ANG II, both at days 3 and 14 (D3 and D14, respectively), to the urinary excretion of Na+ (UNaV) and K+ (UKV).
METHODS
Clearance measurements.
Clearance of endogenous lithium, an indirect estimate of volume flow from the PTs (44), was calculated conventionally, as previously described (45). Analyzing the clearance of endogenous Li+ minimizes the natriuretic, diuretic, and other negative effects of exogenous infused Li+ (21) and is believed to come closer to direct measurements of PT reabsorption than other indirect methods (16). Calculation of the fraction of the filtered load of endogenous Li+ remaining in the tubular fluid indicates that the fraction is near constant after the pars recta, with minor reabsorption (<10%) along the TAL (44). Creatinine was determined by capillary electrophoresis in samples of urine and plasma at the George M. O’Brien Kidney Research Core Center (University of Texas Southwestern). Clearance of creatinine was also calculated conventionally, as previously described (45). Recognizing that there is significant creatinine secretion in the rat (6) and measuring no change in plasma creatinine levels after 3 days of ANG II treatment, creatinine clearance was used to evaluate the relative glomerular filtration rate (GFR).
Model description.
We assessed the impact of ANG II-induced changes in transporter expression using a published model of the superficial nephron of a male rat kidney (18). The model describes the transport of water and 15 solutes along the nephron. The underlying equations express conservation of mass and charge; they are solved at steady state to yield volume, concentrations, and electric potential in the lumen and in each cell type, as a function of position. In each nephron segment, paracellular and transcellular fluxes are computed based on the abundance and activity of the specific apical and basolateral transporters (i.e., channels, exchangers, cotransporters, and pumps) expressed therein. Activity is a function of kinetic constants and the net driving force, determined by the transmembrane gradients in concentration and electric potential. The parameters of the model relate to the structure/morphology of each tubule as well as the expression and kinetics of each transporter. GFR and solute concentrations in plasma and interstitium are specified at the outset.
Since the publication of the full nephron model (18), a few adjustments have been made in the model’s representation of the PT (7); these changes specifically affect phosphate transporter kinetics, the basal expression of NHE3, and the microvillous torque proportionality constant. We consider the impact of flow on the trafficking of PT transporters (via the microvillous torque), but not the flow dependence of transport in distal tubules, because it remains poorly characterized (51). The model now also includes the pH-mediated regulation of ENaC, of the renal outer medullary K+ channel (ROMK), and of the paracellular Cl− permeability in the connecting tubule (CNT) and CD, as described in Ref. 42.
Transporter abundance.
In the simulations below, we examined the impact of changes in transporter abundance on the reabsorption and secretion of Na+ and K+ in each segment and on their urinary excretion. Based on the measurements of Nguyen et al. (29, 30), we assumed the fractional increases or decreases in Na+ transporter abundance at D3 and D14 of ANG II infusion shown in Table 1. In the Nguyen et al. studies, transporter abundance was measured in crude homogenates. We assessed the linearity of the immunodetection system by running ½ and 1 amounts of each sample on each immunoblot and then determining whether the signal density increased by 2 ± 0.3-fold; if out of this range, protein loading was reevaluated and adjusted to meet this metric before abundance was determined (26).
Table 1.
ANG II-induced changes in Na+ transporter abundance
| D3 ANG II | D14 ANG II | |
|---|---|---|
| cNHE3 in proximal tubule | 1.40 | 0.78 |
| mNKCC2-P in medullary thick ascending limb | 1.04 | 0.83 |
| Na+-K+-ATPase in medullary thick ascending limb | 1.00 | 0.70 |
| cNKCC2-P in cortical thick ascending limb | 1.72 | 1.74 |
| NCC-P in distal convoluted tubule | 1.23 | 1.93 |
| ENaC in connecting tubule and collecting duct | 1.00 | 1.21 |
| Na+-K+-ATPase in connecting tubule and collecting duct | 1.00 | 1.00 |
Values are fold changes relative to control, noninfused rats, as measured by Nguyen et al. (29, 30), at 3 days (D3) and 14 days (D14) of angiotensin II (ANG II) treatment. cNHE3, cortical Na+/H+ exchanger 3; cNKCC2-P/mNKCC2-P, phosphorylated cortical/medullary Na+-K+-2Cl− cotransporter 2; ENaC: epithelial Na+ channel; NCC-P, phosphorylated Na+-Cl− cotransporter.
The transport activity of the cotransporters NCC and NKCC2 is activated by kinases, including STE20/SPS1-related proline/alanine-rich kinase (SPAK) and oxidative stress-responsive kinase (OSR1) (12, 35, 36). In fact, phosphorylated (active) NCC-P is localized exclusively to the plasma membrane, whereas unphosphorylated NCC is localized to both subapical endosomes (where it cannot transport) and the plasma membrane (where it is likely inactive) (19, 38). Proper NKCC2 phosphorylation (NKCC2-P) is likewise critical for trafficking to the apical membrane where it can transport (28). Thus we assumed that NKCC2-P and NCC-P are surrogates for active cotransporters in the plasma membrane, and we used the relative change in abundance of NKCC2-P and NCC-P, rather than total NKCC2 and NCC, in the simulations below. We did not consider ANG II-stimulated changes in intrinsic activity that are independent of changes in abundance or covalent modification (phosphorylation, cleavage).
At D14, the measured abundance of β-ENaC, full-length and cleaved α-ENaC, and full-length and cleaved γ-ENaC increased by varying degrees, ranging from 14 to 78% (30). We assumed a net increase of 21%, corresponding to one-half the average increase of 43%; had we assumed a 43% increase, the model would have predicted a decrease in UNaV, in contrast with experimental observations (30).
The abundance of Na+-K+-ATPase at D14 did not change in the renal cortex and decreased in the renal medulla (30). The measurement of cortical Na+-K+-ATPase is dominated by PT Na+-K+-ATPase, since this is the longest and most abundant renal tubule in the cortex. However, when measured in separated tubule segments, the abundance of α- and β-subunits is higher in the cTAL and DCT, and lower in the cortical CD, than in the PT (25). Thus, while we have no direct measurement of Na+-K+-ATPase abundance in individual cortical tubule segments in these data sets, we assumed it is unchanged. The measurement of medullary Na+-K+-ATPase is dominated by TAL Na+-K+-ATPase, since the abundance of α- and β-subunits in the medullary CD and proximal straight tubule is low by comparison (25). Thus we assumed that the observed decrease in medullary Na+-K+-ATPase abundance at D14 reflects a decrease in the mTAL specifically.
At D14, the measured urinary osmolarity was ~2.5 times lower than in control (noninfused) rats (30), which likely reflects greater ANG II-stimulated thirst, water intake, and subsequent suppression of vasopressin secretion. To account for these observations, at D14, the interstitial concentration of urea at the papillary tip was reduced from 500 to 200 mM, and inner medullary CD (IMCD) permeabilities were adjusted as follows. Sands et al. (40) found that arginine vasopressin (AVP) raised the permeability of terminal IMCD to water (Pf) from 70 to 186 μm/s and that of urea (Purea) from 17 × 10−5 to 69 × 10−5 cm/s. Assuming that the tight junction accounts for 30% of the unstimulated permeability (50), we estimated the AVP-induced increase in Pf and Purea as:
Thus, in the D14 simulations below, the apical permeability to water and urea in the IMCD was decreased by a factor of 3.4 and 5.4, respectively.
Other model assumptions.
The single-nephron GFR was taken to be the same (30 nL/min) in control rats and in rats at D3 (see below) and at D14 (47). Following the approach of Weinstein (48, 49), the model accounts for the coalescence of tubules below the DCT, and results are scaled assuming a total of 36,000 nephrons.
The computer code, data, analytic methods, and study materials are available from authors on reasonable request.
RESULTS
Variations in tubular transport during short-term nonpressor ANG II infusion.
The predicted changes in Na+ and K+ reabsorption (or secretion) on D3 of ANG II infusion (200 ng·kg−1·min−1), corresponding to the fractional increases in transporter abundance listed in Table 1, are shown in Fig. 1. The 40% increase in cortical NHE3 (cNHE3) abundance is predicted to raise Na+ reabsorption Na+ (TNa) in the PT (relative to control, noninfused rats), and thus K+ reabsorption as well. In the mTAL, cTAL, and DCT, the predicted TNa and K+ reabsorption (TK) do not change much, even though Na+ transporters are upregulated in these segments, because of the counteracting effects of reduced delivery (i.e., diminished load). In the CNT and CD, reduced delivery is not counterbalanced by increases in Na+ transporter abundance, and TNa is predicted to decrease. The reduced activity of ENaC diminishes the driving force for K+ secretion in the CNT and conversely raises K+ reabsorption in the cortical CD.
Fig. 1.

Predicted transepithelial Na+ (TNa; A) and K+ (TK; B) fluxes across nephron segments in control rats and in rats at 3 days (D3) of angiotensin II treatment, with and without observed changes in cortical Na+/H+ exchanger 3 (cNHE3) or activated Na+-K+-2Cl− cotransporter-2 (NKCC2-P) abundance. Positive values indicate reabsorption; negative values indicate secretion. The last set of columns (“urine”) represents urinary excretion. CCD, cortical collecting duct; CNT, connecting tubule; DCT, distal convoluted tubule; mTAL/cTAL, medullary/cortical thick ascending limb; OMCD/IMCD, outer/inner medullary collecting duct; PT, proximal tubule.
The predicted impact of changes in delivery to the TAL is illustrated in Fig. 2. In these simulations, the load entering the TAL was varied by increasing or decreasing cNHE3 abundance in the PT, while the abundance of mNKCC2 and cortical NKCC2 (cNKCC2) was maintained at control values so as to isolate the effects of changes in delivery. The model predicts that Na+ reabsorption in the TAL decreases with decreasing load, but the relative changes in transport are lower than the relative changes in delivery, as observed experimentally (33). Moreover, the model predicts a weaker dependence of TNa on delivery from the PT in the mTAL than in the cTAL (Fig. 2). Indeed, since Na+ and water are reabsorbed in tandem isosmotically in the PT, varying cNHE3 expression has a small effect on luminal Na+ concentration at the mTAL entrance; thus transport across mNKCC2, the activity of which is modulated by luminal concentrations, does not vary much. However, along the mTAL, which lacks apical water channels, the reabsorption of Na+ is not paralleled by that of water, so changes in delivery have a larger impact on luminal Na+ concentration at the cTAL entrance and, therefore, on the activity of cNKCC2.
Fig. 2.
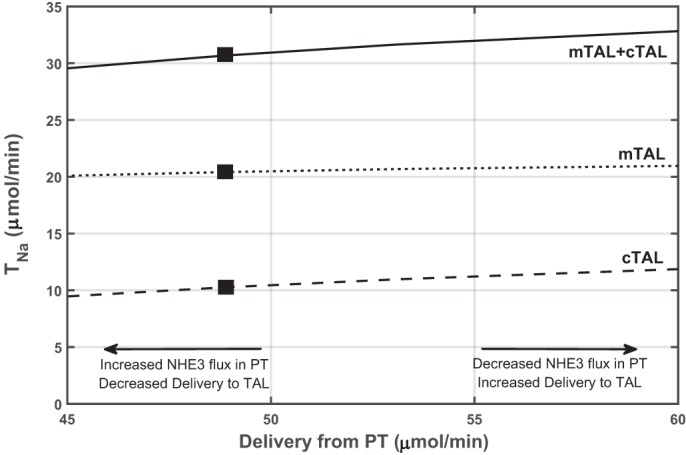
Predicted transepithelial Na+ flux (TNa) across the medullary thick ascending limb (mTAL) and cortical thick ascending limb (cTAL) as a function of Na+ delivery from the proximal tubule (PT). The latter was increased or decreased by varying cortical Na+/H+ exchanger 3 (cNHE3) expression in the PT. The expression of other transporters was maintained constant. Closed rectangles denote base case values. Reabsorption in the TAL is predicted to vary in parallel with delivery, but in lower proportion.
Given the overall transporter pattern, UNaV and UKV at D3 is predicted to decrease by 70 and 60%, respectively, relative to control rats (Table 2). However, no change in UNaV and UKV were observed in vivo in the experimental study (29). This discrepancy suggests that TNa does not increase as much in the PT as would be expected from a 40% increase in cNHE3 abundance. To test this, we measured the clearance of Li+, a measure of volume flow from the PT, and the clearance of creatinine, a surrogate for GFR, at D3 (Fig. 3). The Li+ clearance results indicate that volume flow from the PT did not decrease significantly (P = 0.2), signifying that TNa did not increase in the PT in parallel with the increase in cNHE3 abundance at D3. Moreover, the creatinine clearance measurements do not suggest significant changes in GFR. A prior study showed that the distribution of NHE3 in the brush border may give rise to local pH microdomains that regulate its activity (3), and it is possible that increased cNHE3 abundance favors the development of unfavorable pH gradients that limit the activity of the exchanger.
Table 2.
Predicted urinary excretion of Na+ and K+
| UNaV, μmol/min | FENa, % | UKV, μmol/min | FEK, % | |
|---|---|---|---|---|
| Control (noninfused) rats | 1.44 | 0.92 | 2.57 | 48.5 |
| 3 Days of nonpressor ANG II infusion (D3) | ||||
| D3 original | 0.43 | 0.28 | 1.03 | 19.4 |
| D3 with no upregulation of cNHE3 | 0.99 | 0.64 | 2.12 | 40.0 |
| D3 with no upregulation of NKCC2-P | 0.56 | 0.36 | 1.36 | 25.6 |
| D3 with no upregulation of NCC-P | 0.47 | 0.30 | 1.14 | 21.5 |
| D3 with upregulation of cNHE3 only | 0.61 | 0.40 | 1.47 | 27.8 |
| D3 with upregulation of NKCC2-P only | 1.08 | 0.70 | 2.22 | 42.0 |
| D3 with upregulation of NCC-P only | 1.32 | 0.85 | 2.47 | 46.7 |
| 14 Days of pressor ANG II infusion (D14) | ||||
| D14 original | 2.33 | 1.50 | 3.64 | 68.8 |
| D14 with no downregulation of cNHE3 and mNKCC2-P | 0.74 | 0.47 | 2.00 | 37.8 |
| D14 with no downregulation of cNHE3 | 1.13 | 0.73 | 2.62 | 49.5 |
| D14 with no downregulation of mNKCC2-P | 1.55 | 1.00 | 3.13 | 59.1 |
| D14 with no upregulation of cNKCC2-P, NCC-P and ENaC | 3.99 | 2.57 | 3.91 | 73.8 |
| D14 with no upregulation of NCC-P | 2.84 | 1.82 | 3.87 | 73.1 |
| D14 with no upregulation of NKCC2-P | 2.95 | 1.90 | 3.94 | 74.5 |
| D14 with no upregulation of ENaC | 2.75 | 1.77 | 3.45 | 65.1 |
| D14 with upregulation of ENaC only | 3.51 | 2.26 | 4.14 | 78.2 |
| D14 with upregulation of Na+-K+-ATPase in the CNT and CD | 1.73 | 1.11 | 3.91 | 73.9 |
| D14 with upregulation of mNKCC2-P | 1.28 | 0.82 | 2.87 | 54.3 |
| D14 with downregulation of cNKCC2-P | 3.36 | 2.16 | 4.11 | 77.7 |
The filtered loads of Na+ and K+ are, respectively, 155.5 and 5.29 μmol/min. ANG II, angiotensin II; cNHE3, cortical Na+/H+ exchanger 3; cNKCC2-P/mNKCC2-P, phosphorylated cortical/medullary Na+-K+-2Cl− cotransporter 2; ENaC, epithelial Na+ channel; FENa and FEK, fractional urinary excretion of Na+ and K+, respectively; NCC-P, phosphorylated Na+-Cl− cotransporter; NKCC2-P, phosphorylated Na+-K+-2Cl− cotransporter 2; UNaV and UKV, urinary excretion of Na+ and K+, respectively.
Fig. 3.
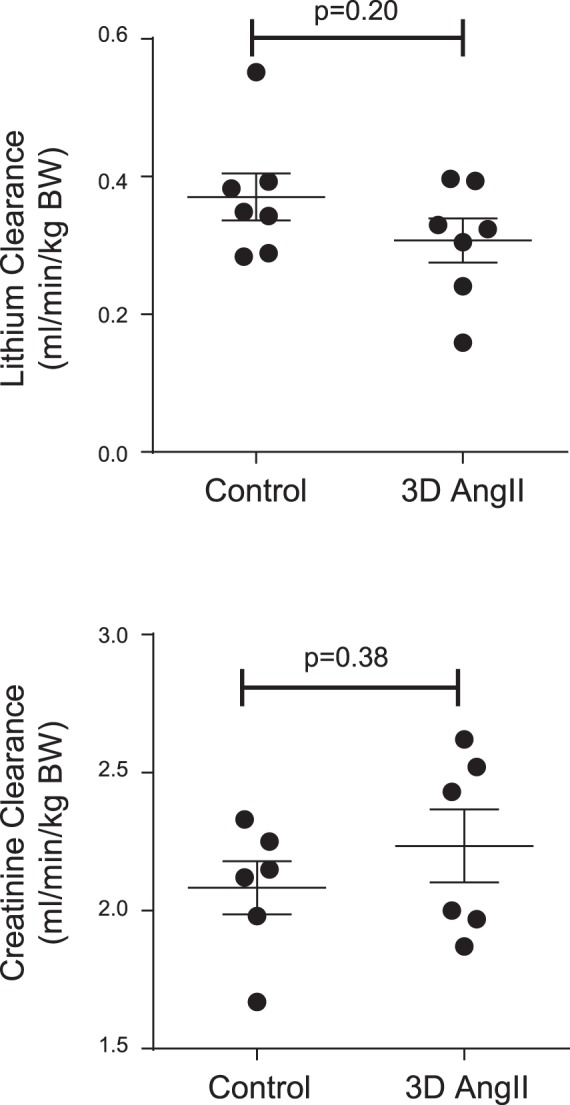
Lithium clearance, a measure of volume flow from the proximal tubule (top), and creatinine clearance, an estimate of glomerular filtration rate (bottom), measured conventionally (as urine concentration times overnight urine volume divided by plasma concentration) in control rats and in rats infused for 3 days (D3) with a nonpressor dose of angiotensin II (ANG II). Results were assessed by unpaired t test using GraphPad prism. BW, body weight.
Assuming no change in cNHE3 activity, the computed UNaV and UKV are, respectively, 31 and 17% below control levels, as shown in Fig. 1. In this case, the model predicts enhanced Na+ reabsorption predominantly in the cTAL, owing to the 72% increase in cortical NKCC2-P (cNKCC2-P) abundance, and resulting in reduced delivery downstream. However, this scenario is unlikely, since the absence of changes in creatinine clearance (which reflects the GFR) suggests that Na+ delivery to the macula densa remained unaffected at D3, despite increased cNKCC2-P abundance. In the absence of changes in cNHE3 and cNKCC2-P activity, that is, considering only a 23% increase in NCC-P abundance, the predicted UNaV and UKV at D3 are <10% lower than in control rats (Table 2), in agreement with experimental observations (29). Taken together, these results suggest that, at 3 days, even though ANG II stimulates the accumulation of Na+ transporters, there is not proportional activation of Na+ transport, as further discussed below.
Variations in tubular transport during chronic ANG II infusion.
On D14 of ANG II infusion (400 ng·kg−1·min−1), TNa is predicted to decrease (relative to control, noninfused rats) in the PT and mTAL and to increase in the segments downstream up to the cortical CD (Fig. 4), in tandem with the changes in Na+ transporter abundance. In parallel, TK fluxes are reduced in the proximal nephron and augmented in the DCT and CNT. Relative to control (noninfused) rats, UNaV and UKV at D14 are predicted to increase by 62 and 42%, respectively (Table 2); in the experimental study, UNaV and UKV at D14 were 67 and 21%, respectively, higher than in control rats (30). Overall, these results confirm that proximal Na+ transport inhibition counteracts ANG II-induced distal stimulation of Na+ reabsorption. In the simulations below, we sought to examine the specific contribution of each Na+ transporter to these regulatory mechanisms.
Fig. 4.

Predicted transepithelial Na+ (TNa; A) and K+ (TK; B) fluxes in control rats and in rats at 14 days (D14) of angiotensin II treatment, with and without the observed downregulation of cortical Na+/H+ exchanger 3 (cNHE3) or activated medullary Na+-K+-2Cl− cotransporter-2 (mNKCC2-P). CCD, cortical collecting duct; CNT, connecting tubule; DCT, distal convoluted tubule; mTAL/cTAL, medullary/cortical thick ascending limb; OMCD/IMCD, outer/inner medullary collecting duct; PT, proximal tubule.
Coordinated regulation.
As discussed above, we assumed no change in Na+-K+-ATPase abundance in the CNT and CD. Other studies have found evidence for the coordinated regulation of apical and basolateral Na+ transporters (9, 41, 53). According to our model, if the abundance of Na+-K+-ATPase in the CNT and CD increased to the same extent as that of ENaC at D14, Na+ reabsorption and K+ secretion would each rise more in the CNT. However, due to diminished Na+ delivery downstream, TNa would slightly decrease in the CD; conversely, due to elevated K+ delivery, K+ reabsorption would increase in the CD (results not shown). Overall, relative to the original D14 scenario, UNaV would be 25% lower and UKV would be 7% higher (Table 2); relative to control rats, the predicted UNaV and UKV would be 20 and 52% higher, respectively. In other words, assuming identical increases in Na+-K+-ATPase and ENaC abundance yields poor agreement with the measured changes in urinary excretion rates at D14.
Note that the activity of Na+-K+-ATPase is regulated in part by the intracellular concentration of Na+ ([Na+]i), for which the model accounts. This in itself provides a mechanism to coordinate transporter activity at the apical and basolateral membranes: an increase in apical Na+ entry will raise [Na+]i, which will in turn stimulate basolateral Na+ efflux across the pump (9).
Proximal inhibition.
Next, we evaluated the impact of the depression in proximal NHE3-mediated transport by assuming no decrease in cNHE3 abundance at D14. The predicted UNaV was then reduced by 51% (relative to the original D14), and UKV by 28% (Table 2). Stated differently, if cNHE3 was not downregulated, urinary Na+ excretion at D14 would be reduced by 21% (instead of raised by 62% as observed experimentally) relative to control rats, thus underlining the essential role of NHE3 inhibition in counterbalancing the effects of ANG II (Fig. 4). Similarly, assuming no decrease in medullary NKCC2-P (mNKCC2-P) abundance at D14, urinary Na+ excretion at D14 would increase by only 8% above that in control rats (Table 2), demonstrating that mNKCC2-P inhibition also contributes significantly to reducing Na+ reabsorption along the nephron.
Differential regulation of mNKCC2 and cNKCC2.
Pressure-natriuresis mechanisms appear to override the effects of ANG II only in the PT and mTAL. At D14, the abundance of cNKCC2-P is augmented, whereas that of mNKCC2-P is reduced (30). To assess the impact of this differential regulation, we performed simulations in which NKCC2-P abundance was varied to the same extent in the mTAL and cTAL. If cNKCC2-P abundance were reduced by 17% at D14 (as is mNKCC2-P abundance), TNa would decrease in the cTAL. The elevated load would raise TNa in all downstream segments (Fig. 5). Despite these compensatory antinatriuretic effects, UNaV and UKV would be 44 and 13% higher, respectively, relative to the original D14 (Table 2). In other words, cNKCC2-P upregulation contributes significantly to Na+ and K+ retention during chronic ANG II infusion.
Fig. 5.

Predicted transepithelial Na+ (TNa; A) and K+ (TK; B) fluxes in control rats and in rats at 14 days (D14) of angiotensin II treatment, assuming that activated cortical Na+-K+-2Cl− cotransporter-2 (cNKCC2-P) is downregulated instead of upregulated at D14, and assuming that activated medullary NKCC2-P (mNKCC2-P) is upregulated instead of downregulated at D14. CCD, cortical collecting duct; CNT, connecting tubule; DCT, distal convoluted tubule; mTAL/cTAL, medullary/cortical thick ascending limb; OMCD/IMCD, outer/inner medullary collecting duct; PT, proximal tubule.
Conversely, if mNKCC2-P abundance were augmented by 74% at D14 (as is cNKCC2-P abundance), TNa would greatly increase in the mTAL (Fig. 5), and, due to reduced delivery, TNa would then decrease downstream. Nevertheless, overall, Na+ reabsorption would be greater. In addition, owing to diminished Na+ delivery to the CNT, K+ secretion would decrease therein, and overall K+ reabsorption would increase. Relative to the original D14, UNaV and UKV would be 45 and 21% lower, respectively (Table 2). In fact, urinary Na+ excretion would be 11% below that in control rats (Fig. 5). These simulations support the importance of both the depression in mNKCC2-P and the reciprocal stimulation of cNKCC2-P to the ANG II-hypertension response.
Upregulation of NCC.
A previous study from our group has suggested that the increase in NCC abundance and phosphorylation at D14 is secondary to urinary K+ loss driven by ENaC stimulation (46). To test this hypothesis, we performed simulations assuming no NCC-P upregulation in the DCT. As shown in Fig. 6, Na+ delivery downstream would be increased, K+ secretion in the CNT would rise, and K+ reabsorption in the CD would decrease. Fractional urinary K+ excretion would increase from 69% (D14 with NCC-P upregulation) to 73% (D14 without NCC-P upregulation) versus 49% in control rats (Table 2). In other words, the model predicts that NCC-P upregulation acts to mitigate urinary K+ loss, in addition to enhancing Na+ retention. Interestingly, our results also suggest that the upregulation of cNKCC2-P impacts urinary K+ loss similarly to that of NCC-P (Fig. 6). If cNKCC2-P abundance were not increased at D14, fractional urinary K+ excretion would rise from 49 to 74% (Table 2).
Fig. 6.

Predicted transepithelial Na+ (TNa; A) and K+ (TK; B) fluxes in control rats and in rats at 14 days (D14) of angiotensin II treatment, with and without the observed upregulation of activated cortical Na+-K+-2Cl− cotransporter-2 (cNKCC2-P), phosphorylated Na+-Cl− cotransporter (NCC-P), or epithelial Na+ channel (ENaC). CCD, cortical collecting duct; CNT, connecting tubule; DCT, distal convoluted tubule; mTAL/cTAL, medullary/cortical thick ascending limb; OMCD/IMCD, outer/inner medullary collecting duct; PT, proximal tubule.
Upregulation of ENaC.
To assess the specific impact of changes in ENaC abundance in the CNT and CD, we assumed no increase at D14. Without ENaC upregulation, TNa would decrease in the CNT and cortical CD, as would net K+ secretion, relative to the original D14. However, Na+ reabsorption would increase in the IMCD, given the higher load (Fig. 6). As a net result, if ENaC were not upregulated, UNaV would be 18% higher, and UKV would be 5% lower, relative to the original D14 (Table 2). Thus a 21% increase in ENaC abundance is sufficient to significantly impact renal Na+ handling.
Conversely, if ENaC were the only apical Na+ transporter upregulated by ANG II (that is, in the absence of cNKCC2-P and NCC-P upregulation), Na+ reabsorption would increase more in the CNT and CD (relative to the original D14), owing to enhanced delivery, and net K+ secretion would also increase in these segments. Nevertheless, overall Na+ and K+ reabsorption would be diminished: UNaV and UKV would be 50 and 14% higher, respectively, relative to the original D14 (Table 2).
DISCUSSION
Previously, we reported changes in Na+ transporter abundance, as well as UNaV and UKV, during short-term nonpressor 3-day (D3) ANG II infusion (29) and 14-day (D14) ANG II infusion, with accompanying hypertension and pressure natriuresis (30). The objective of the present study was to gain more insight into the physiological impact of the measured changes in renal transporter abundance induced by ANG II. For this purpose, we used a computational model of water and solute transport along the nephron to quantitatively assess the effects of these changes on Na+ and K+ reabsorption, secretion, and urinary excretion. The overall impact of transporter regulation is difficult to gauge without a mathematical model for several reasons. First, changes in transporter abundance affect concentrations, driving forces, the coupling between water and solutes, as well as delivery downstream. As described in results, accumulation of a given transporter may not translate into a higher flux if the delivered load is concomitantly reduced; that is, changes in transporter abundance may be “overridden” by changes in delivery. Second, transporter pools can accumulate both in the plasma membrane (in both active and inactive configurations) and in subapical membrane pools (inactive).
Note that the model is based on a population of identical, superficial nephrons that represent two-thirds of total nephrons in the rat (2). Transport rates vary along juxtamedullary nephrons, depending on their length (17, 51). In addition, since the model does not explicitly represent the renal vasculature, which contributes to the cortico-medullary gradient, interstitial concentrations are taken to be fixed. It is likely that changes in the abundance and/or activity of medullary transporters (such as mNKCC2-P and ENaC) affect the interstitial concentration gradient. Note too that we did not assess the impact of transporters other than Na+ transporters. In more recent studies, we did not detect ANG II-induced regulation of ROMK abundance, even though we know that this K+ secretory pathway is regulated by trafficking between the plasma membrane and subapical pools (22).
Short-term nonpressor ANG II infusion.
Combining our experimental and theoretical results leads to the conclusion that, at 3 days, ANG II infusion stimulates the accumulation of Na+ transporters without proportional activation. Moreover, this pattern of accumulation without activation appears to occur along the entire nephron. First, even though there is significant accumulation of each of the three full-length ENaC subunits at D3, the cleaved forms that track channel activation increase much less (29); thus we assumed no change in the abundance of active plasma membrane-associated ENaC at D3 in our model.
Second, if we assume that transport capacity varies in direct proportion with transporter abundance, our model predicts that the measured changes in the abundance of cNHE3, cNKCC2-P, and NCC-P at D3 should lower UNaV by 70% and UKV by 60%, relative to control, noninfused rats (Table 2), whereas in vivo UNaV and UKV at D3 remained unchanged (29). We also performed simulations assuming that only subsets of transporters were activated: the measured changes in abundance of cNHE3 or NKCC2-P predicted significant reductions in UNaV (by 57 or 25%, respectively; Table 2), which conflicts with experimental observations. That there is no enhanced Na+ reabsorption across either of these two transporters, despite their accumulation, was supported by the absence of variations in the clearance of Li+ and creatinine (Fig. 3).
The activity of NHE3 is regulated by its distribution along the PT apical membrane microvilli. In fact, there are no discernible subapical pools of NHE3 in the PT (55). A rise in blood pressure induces the translocation of NHE3 within the plane of the membrane to the base of the microvilli, where its activity is diminished (55). To explain this, Brasen et al. (3) showed that this clustering of NHE3 gives rise to local pH microdomains that are predicted to significantly alter the driving force across the exchanger.1 We did not explore the precise localization of NHE3 during the nonpressor 3-day ANG II treatment by immunohistochemistry. NHE3 phosphorylated at serine-552 is associated with NHE3 localization at the base of the microvilli where it is less active (14, 15). After 3-day ANG II treatment, the abundance of NHE3pS552 was measured by immunoblot at 1.18 ± 0.09 versus control = 1 (P = 0.09 by unpaired t test), which may indicate a propensity for NHE3 to cluster at the base of the microvilli at this time point.
The results of the 3-day ANG II simulations indicate that transporter abundance is not always a proxy for transporter flux. Activity per transporter can be modulated by local signals (nitric oxide, 20-HETE, dopamine, superoxide) that can override changes in transporter pool size (5, 11), for example, by stimulating trafficking into and out of active domains in the plasma membrane (24). Lack of correlation between overall abundance and transport capacity is also evident in ENaC activation mediated by subunit cleavage, which is associated with apparent decreases in pool size of full-length subunits with the appearance of pools of smaller subunits (43).
The abundance of cNKCC2 and that of its phosphorylated form were both increased by ~50% at D3, consistent with increased TNa in the cTAL. Such an increase is predicted to signal through the macula densa to reduce GFR. However, we did not detect a significant change in creatinine clearance, a surrogate marker for GFR. This evidence may be insufficient to conclude that NKCC2 activity is not elevated in the cTAL: we do not know if ANG II regulates NKCC2 similarly in cTAL cells and in macula densa cells, and we do not know whether or how tubuloglomerular feedback is altered after 3 days of ANG II infusion. Stimulation of NKCC2-P alone is predicted to reduce UNaV and UKV by 25 and 13% (Table 2), leading us to speculate that NKCC2-P activation must be less than that indicated from the change in its abundance. NKCC2 is known to undergo robust constitutive recycling, so the change in abundance of NKCC2-P in the apical membrane may be significantly less than suggested by the 72% increase in total NKCC2-P abundance. In contrast to NKCC2, it is possible that NCC is in fact activated in proportion to the measured 23% change in NCC-P abundance: in isolation, this change is predicted to reduce UNaV and UKV by 8 and 4%, respectively (Table 2); such small changes are within measurement errors. Thus we cannot exclude NCC activation at D3.
Chronic ANG II infusion.
NHE3 abundance is increased at 3 days of ANG II infusion and suppressed at 14 days of ANG II infusion compared with baseline levels. Can this be attributed to the previously reported biphasic effect of ANG II on NHE3 in the PT? Evidence suggests this is quite unlikely, since the concentration at which NHE3 inhibition has been observed is 10−7 M or above (13), whereas the concentration measured in the tubular fluid during ANG II hypertension (rats infused with 300 ng·kg−1·min−1 for 13 days) is 1,000-fold less, in the picomolar range (47).
Our modeling results confirm that the changes in transporter abundance seen at D14 favor decreased Na+ reabsorption in the proximal nephron and enhanced reabsorption in the distal nephron (Fig. 4). ANG II-induced stimulation of transporters from the cTAL to the medullary CD is counteracted by the inhibition of transporters in the PT and mTAL. Specifically, the model predicts that, without the latter inhibition, UNaV and UKV at D14 would be, respectively, 49 and 22% lower than in control (noninfused) rats (Table 2), instead of 62 and 42% higher, as measured in vivo (30). As expected, the 22% decrease in cNHE3 abundance contributes more to raising urinary Na+ excretion than the 17% decrease in mNKCC2-P abundance, but their effects are not additive, given that changes in delivery also impact transport (Table 2, Fig. 2).
Conversely, with no ANG II-induced upregulation of cNKCC2-P, NCC-P, and ENaC, the predicted UNaV and UKV at D14 would be, respectively, 178 and 52% higher than in control (noninfused) rats and 71 and 7% higher than in the original D14 scenario (Table 2). This simulation demonstrates the impact of depressing proximal NHE3 and mNKCC2, the pressure-natriuresis response, in the absence of distal activation. Our simulations also suggest that cNKCC2, NCC, and ENaC each contribute to Na+ retention to a similar extent (Fig. 6). However, whereas upregulation of cNKCC2-P and NCC-P acts to enhance K+ reabsorption, upregulation of ENaC favors K+ secretion. Indeed, our model supports the hypothesis that NCC-P accumulation serves to counteract ENaC-induced urinary K+ loss (46). According to our simulations, fractional urinary K+ excretion would be 4% higher if NCC-P abundance were not increased (Table 2). The present results provide strong complementary support for the idea that the rise in NCC abundance is driven by K+ loss, not by ANG II alone (Fig. 7), as discussed below.
Fig. 7.

Schematic representation of the impact of angiotensin II (ANG II) on Na+ and K+ transport in the distal nephron. The proposed chain of events is illustrated, connecting direct ANG II activation of epithelial Na+ channel (ENaC) (1) with indirect activation of Na+-Cl− cotransporter (NCC) phosphorylation mediated by coupling of Na+ reabsorption (2) with K+ secretion (3) in the connecting tubule (CNT) and cortical collecting duct (CCD) driving urinary K+ loss (4), which activates NCC via phosphorylation (NCC-P) (5) to minimize Na+ delivery to ENaC to blunt further K+ loss. AT1R, ANG II type 1 receptor; BK, large-conductance Ca2+-activated K+ channel; DCT, distal convoluted tubule; ROMK, renal outer medullary K+ channel.
Is the increase in NCC abundance and phosphorylation due to direct stimulation by ANG II and aldosterone or secondary to the ANG II and aldosterone stimulation of ENaC known to drive K+ secretion and loss? DCT cells are K+ sensors that increase or decrease NCC activation in the same sensor cells in response to reduced or elevated extracellular K+ concentration, respectively (27). Additionally, ANG II increases aldosterone production, and aldosterone and ANG II can independently activate ENaC (56), which will drive K+ secretion and loss. Our laboratory recently examined whether the activation of NCC observed during 14-day ANG II infusion was a primary effect of ANG II and/or aldosterone or secondary to K+ deficiency, by doubling K+ intake during ANG II infusion. In shams without ANG II infusion, this mild supplementation did not change NCC abundance or phosphorylation, but it prevented significant activation of NCC during ANG II infusion (46), supporting the conclusion that accumulation of NCC-P was secondary to ANG II- and aldosterone-induced ENaC activation (>1.5-fold increase in the abundance of cleaved subunits) driving K+ loss. The signals driving the suppression of NHE3 and NKCC2 are not viewed as secondary to K+ loss, as hypokalemia either does not change (31) or increases (8) NHE3 abundance, whereas elevated plasma K+ concentration suppresses these transporters (51). Is ENaC activation also responsible for NCC activation at 3-day ANG II infusion? At this earlier time point, cleavage of ENaC α- and γ-subunits did not reach statistical significance, whereas NCC abundance was increased by 23% (Table 1). Thus NCC activation at this early time point may reflect a direct impact of ANG II and aldosterone.
Interestingly, the model predicts that the measured increase in cNKCC2-P abundance reduces urinary K+ excretion to a greater extent than the measured increase in NCC-P abundance (Fig. 6), suggesting that cNKCC2-P accumulation also plays a key role in preserving K+ homeostasis during chronic ANG II infusion. However, doubling dietary K+ during 14-day ANG II infusion did not alter cNKCC2-P accumulation; thus this response is more likely driven by ANG II than by plasma K+ concentration (46). Our findings can be viewed as the “mirror image” of those of Yang et al. (54), who assessed the impact of transporter downregulation in mice with increased dietary K+ intake. The observed 80% decrease in NCC-P abundance was predicted to have a smaller effect on K+ excretion than the observed 50% decrease in NKCC2-P abundance; in fact, the predominant effect on UKV stemmed from the 40% decrease in NHE3 abundance (54). Taken together, these studies provide evidence that K+ excretion is controlled all along the nephron from the PT and loop of Henle, which impacts TK and K+ delivery downstream, to the K+-sensing DCT that regulates NCC activity, and the CNT and CD where ENaC activity drives K+ secretion (Fig. 7).
The differential regulation of mNKCC2-P and cNKCC2-P at D14, observed in both rats (30) and mice (unpublished results), appears essential to maintain Na+ homeostasis during chronic ANG II infusion. Note that, while the nonpressor ANG II at D3 increased cNKCC2-P 1.72-fold, it did not change mNKCC2-P, so we provide no evidence for ANG II stimulation of mNKCC2 at either time point. According to our simulations, if the abundance of mNKCC2-P were increased to the same extent as that of cNKCC2-P at D14, Na+ reabsorption would increase to the point where urinary Na+ excretion would be lower (instead of higher) than in control rats. Conversely, if the abundance of cNKCC2-P were decreased to the same extent as that of mNKCC2-P, both UNaV and UKV would rise significantly more (Table 2). Altogether, our results suggest that the differential regulation of mNKCC2 and cNKCC2 is important to preserve K+ while minimizing Na+ retention. The molecular mechanisms responsible for ANG II-induced stimulation of NKCC2 in the cortex but not in the medulla remain to be elucidated, but one clue comes from the cotransporter activating kinase SPAK, which, like NKCC2, is differentially suppressed in the medulla and activated in the cortex at D14 (30). Cowley (5) summarized the many studies indicating that depression of mTAL Na+ transport plays a key role in driving pressure natriuresis (as evident during ANG II hypertension), specifically mediated by changes in medullary blood flow driven by renal interstitial hydrostatic pressure and in medullary redox state influenced by nitric oxide production. The regulation of NKCC2 along the TAL was recently reviewed by Gonzalez-Vicente et al. (11), who concluded that the effects of ANG II on Na+ transport in this region, including the mediating signaling cascades, are not well understood.
Our results also emphasize the essential role of ENaC in promoting Na+ retention (Fig. 6). ANG II stimulation of ENaC channel activity has been demonstrated directly in rats, both acutely and chronically (23, 34). Using ENaC abundance as a surrogate measure of ENaC activity is problematic because the three subunits can accumulate in inactive pools. However, the change in the cleaved (activated) forms of both α- and γ-subunits, as well as increased glycosylation of the β-subunit, provides evidence for activation. Interestingly, the abundance of the cleaved ENaC subunits (α, γ) was not significantly increased at D3; thus we interpret the increased abundance of the full-length subunits as evidence for accumulation of inactive ENaC (29). In contrast, at D14, the cleaved forms of α- and γ-subunits increased >50% (30). To assess the impact of ENaC in this simulation, we averaged the increase in the three subunits and their cleaved forms and divided by 2 to account for inactive pools. In our interpretation, ENaC stimulation drives K+ secretion, urinary K+ loss, and subsequent stimulation of NCC (Fig. 7). In subsequent studies, we did detect hypokalemia at D14, but it was only evident after overnight fast (46).
In future studies, the model should be expanded to take into account signaling cascades that modulate transporter activity via phosphorylation, trafficking, and protein-protein interactions. NKCC2 is phosphorylated by kinases such as PKA, SPAK, and OSR1, which mediate the response to hormones and cyclic nucleotides such as cAMP and cGMP (1, 10, 32). When sufficiently (and quantitatively) characterized, such regulatory pathways could be incorporated in the model.
In conclusion, our results indicate that, at 3 days, ANG II stimulates the accumulation of transporters without proportional activation. At 14 days, ANG II stimulates ENaC to promote Na+ retention. The concomitant increase in K+ secretion drives accumulation of NCC-P and possibly that of cNKCC2-P, so as to reduce K+ loss. The parallel decrease in cNHE3 and mNCCK2-P abundance acts to counteract the increase in distal Na+ reabsorption.
GRANTS
This work was supported by the NIH via National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-083785 (to A. A. McDonough).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.E. and A.A.M. conceived and designed research; A.E. and A.A.M. performed experiments; A.E. and A.A.M. analyzed data; A.E. and A.A.M. interpreted results of experiments; A.E. and A.A.M. prepared figures; A.E. and A.A.M. drafted manuscript; A.E. and A.A.M. edited and revised manuscript; A.E. and A.A.M. approved final version of manuscript.
Footnotes
Note that the spatiotemporal model developed by Brasen et al. (3) describes a single microvillus and the underlying part of the cell, and concentration profiles within that domain are obtained by solving the diffusion-reaction equation at each mesh point. Such a local model cannot be readily expanded to the entire cell, let alone to a full nephron segment.
REFERENCES
- 1.Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol Renal Physiol 301: F1143–F1159, 2011. doi: 10.1152/ajprenal.00396.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bankir L, de Rouffignac C. Urinary concentrating ability: insights from comparative anatomy. Am J Physiol Regul Integr Comp Physiol 249: R643–R666, 1985. doi: 10.1152/ajpregu.1985.249.6.R643. [DOI] [PubMed] [Google Scholar]
- 3.Brasen JC, Burford JL, McDonough AA, Holstein-Rathlou N-H, Peti-Peterdi J. Local pH domains regulate NHE3-mediated Na+ reabsorption in the renal proximal tubule. Am J Physiol Renal Physiol 307: F1249–F1262, 2014. doi: 10.1152/ajprenal.00174.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burns KD, Li N. The role of angiotensin II-stimulated renal tubular transport in hypertension. Curr Hypertens Rep 5: 165–171, 2003. doi: 10.1007/s11906-003-0074-1. [DOI] [PubMed] [Google Scholar]
- 5.Cowley AW., Jr Renal medullary oxidative stress, pressure-natriuresis, and hypertension. Hypertension 52: 777–786, 2008. doi: 10.1161/HYPERTENSIONAHA.107.092858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darling IM, Morris ME. Evaluation of “true” creatinine clearance in rats reveals extensive renal secretion. Pharm Res 8: 1318–1322, 1991. doi: 10.1023/A:1015820316660. [DOI] [PubMed] [Google Scholar]
- 7.Edwards A, Bonny O. A model of calcium transport and regulation in the proximal tubule. Am J Physiol Renal Physiol 315: F942–F953, 2018. doi: 10.1152/ajprenal.00129.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elkjaer ML, Kwon TH, Wang W, Nielsen J, Knepper MA, Frøkiaer J, Nielsen S. Altered expression of renal NHE3, TSC, BSC-1, and ENaC subunits in potassium-depleted rats. Am J Physiol Renal Physiol 283: F1376–F1388, 2002. doi: 10.1152/ajprenal.00186.2002. [DOI] [PubMed] [Google Scholar]
- 9.Feraille E, Dizin E. Coordinated control of ENaC and Na+,K+-ATPase in renal collecting duct. J Am Soc Nephrol 27: 2554–2563, 2016. doi: 10.1681/ASN.2016020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giménez I, Forbush B. Short-term stimulation of the renal Na-K-Cl cotransporter (NKCC2) by vasopressin involves phosphorylation and membrane translocation of the protein. J Biol Chem 278: 26946–26951, 2003. doi: 10.1074/jbc.M303435200. [DOI] [PubMed] [Google Scholar]
- 11.Gonzalez-Vicente A, Saez F, Monzon CM, Asirwatham J, Garvin JL. Thick ascending limb sodium transport in the pathogenesis of hypertension. Physiol Rev 99: 235–309, 2019. doi: 10.1152/physrev.00055.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadchouel J, Ellison DH, Gamba G. Regulation of renal electrolyte transport by WNK and SPAK-OSR1 kinases. Annu Rev Physiol 78: 367–389, 2016. doi: 10.1146/annurev-physiol-021115-105431. [DOI] [PubMed] [Google Scholar]
- 13.Harris PJ, Navar LG. Tubular transport responses to angiotensin. Am J Physiol Renal Physiol 248: F621–F630, 1985. doi: 10.1152/ajprenal.1985.248.5.F621. [DOI] [PubMed] [Google Scholar]
- 14.Kocinsky HS, Dynia DW, Wang T, Aronson PS. NHE3 phosphorylation at serines 552 and 605 does not directly affect NHE3 activity. Am J Physiol Renal Physiol 293: F212–F218, 2007. doi: 10.1152/ajprenal.00042.2007. [DOI] [PubMed] [Google Scholar]
- 15.Kocinsky HS, Girardi AC, Biemesderfer D, Nguyen T, Mentone S, Orlowski J, Aronson PS. Use of phospho-specific antibodies to determine the phosphorylation of endogenous Na+/H+ exchanger NHE3 at PKA consensus sites. Am J Physiol Renal Physiol 289: F249–F258, 2005. doi: 10.1152/ajprenal.00082.2004. [DOI] [PubMed] [Google Scholar]
- 16.Koomans HA, Boer WH, Dorhout Mees EJ. Evaluation of lithium clearance as a marker of proximal tubule sodium handling. Kidney Int 36: 2–12, 1989. doi: 10.1038/ki.1989.153. [DOI] [PubMed] [Google Scholar]
- 17.Layton AT, Vallon V, Edwards A. A computational model for simulating solute transport and oxygen consumption along the nephrons. Am J Physiol Renal Physiol 311: F1378–F1390, 2016. doi: 10.1152/ajprenal.00293.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Layton AT, Vallon V, Edwards A. Predicted consequences of diabetes and SGLT inhibition on transport and oxygen consumption along a rat nephron. Am J Physiol Renal Physiol 310: F1269–F1283, 2016. doi: 10.1152/ajprenal.00543.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee DH, Maunsbach AB, Riquier-Brison AD, Nguyen MT, Fenton RA, Bachmann S, Yu AS, McDonough AA. Effects of ACE inhibition and ANG II stimulation on renal Na-Cl cotransporter distribution, phosphorylation, and membrane complex properties. Am J Physiol Cell Physiol 304: C147–C163, 2013. doi: 10.1152/ajpcell.00287.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lerman LO, Kurtz TW, Touyz RM, Ellison DH, Chade AR, Crowley SD, Mattson DL, Mullins JJ, Osborn J, Eirin A, Reckelhoff JF, Iadecola C, Coffman TM. Animal models of hypertension: a scientific statement from the American Heart Association. Hypertension 73: e87–e120, 2019. doi: 10.1161/HYP.0000000000000090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leyssac PP, Christensen P. A comparison between endogenous and exogenous lithium clearance in the anaesthetized rat. Acta Physiol Scand 151: 173–179, 1994. doi: 10.1111/j.1748-1716.1994.tb09735.x. [DOI] [PubMed] [Google Scholar]
- 22.Lin DH, Sterling H, Wang WH. The protein tyrosine kinase-dependent pathway mediates the effect of K intake on renal K secretion. Physiology (Bethesda) 20: 140–146, 2005. doi: 10.1152/physiol.00044.2004. [DOI] [PubMed] [Google Scholar]
- 23.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem 287: 660–671, 2012. doi: 10.1074/jbc.M111.298919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McDonough AA. ISN Forefronts Symposium 2015: maintaining balance under pressure-hypertension and the proximal tubule. Kidney Int Rep 1: 166–176, 2016. doi: 10.1016/j.ekir.2016.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDonough AA, Magyar CE, Komatsu Y. Expression of Na+-K+-ATPase alpha- and beta-subunits along rat nephron: isoform specificity and response to hypokalemia. Am J Physiol Cell Physiol 267: C901–C908, 1994. doi: 10.1152/ajpcell.1994.267.4.C901. [DOI] [PubMed] [Google Scholar]
- 26.McDonough AA, Veiras LC, Minas JN, Ralph DL. Considerations when quantitating protein abundance by immunoblot. Am J Physiol Cell Physiol 308: C426–C433, 2015. doi: 10.1152/ajpcell.00400.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McDonough AA, Youn JH. Potassium homeostasis: the knowns, the unknowns, and the health benefits. Physiology (Bethesda) 32: 100–111, 2017. doi: 10.1152/physiol.00022.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mutig K. Trafficking and regulation of the NKCC2 cotransporter in the thick ascending limb. Curr Opin Nephrol Hypertens 26: 392–397, 2017. doi: 10.1097/MNH.0000000000000351. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen MT, Han J, Ralph DL, Veiras LC, McDonough AA. Short-term nonpressor angiotensin II infusion stimulates sodium transporters in proximal tubule and distal nephron. Physiol Rep 3: e12496, 2015. doi: 10.14814/phy2.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen MT, Lee DH, Delpire E, McDonough AA. Differential regulation of Na+ transporters along nephron during ANG II-dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305: F510–F519, 2013. doi: 10.1152/ajprenal.00183.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen MT, Yang LE, Fletcher NK, Lee DH, Kocinsky H, Bachmann S, Delpire E, McDonough AA. Effects of K+-deficient diets with and without NaCl supplementation on Na+, K+, and H2O transporters’ abundance along the nephron. Am J Physiol Renal Physiol 303: F92–F104, 2012. doi: 10.1152/ajprenal.00032.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ortiz PA, Garvin JL. NO inhibits NaCl absorption by rat thick ascending limb through activation of cGMP-stimulated phosphodiesterase. Hypertension 37: 467–471, 2001. doi: 10.1161/01.HYP.37.2.467. [DOI] [PubMed] [Google Scholar]
- 33.Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol 10: 676–687, 2015. doi: 10.2215/CJN.12391213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol 13: 1131–1135, 2002. doi: 10.1097/01.ASN.0000013292.78621.FD. [DOI] [PubMed] [Google Scholar]
- 35.Richardson C, Rafiqi FH, Karlsson HK, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci 121: 675–684, 2008. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 36.Richardson C, Sakamoto K, de los Heros P, Deak M, Campbell DG, Prescott AR, Alessi DR. Regulation of the NKCC2 ion cotransporter by SPAK-OSR1-dependent and -independent pathways. J Cell Sci 124: 789–800, 2011. doi: 10.1242/jcs.077230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riquier-Brison ADM, Leong PKK, Pihakaski-Maunsbach K, McDonough AA. Angiotensin II stimulates trafficking of NHE3, NaPi2, and associated proteins into the proximal tubule microvilli. Am J Physiol Renal Physiol 298: F177–F186, 2010. doi: 10.1152/ajprenal.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rosenbaek LL, Assentoft M, Pedersen NB, MacAulay N, Fenton RA. Characterization of a novel phosphorylation site in the sodium-chloride cotransporter, NCC. J Physiol 590: 6121–6139, 2012. doi: 10.1113/jphysiol.2012.240986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandberg MB, Riquier ADM, Pihakaski-Maunsbach K, McDonough AA, Maunsbach AB. ANG II provokes acute trafficking of distal tubule Na+-Cl− cotransporter to apical membrane. Am J Physiol Renal Physiol 293: F662–F669, 2007. doi: 10.1152/ajprenal.00064.2007. [DOI] [PubMed] [Google Scholar]
- 40.Sands JM, Nonoguchi H, Knepper MA. Vasopressin effects on urea and H2O transport in inner medullary collecting duct subsegments. Am J Physiol Renal Physiol 253: F823–F832, 1987. doi: 10.1152/ajprenal.1987.253.5.F823. [DOI] [PubMed] [Google Scholar]
- 41.Schultz SG, Lapointe J-Y. Membrane cross-talk in sodium-absorbing epithelial cells. : The Kidney: Physiology and Pathology (3rd ed.), edited by Seldin DW, Giebisch G. Philadelphia, PA: Lippincott Williams & Wilkins, 2000. [Google Scholar]
- 42.Strieter J, Stephenson JL, Giebisch G, Weinstein AM. A mathematical model of the rabbit cortical collecting tubule. Am J Physiol Renal Physiol 263: F1063–F1075, 1992. doi: 10.1152/ajprenal.1992.263.6.F1063. [DOI] [PubMed] [Google Scholar]
- 43.Svenningsen P, Friis UG, Bistrup C, Buhl KB, Jensen BL, Skøtt O. Physiological regulation of epithelial sodium channel by proteolysis. Curr Opin Nephrol Hypertens 20: 529–533, 2011. doi: 10.1097/MNH.0b013e328348bcc7. [DOI] [PubMed] [Google Scholar]
- 44.Thomsen K, Shirley DG. The validity of lithium clearance as an index of sodium and water delivery from the proximal tubules. Nephron 77: 125–138, 1997. doi: 10.1159/000190264. [DOI] [PubMed] [Google Scholar]
- 45.Veiras LC, Girardi ACC, Curry J, Pei L, Ralph DL, Tran A, Castelo-Branco RC, Pastor-Soler N, Arranz CT, Yu ASL, McDonough AA. Sexual dimorphic pattern of renal transporters and electrolyte homeostasis. J Am Soc Nephrol 28: 3504–3517, 2017. doi: 10.1681/ASN.2017030295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Veiras LC, Han J, Ralph DL, McDonough AA. Potassium supplementation prevents sodium chloride cotransporter stimulation during angiotensin II hypertension. Hypertension 68: 904–912, 2016. doi: 10.1161/HYPERTENSIONAHA.116.07389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang CT, Navar LG, Mitchell KD. Proximal tubular fluid angiotensin II levels in angiotensin II-induced hypertensive rats. J Hypertens 21: 353–360, 2003. doi: 10.1097/00004872-200302000-00027. [DOI] [PubMed] [Google Scholar]
- 48.Weinstein AM. A mathematical model of distal nephron acidification: diuretic effects. Am J Physiol Renal Physiol 295: F1353–F1364, 2008. doi: 10.1152/ajprenal.90356.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weinstein AM. A mathematical model of rat collecting duct. I. Flow effects on transport and urinary acidification. Am J Physiol Renal Physiol 283: F1237–F1251, 2002. doi: 10.1152/ajprenal.00162.2002. [DOI] [PubMed] [Google Scholar]
- 50.Weinstein AM. A mathematical model of the inner medullary collecting duct of the rat: acid/base transport. Am J Physiol Renal Physiol 274: F856–F867, 1998. doi: 10.1152/ajprenal.1998.274.5.F856. [DOI] [PubMed] [Google Scholar]
- 51.Weinstein AM. A mathematical model of the rat kidney: K+-induced natriuresis. Am J Physiol Renal Physiol 312: F925–F950, 2017. doi: 10.1152/ajprenal.00536.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weinstein AM, Weinbaum S, Duan Y, Du Z, Yan Q, Wang T. Flow-dependent transport in a mathematical model of rat proximal tubule. Am J Physiol Renal Physiol 292: F1164–F1181, 2007. doi: 10.1152/ajprenal.00392.2006. [DOI] [PubMed] [Google Scholar]
- 54.Yang L, Xu S, Guo X, Uchida S, Weinstein AM, Wang T, Palmer LG. Regulation of renal Na transporters in response to dietary K. Am J Physiol Renal Physiol 315: F1032–F1041, 2018. doi: 10.1152/ajprenal.00117.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang LE, Maunsbach AB, Leong PK, McDonough AA. Differential traffic of proximal tubule Na+ transporters during hypertension or PTH: NHE3 to base of microvilli vs. NaPi2 to endosomes. Am J Physiol Renal Physiol 287: F896–F906, 2004. doi: 10.1152/ajprenal.00160.2004. [DOI] [PubMed] [Google Scholar]
- 56.Zaika O, Mamenko M, Staruschenko A, Pochynyuk O. Direct activation of ENaC by angiotensin II: recent advances and new insights. Curr Hypertens Rep 15: 17–24, 2013. doi: 10.1007/s11906-012-0316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]