

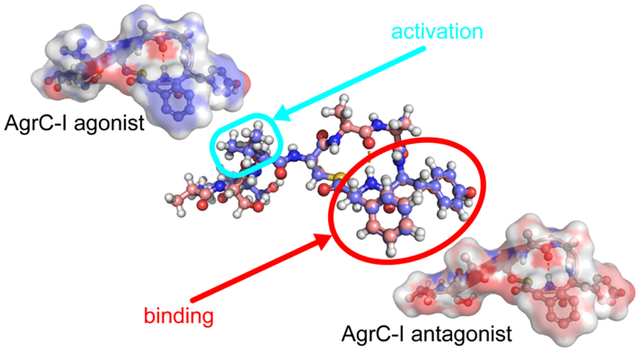
Abstract
We report the solution-phase structures of native signal peptides and related analogs capable of either strongly agonizing or antagonizing the AgrC quorum sensing (QS) receptor in the emerging pathogen Staphylococcus epidermidis. Chronic S. epidermidis infections are often recalcitrant to traditional therapies due to antibiotic resistance and formation of robust biofilms. The accessory gene regulator (agr) QS system plays an important role in biofilm formation in this opportunistic pathogen, and the binding of an autoinducing peptide (AIP) signal to its cognate transmembrane receptor (AgrC) is responsible for controlling agr. Small molecules or peptides capable of modulating this binding event are of significant interest as probes to investigate both the agr system and QS as a potential antivirulence target. We used NMR spectroscopy to characterize the structures of the three native S. epidermidis AIP signals and five non-native analogs with distinct activity profiles in the AgrC-I receptor from S. epidermidis. These studies revealed a suite of structural motifs critical for ligand activity. Interestingly, a unique β-turn was present in the macrocycles of the two most potent AgrC-I modulators—in both an agonist and an antagonist—that was distinct from the macrocycle conformation in the less-potent AgrC-I modulators and in the native AIP-I itself. This previously unknown β-turn provides a structural rationale for these ligands’ respective biological activity profiles. Development of analogs to reinforce the β-turn resulted in our first antagonist with subnanomolar potency in AgrC-I, while analogs designed to contain a disrupted β-turn were dramatically less potent relative to their parent compounds. Collectively, these studies provide new insights into the AIP:AgrC interactions crucial for QS activation in S. epidermidis and advance the understanding of QS at the molecular level.
Keywords: Staphylococcus epidermidis, autoinducing peptide, AgrC receptor, quorum sensing, virulence
Graphical Abstract
INTRODUCTION
Staphylococcus epidermidis is a ubiquitous, skin-colonizing Gram-positive bacterium1 that has emerged as an opportunistic pathogen in many hospital acquired infections.2–3 This bacterium is now a leading cause of biomedical device-associated infections due to its propensity to form robust biofilms on abiotic surfaces.3–8 This biofilm lifestyle, combined with its increasing antibiotic resistance, makes treating S. epidermidis infections with traditional antibacterial drugs difficult.2–3, 6, 9–11 Playing an important role in the pathogenesis of S. epidermidis infections is the accessory gene regulator (agr) system,12–17 a cell-cell communication system that allows the bacteria to sense population density and coordinate gene expression at high cell numbers to initiate group behaviors.18–19 This cell-cell signaling process is a type of quorum sensing (QS, Figure 1) and is mediated in S. epidermidis by a macrocyclic peptide pheromone called an autoinducing peptide (AIP).20–23 As the bacterial population density increases, so does the concentration of the AIP. Once a threshold concentration is reached, the AIP can productively bind with its cognate receptor AgrC, a transmembrane histidine kinase, which subsequently initiates the signaling cascade that leads to changes in gene expression.22
Figure 1.

Schematic of the QS process in S. epidermidis. At low cell density, the QS signal (AIP) is produced at a low level. As the population grows, the corresponding AIP concentration increases until the AIP reaches a threshold level at which it productively binds to AgrC. This AIP:AgrC binding event then activates the response regulator AgrA to simultaneously amplify expression of agr and alter gene expression to drive group-beneficial behaviors.
As a number of phenotypes associated with S. epidermidis virulence, such as the production of phenol-soluble modulins and biofilms, are under the direct control of QS,11–14, 24–28 targeting the agr system represents an attractive approach for studying the role of QS in infection and even possibly attenuating S. epidermidis infections.25, 29–32 This strategy has been used in Staphylococcus aureus, a pathogen closely related to S. epidermidis with an analogous agr QS system, and prior studies by our lab and others have revealed several highly potent, non-native antagonists of the agr system in S. aureus.33–48 Similar to S. aureus, S. epidermidis has evolved into different agr specificity groups (I–III), each with a different AIP signal and some variability in the other components of the agr system (i.e., in proteins AgrB–D).30, 49 Interestingly, these AIP signals are also capable of either inhibiting or activating the receptors of the other S. epidermidis groups, motivating hypotheses about cross-group interactions mediated by QS. For example, AIP-II and AIP-III each inhibit AgrC-I, while AIP-I inhibits both AgrC-II and AgrC-III (AIP signals shown in Figure 2).30 Synthetic ligands that selectively activate or inhibit each of these receptors, or that pan-activate or pan-inhibit all receptors, would represent valuable chemical probes to interrogate the nature of such possible cross-group interactions, and to study agr-based QS in general.
Figure 2.

Structures of the native S. epidermidis AIP signals (I–III) and AIP-I analogs examined in this study. Relative potency in the QS receptor AgrC-I is indicated with the arrow for AIP-I analogs. Relative potencies indicated are from cell-based assays of AgrC-I activity reported in our previous work.50
Recently, our laboratory performed a systematic study of the structure of the AIP-I signal from group-I S. epidermidis, and using cell-based assays reporting agr activity, delineated the structure-activity relationships (SARs) governing its ability to activate its cognate receptor, AgrC-I.50 This study revealed the first set of non-native AIP analogs that are capable of potently agonizing or antagonizing the agr system in S. epidermidis (selected peptides shown in Figure 2). However, we lack an understanding of how these activity data connect to the three-dimensional (3-D) structures of these peptides. Such a connection, assuming that these small and rigidified macrocyclic peptides can adopt a similar conformation upon binding to AgrC-I, would illuminate the chemical features crucial to AgrC-I receptor agonism or antagonism by these peptides, and provide new insights into their mechanisms of action. To date, only the solution-phase structure of the S. epidermidis AIP-I has been reported in a mixed-solvent system,51 and the analysis of this structure focused on the motifs important for interaction with the S. aureus AgrC receptors, not with S. epidermidis AgrC-I. To the best of our knowledge, no structural information on any S. epidermidis AIP analogs has been reported.
Herein, we report the first detailed characterization of the 3-D solution-phase structures of the S. epidermidis AIP-I signal, several non-native AIP-I analogs capable of strongly modulating AgrC-I, and the S. epidermidis AIP-II and AIP-III signals using NMR spectroscopy. Comparison of these peptide structures revealed several features that we propose, when aligned with their cell-based activity profiles, to be critical to receptor binding and activation, including a β-turn motif that was present in the macrocycles of both the most potent agonist and the most potent antagonist ligands, yet lacking in less potent ligands, including the native AIP-I signal. Our structural data suggest that a minimum of two endocyclic hydrophobic residues and the presence and specific orientation of a C-terminal hydrophobic group are necessary for activation of AgrC-I. Together, these structural analyses illuminate the mechanism of both AgrC-I agonism and antagonism by peptide ligands, and motivate new hypotheses on the modularity of the receptor-binding and receptor-activating motifs of AIP-I. A small set of second-generation analogs designed to strengthen or weaken the β-turn were synthesized based upon these findings, resulting in analogs with comparable or greatly diminished potency relative to the parent peptides, respectively. These studies also revealed the first AgrC-I antagonist with subnanomolar potency. The effects of these β-turn alterations on potency represent proof-of-concept and validation for structure-function studies such as this one.
RESULTS AND DISCUSSION
Selection of peptides for structural analyses.
To start, we selected a series of non-native AIP-I analogs from our previous study that displayed a range of agonistic and antagonistic activity in AgrC-I as determined utilizing a S. epidermidis GFP reporter strain (Figure 2; Table 1).50 We chose two agonists that were more potent than the native AIP-I signal: AIP-I D1A, in which a single Asp to Ala modification gave a four-fold increase in potency, and AIP-I D1AS6A, in which the double Ala modification gave a 20-fold increase in potency. We envisioned that comparisons between these two AIP-I analogs and the native AIP-I could provide insights into how the Ala substitutions increased agonism potency. We also selected three AgrC-I antagonists that displayed moderate to strong antagonism potencies (Table 1). This trio included t-AIP-I, in which the N-terminal tail was removed and the Cys residue was simply acetylated; AIP-I V3A, in which a single modification to the native AIP-I (Val to Ala) mode-switched its activity from agonist to antagonist; and the triple Ala modified peptide, AIP-I D1AV3AS6A (hereafter referred to as AAA), which is a highly potent antagonist with a single-digit nanomolar IC50 value. As the S. epidermidis AIP-II and AIP-III signals are also relatively strong antagonists of AgrC-I (Table 1), we included these native peptides in our structural studies. To facilitate these comparative studies, we also chose to determine the NMR structure of the native AIP-I under comparable experimental conditions. All of these native and non-native peptides were prepared and purified to homogeneity using the methods in our prior study.50
Table 1.
Structures and cell-based reporter activities in AgrC-I of the peptides evaluated in this studya
| peptide name | sequence | IC50 in nM, (95% CI) | EC50 in nM, (95% CI) |
|---|---|---|---|
| AIP-I | D-S-V-(C-A-S-Y-F) | - | 196 (162–238) |
| AIP-I D1A | A-S-V-(C-A-S-Y-F) | - | 49.3 (43.3–56.2) |
| AIP-I D1AS6A | A-S-V-(C-A-A-Y-F) | - | 10.3 (6.18–17.2) |
| t-AIP-I | Ac-(C-A-S-Y-F) | 192 (150–245) | - |
| AIP-I V3A | D-S-A-(C-A-S-Y-F) | 51.9 (37.9–71.0) | - |
| AAA | A-S-A-(C-A-A-Y-F) | 2.84 (1.95–4.11) | - |
| AIP-II | N-A-S-K-Y-N-P-(C-S-N-Y-L) | 9.64 (7.99–11.6) | - |
| AIP-III | N-A-A-K-Y-N-P-(C-A-S-Y-L) | 34.3 (31.4–37.4) | - |
Values from reference.50
CI = 95% confidence interval.
NMR analyses.
We used NMR spectroscopy to characterize the solution-phase structures of the peptides, using methods similar to our prior reports (see SI for full details).43, 51–52 The 1-D and 2-D (TOCSY and ROESY) NMR spectra were obtained on a 750 MHz spectrometer equipped with a cryoprobe at ambient temperature in H2O/D2O (95:5) except for AIP-I and t-AIP-I, which were dissolved in H2O/CD3CN (8:1 and 7:3, respectively) due to solubility limitations. Previous studies of similar S. aureus AgrC modulators showed that the presence of these amounts of acetonitrile did not cause significant structural changes relative to structures obtained in water alone.43 The 1-D proton and 2-D TOCSY and ROESY spectra were used to determine chemical shifts via sequential assignments of each amino acid residue. Internuclear proton distances were determined from 2-D ROESY cross peaks and converted to distance constraint files. These files were used to calculate and refine 3-D structures using torsion angle simulated annealing through Xplor-NIH,53–54 and an ensemble of 20 lowest-energy structures was utilized to determine the most representative low energy structure (ensembles in SI Figures S1–S8). Structure comparisons were performed with alignment fitting using PyMOL.55
Structural comparison of AIP-I to the antagonist t-AIP-I.
The NMR experimental results for AIP-I and t-AIP-I were compared first. Again, t-AIP-I lacks the N-terminal tail and is a moderate antagonist of AgrC-I (Table 1). Analogous ROESY cross peaks appeared in both spectra, with extremely similar coupling constants and chemical shifts for all analogous hydrogens (see SI; Tables S1, S2, S9, and S10). The ROEs observed for AIP-I revealed an almost identical macrocycle conformation compared to t-AIP-I, as illustrated in the overlay in Figure 3C. The structures align with an RMS difference of 0.3 Å for all analogous atoms. This close structural similarity between native AIP-I and the antagonist t-AIP-I suggests that while the tail in AIP-I has little impact on the structure of the macrocycle, it contains a contact critical for AgrC activation. When missing, as in t-AIP-I, the ligand is capable of competitively binding to AgrC-I yet fails to activate the receptor.
Figure 3.

Representative structures of AIP-I (A) and t-AIP-I (B). Oxygen is shown in red, nitrogen in blue, hydrogen in white, and sulfur in gold. (C) Overlay of AIP-I (in yellow) and t-AIP-I (in orange, with an all-atom RMS difference of 0.3 Å (72 atoms).
Several features from the comparative analyses of AIP-I and t-AIP-I are important and warrant discussion. First, Asp1 and Ser2 of the AIP-I tail do not have well-defined conformations (see ensembles in SI Figure S1), despite the rigidified conformation of its macrocycle (Figure 3; SI Figure S1). Second, the spectra suggest that the Ser in the macrocycle of both peptides adopts a conformation where the hydroxyl oxygen forms a hydrogen bond to the adjacent Tyr amide hydrogen. This finding is aligned with the well-established propensity of short, polar sidechains such as Ser and Thr for this interaction.56–57 The result of this hydrogen bond is the formation of a half-chair six-member ring, and we reason that this ring acts to stabilize the macrocycle. Third, our structures of AIP-I and t-AIP show the side chains of both of the endocyclic hydrophobic residues (Phe and Tyr) oriented away from the macrocycle to form a hydrophobic face (Figure 3). This observation supports the conclusions of prior SAR studies of AIPs in both S. aureus and S. epidermidis that implicate a hydrophobic face as critical for productive AgrC binding.36, 42, 50, 52, 58 Indeed, removal of these hydrophobic residues from the macrocycle completely ablates AIP-I activity.50 Fourth, the Cys and Phe sidechains adopt well-defined rotational conformations, while the Tyr sidechain appears to be less restricted. This last trend was observed for all the peptides studied in this work.
Structural comparison of AIP-I to the antagonist AIP-I V3A.
We next compared the structure of native AIP-I to the antagonist AIP-I V3A (Figure 4), which is approximately four-fold more potent than t-AIP-I (Table 1) and has a single amino acid modification (Val to Ala) relative to native AIP-I.50 This activity mode-switch (agonist to antagonist) suggests that Val3 may play an important role in dictating the active conformation of the native signal or makes a crucial contact with the receptor. Interestingly, when examining the structure of AIP-I, Val3 is directed in the opposite direction of its endocyclic hydrophobic face (composed of Tyr7 and Phe8), as shown in Figures 3A, C.
Figure 4.

(A) Representative structure of AIP-I V3A. Atom colors analogous to Figure 3. (B) Overlay of AIP-I (in yellow) and AIP-I V3A (in green), having an all-atom RMS difference of 2.1 Å (79 atoms) for residues 3–8.
The ensemble of AIP-I V3A from Xplor has the highest RMSD of the peptides analyzed in this study, but like AIP-I, the structure of the macrocycle and relative position of residue 3 were highly ordered while Asp1 and Ser2 were the most disordered (SI Figure S3). Also similar to AIP-I, the hydrophobic residues (Phe8 and Tyr7) of AIP-I V3A were extended towards one face of the macrocycle and create a hydrophobic surface (Figure 4A), and the Ser6 hydroxyl to Tyr7 amide hydrogen bond appears to be maintained as a stabilizing interaction of the macrocycle. While the isopropyl side chain of Val3 is replaced with a methyl side chain in AIP-I V3A, the orientation of the Ala side chain relative to the hydrophobic face of the macrocycle is conserved (Figure 4B). Therefore, the structures of native AIP-I and AIP-I V3A are highly similar, having an all-atom RMS difference for residues 3–8 (excluding the more disordered hydrophilic N-terminal residues) of 2.1 Å (79 atoms), and an RMS difference for the backbone of only 0.8 Å (24 atoms). In view of these data, we propose that the lack of the branched hydrophobic side chain of residue Val3 (as in AIP-I V3A) is the primary cause for switching AIP-I from an agonist to an antagonist, with little or no dependence on conformational changes. We reason that the Val3 side chain in AIP-I makes a key contact with AgrC-I that is essential for activation, and in its absence, AIP-I V3A competitively inhibits AgrC-I.
Structural comparison of AIP-I to agonists with increased potency.
Next, we turned our attention to the structures of two non-native agonists of AgrC-I, AIP-I D1A and AIP-I D1AS6A (Figures 5A and 5B). Both analogs are more potent agonists than the native agonist AIP-I (Table 1), with AIP-I D1AS6A being the most potent non-native agonist targeting S. epidermidis AgrC-I reported to date.50 Counterintuitively in view of their less polar primary structures, AIP-I D1A and AIP-I D1AS6A were found to be more soluble in water than AIP-I, and their corresponding NMR spectra reflected that difference with higher S/N. Regardless, representative structures of the native AIP-I and AIP-I D1A were found to be exceptionally similar (overlay in Figure 5C), having an all-atom RMS difference of 1.3 Å (99 atoms) and a backbone-atom RMS difference of 0.5 Å (28 atoms). While the ROESY spectrum of AIP-I indicated disorder in Asp1 and Ser2 (see above), the ROESY spectrum of AIP-I D1A had more cross peaks related to Ala1 and Ser2, yielding a more defined structure from Xplor (SI Figure S4). It seemed possible that this was simply due to the greater S/N of the AIP-I D1A sample, but a closer examination of the AIP-I spectrum revealed that other small ROE cross peaks were visible, suggesting the issue was not S/N, but rather a true disorder in Asp1 and Ser2. In view of their highly similar structures, we speculate that the more ordered tail of AIP-I D1A relative to AIP-I contributes to its heightened agonistic activity, presumably better positioning an activation motif therein.
Figure 5.

Representative structures of AIP-I D1A (A) and AIP-I D1AS6A (B) Atom colors analogous to Figure 3. The characteristic hydrogen bond of a β-turn is indicated, with i+1 φ = 63° and ψ = −88°, i+2 φ = −125° and ψ = 16° (in B). (C) Overlay of AIP-I (in yellow) and AIP-I D1A (in white), with an all-atom RMS difference of 1.3 Å (99 atoms). (D) Overlay of AIP-I (in yellow) and AIP-I D1AS6A (in dark blue), with an all-atom RMS difference of 3.6 Å (97 atoms).
The structural similarity between AIP-I and the more potent agonist AIP-I D1AS6A, with an all-atom RMS difference of 3.6 Å (97 atoms), was less than that between AIP-I and AIP-I D1A. The differences originate from conformational changes in the AIP-I D1AS6A macrocycle. AIP-I D1AS6A has an Ala residue in place of Ser6, and thus no hydrogen bond is possible between the Ser6 hydroxyl and Tyr7 amide hydrogen. Instead, the AIP-I D1AS6A macrocycle was observed to adopt a β-turn motif with the Ala5 carbonyl H-bonded to the Phe8 HN (Figure 5B). Analysis of the appropriate φ and y angles revealed the turn was a type II’ β-turn.59 The ROESY spectrum for AIP-I D1AS6A also featured several major differences in cross peaks that were analogous in the spectra of each of the other peptides compared up to this point. For example, in these latter spectra, an ROE existed between Ser6 Hα and Ser6 NH that was calculated at 2.5 or 2.6 Å. In the spectrum of AIP-I D1AS6A, however, this ROESY cross peak was much more intense, with a calculated value of 2.2 Å. This difference indicates a dissimilar φ-dihedral angle at Ala6 in AIP-I D1AS6A. Additionally, what was observed as a moderate cross peak between Ser6 HN and Phe8 HN in the AIP-I D1A spectrum, which only differs by a single amino acid from AIP-I D1AS6A, was a weak cross peak between Ala6 HN and Phe8 HN in the AIP-I D1AS6A spectrum, indicating a difference in the plane of the amide bond of Ala5 and Ala6. This difference brings the carbonyl of the amide bond into the center of the AIP-I D1AS6A macrocycle, allowing for formation of the hydrogen bond to the Phe8 HN (Figure 5B). This hydrogen bond then stabilizes the β-turn in the macrocycle of AIP-I D1AS6A.
This alteration to the macrocycle in AIP-I D1AS6A caused a change in the way the exocyclic tail protrudes from the macrocycle, and brought the Val3 sidechain to the same face as the hydrophobic endocyclic sidechains. The tails of AIP-I and related analogs studied so far form a short β-strand that is connected to the macrocycle, comprised of Ser2 to Ala5. As Ala5 in AIP-I D1AS6A has a major change in its conformation, the strand exiting the macrocycle is rotated by roughly 60° relative to AIP-I. Given that the hydrophobic sidechain of Val3 is strongly implicated as an effector of agonism in these peptides (see above), and also that AIP-I D1AS6A is 20-fold more potent than AIP-I and 5-fold more potent than AIP-I D1A, we reason that the macrocyclic fold and corresponding change of orientation of Val3 in AIP-I D1AS6A make this ligand more efficacious for binding and activating AgrC-I.
Structural comparison of AIP-I to the antagonist AAA.
To further examine the relationships between the macrocycle conformation, the potency of the peptide, and the presence of the hydrophobic sidechain of Val3, we determined the structure of the most potent AgrC-I antagonist from our previous study, peptide AAA (Table 1). Xplor outputted a very ordered ensemble from the AAA constraint file (SI Figure S6), and strikingly, a β-turn in the macrocycle was observed that was closely analogous to that of agonist AIP-I D1AS6A. Indeed, just as many ROESY cross peaks were analogous between the spectra of AIP-I, t-AIP-I, AIP-I V3A, and AIP-I D1A, the ROESY cross peaks of the antagonist AAA and the agonist AIP-I D1AS6A were equally analogous, with patterns suggesting the same type II’ β-turn in the macrocycle and the same short β-strand exiting the macrocycle in a similar manner. A representative structure of AAA is shown in Figure 6A, with an overlay of AIP-I and AAA having a relatively large all-atom RMS difference of 3.4 Å (88 atoms) shown in Figure 6B. This overlay is suggestive that the antagonist AAA, like the agonist AIP-I D1AS6A, adopts a structure quite different from AIP-I.
Figure 6.

(A) Representative structure of AAA. Atom colors analogous to Figure 3. For the macrocycle β-turn, i+1 φ = 62° and ψ = −89°, i+2 φ = −123° and ψ = 18°. (B) Overlay of AIP-I and AAA, with an all-atom RMS difference of 3.4 Å (88 atoms). AIP-I is shown in yellow, and AAA is shown in pink.
Structures of S. epidermidis AIP-II and AIP-III.
Because both native AIP-II and native AIP-III are also relatively potent AgrC-I antagonists, we investigated the structures of these two AIPs for comparison to the native AIP-I and antagonist AAA. AIP-II and AIP-III have similar primary sequences (Figure 2, Table 1), and their corresponding 2-D NMR spectra showed many similar cross peaks in the macrocycle region. However, a single amino acid difference in the tail sequence yielded a dramatic difference in the calculated tail structures. AIP-II has a sequence in its tail region (S-K-Y-N) that formed a β-turn (Figure 7A), indicated strongly by a robust series of cross peaks in the ROESY spectrum. In AIP-III, the corresponding sequence is A-K-Y-N, and the ROESY spectrum in this region instead indicated a β-stand from residue 2 through residue 9 within the ring, with no i to i+3 cross peaks analogous to the β-turn as indicated in AIP-II (Figure 7B). Overall, AIP-II was more globular, with the tail folding around the macrocycle but not quite contacting it, and AIP-III was more extended, with the strand of the tail pointing directly out from the ring. Additionally, in a comparison of the macrocycles, the plane of the amide bond between residue-10 (Asn in AIP-II and Ser in AIP-III) and Tyr11 was inverted by ~180° (Figure 7C).
Figure 7.

Representative structures of AIP-II (A) and AIP-III (B). Atom colors analogous to Figure 3. C: Overlay of AIP-II (cyan) and AIP-III (magenta) for the macrocycle only, with an all-atom RMS difference of 1.5 Å (68 atoms).
An alignment of the macrocycle of AIP-I with either AIP-II or AIP-III yields only modest overlap, having all-atom RMS differences of 2.2 Å (PyMOL fits 49 atoms) or 2.1 Å (PyMOL fits 52 atoms) respectively, for most-similar atom matching (results not shown), indicating that AIP-II and AIP-III have major 3-D structural differences in their macrocycles relative to AIP-I. In contrast, there is better overlap of the macrocycle of the antagonist AAA with that of AIP-II (all-atom RMS difference for macrocycle of 1.9 Å, of 49 atoms) and AIP-III (all-atom RMS difference of macrocycle of 1.2 Å, 50 atoms) (SI Figure S9), which is in accord with all three peptides acting as AgrC-I antagonists. We return to this below.
Comparison of the most potent AgrC-I agonist and antagonist.
Given the remarkable, and unexpected, similarity of the ROESY spectra of the most potent AgrC agonist AIP-I D1AS6A and the most potent AgrC antagonist AAA, a direct comparison of these two structures was warranted. The structural overlay is shown in Figure 8. Although the slight differences in ROE intensity from the spectra lead to subtle differences between the two structures, they are very similar overall, with an all-atom RMS difference of 1.0 Å (101 atoms) and a backbone RMS difference of 0.8 Å for all residues (32 atoms), or an all-atom RMS difference of just 0.2 Å (78 atoms) and a backbone RMS difference of just 0.1 Å (24 atoms) when considering only residues 3–8. To further validate the interpretation of these two solution NMR structures, they were each analyzed using MolProbity, and both AIP-I D1AS6A and AAA have very good relative clash and MolProbity scores (see MolProbity validation in SI).60–61
Figure 8.

Overlay of AIP-I D1AS6A (dark blue) and AAA (pink), with an all-atom RMS difference of 1.0 Å (101 atoms), and all-atom RMS difference for residues 3–8 of 0.2 Å (78 atoms). Atom colors analogous to Figure 3.
These structural data suggest that the β-turn in the macrocycles of AIP-I D1AS6A and AAA arranges them for optimal binding to AgrC-I relative to the macrocycle of the native ligand AIP-I, which lacks the same β-turn. We then speculate that the stabilizing Ser6 hydroxyl to Tyr7 amide hydrogen bond in the AIP-I macrocycle impairs it from adopting the optimal β-turn conformation for binding its own cognate receptor. This difference is correlated with the heightened potencies of AIP-I D1AS6A (as an agonist) and AAA (as an antagonist) relative to AIP-I (Table 1). The opposite activity profiles for AIP-I D1AS6A and AAA, despite their very similar structures, indicates that their only point of real structural divergence—the branched hydrophobic motif of the Val3 side chain—engages in critical interactions with the AgrC-I receptor, with its position dictated by the fold of the macrocycle. The Val3 sidechain is oriented more toward the hydrophobic endocyclic face of AIP-I D1AS6A relative to that of AIP-I, and this positioning of both Val3 and the hydrophobic face in AIP-I D1AS6A presumably facilitates receptor binding and activation. This model then implicates Val, and only Val positioned properly next to the ring, as essential for receptor activation. If the model is accurate, introducing a Val residue or other appropriate hydrophobic moiety next to a different AIP macrocycle or even a β-turn mimetic could lead to new modulators that activate S. epidermidis AgrC-I.
Exploration of the β-turn motif through new analogs.
We sought to further examine the importance of the β-turn motif for peptide:AgrC-I interactions in S. epidermidis and designed eight new analogs of AIP-I D1AS6A and AAA to modify the type II’ β-turn. Substitution of Ala6 with D-Ala or Gly should strengthen the β-turn, and we hypothesized that, if the β-turn motif was indeed critical, such substitutions should either maintain or possibly enhance the agonism or antagonism potency of the two parent peptides.59, 62–63 Conversely, substitution of Phe8 with N-MePhe or phenyllactic acid (PLA) should destabilize the β-turn by removing the key hydrogen bond between the Ala5 carbonyl and Phe8 NH. We hypothesized that destabilization of the β-turn would reduce the potency of these new analogs relative to the parent peptides where the β-turn is intact. We synthesized new AIP-I D1AS6A and AAA analogs containing these modifications (shown in Figure 9) using standard methods and screened them in agr reporter assays (side-by-side with their parent peptides) in group-I S. epidermidis to allow for comparisons of their activity profiles (Figure 10, see SI for synthetic protocols and assay methods).50
Figure 9.

Structural modifications of lead agonist AIP-I D1AS6A and lead antagonist AAA to provide β-turn modifying analogs.
Figure 10.

Group-I AgrC activity agonism (closed shapes) and antagonism (open shapes) dose-response curves for the new peptide analogs with modifications to the β-turn. Curves depicted for agonist (A) and antagonist (B) analogs designed to strengthen the β-turn, and agonists (C) and antagonists (D) designed to weaken the β-turn. The parent agonist (AIP-I D1AS6A) or antagonist (AAA) are shown alongside the new analogs (in blue) for comparison.
The dose-response assays for the new analogs with D-Ala and Gly substitutions, which should strengthen or maintain the β-turn, showed only slight changes in potency relative to their parent compounds. As the parents already contains the critical β-turn, this result is not altogether surprising. The AgrC-I agonism assays revealed AIP-I D1AS6G and AIP-I D1AS6dA to be ~2-fold and ~4 fold less potent, respectively, than the parent AIP-I D1AS6A (Figure 10A, see SI Table S18 for potency data for all new analogs). These reductions in potency, especially for AIP-I D1AS6G, are relatively minor; indeed, both peptides are still highly active, with EC50 values in the low nanomolar range. This only minor reduction in activity relative to AIP-I D1AS6A reinforces the β-turn as important for AgrC-I binding, and suggests that these particular type II’ β-turn mimics are simply not optimal for AgrC-I activation. Turning to the AgrC-I antagonism assay data, AAG and AAdA both maintained their high potencies, with IC50 values comparable or lower than the parent AAA (Figure 10B). These results provide further support for the β-turn as a critical element for the interactions of these AIP-I analogs with AgrC-I. Moreover, they revealed the most potent AgrC-I antagonist that we have observed to date in S. epidermidis (AAdA), with a subnanomolar IC50 value (0.908 nM).
In contrast to the data for the D-Ala and Gly substituted analogs, the incorporation of N-MePhe and PLA, hypothesized to eliminate the key hydrogen bond necessary for the β-turn, obliterated activity in AgrC-I relative to their parent compounds. Dose-response agonism assays of AIP-I D1AS6A [N-MeF8] and AIP-I D1AS6A [F8PLA] indicated a complete lack of activity (Figure 10C), while the antagonism assays of AAA [N-MeF8] and AAA [F8PLA] revealed a potency loss of over three orders of magnitude relative to the parent antagonist AAA (Figure 10D). These major shifts in potency due to the elimination of a single hydrogen bond strongly supports the importance of the β-turn for optimal binding interactions with AgrC-I.
To further examine the structures of these β-turn modified analogs and their activity profiles in AgrC-I, we performed additional 1-D and 2-D NMR experiments on a set of peptides with varying agonistic activities: AIP-I, AIP-I D1AS6A, AIP-I D1AS6dA, and AIP-I D1AS6A [N-MeF8]. AIP-I D1AS6A and its second-generation analogs were effective surrogates for the AAA family as well, as the chemical shifts of AIP-I D1AS6A and AAA were highly conserved and their solution NMR structures were analogous (again, both shown to adopt a type II’ β-turn; see above). Chemical shifts are well-known to be sensitive to local structural changes in peptide/protein conformation, including the torsion angles used to classify β-turns.64–67 We initially sought to use NMR chemical shift data to generate backbone torsion angles using the established TALOS-N computer program,68 in order to gauge similarities in the local conformations of the peptides.
We performed 1-D 1H, 2-D TOCSY and 2-D 1H-13C HSQC NMR experiments in D2O on an 800 MHz spectrometer (see SI for full details of NMR experiments and data) on the family of peptides, and first inputted chemical shift data into TALOS-N for AIP-I and AIP-I D1AS6A to compare the software’s predicted torsion angles to those determined in our solution-phase NMR structures. We found that TALOS-N could not make torsion angle predictions for all the amino acids in these macrocyclic peptides, most likely because it relies on a training set based on natural linear peptides/proteins, and it was unable to predict torsion angles for all of the residues in the β-turn in AIP-I D1AS6A (see SI for the TALOS-N predictions and associated analysis). TALOS-N could make predictions with high confidence for residues in the exocyclic tails, and these predictions fell in the same regions of the Ramachandran plots as the observed torsion angles from the representative solution NMR structures, adding a second measure of validation for these NMR structures in addition to the validation with MolProbity (Tables S23 and S24). In view of this incomplete data set, however, we turned to comparative NMR chemical shift analysis, a technique commonly used for analyzing conformations of proteins and peptides,65–66 to identify putative structural changes between the agonist AIP-I D1AS6A and the two second-generation analogs, AIP-I D1AS6dA and AIP-I D1AS6A [N-MeF8].
We performed a systematic analysis of the Hα, Cα, Hβ, and Cβ chemical shifts associated with each residue in AIP-I D1AS6A, AIP-I D1AS6dA, and AIP-I D1AS6A [N-MeF8] to obtain a gauge of their overall structural similarity (see SI for full assignments [Tables S20–S22] and additional detailed text). A side-by-side comparison of the chemical shifts for AIP-I D1AS6A versus AIP-I D1AS6dA (Table S25) revealed no significant changes in chemical shifts between the two peptides (i.e., no chemical shift change was greater than two standard deviations above the average shift difference).69 This lack of significant chemical shift changes suggests that the conformation of AIP-I D1AS6dA is very similar to the parent AIP-I D1AS6A. This result correlates with the comparable agonistic activities of the two peptides in the cell-based agr reporter assay. An analogous comparison was performed on the NMR data for AIP-I D1AS6A and its the N-methyl amino acid, AIP-I D1AS6A [N-MeF8] (Table S25). While there were only small differences in chemical shifts in many of the residues between the two peptides, in the residues near the β-turn there were four significant chemical shift changes between AIP-I D1AS6A and AIP-I D1AS6A [N-MeF8], signifying that the local conformation around these residues was perturbed.69 These chemical shift changes suggest that the N-methyl amino acid modification not only removes the key hydrogen necessary for a β-turn hydrogen bond, but also causes changes in the local conformation of AIP-I D1AS6A [N-MeF8] relative to its parent AIP-I D1AS6A. This result correlates with the dramatic loss in agonistic activity for AIP-I D1AS6A [N-MeF8] relative to AIP-I D1AS6A in the cell-based agr reporter assay. These new NMR data and comparative analyses serve to support the claim that the structure of the macrocycle in this class of AIP-I mimics is critical to strong binding interactions with AgrC-I.
SUMMARY AND CONCLUSIONS
We have determined the 3-D structures of the three S. epidermidis AIPs (I–III) and five AIP-I analogs to gain insight into the modes by which these compounds activate and inhibit S. epidermidis AgrC-I. Comparisons of the solution-phase NMR structures revealed chemical features essential to AgrC-I agonism and antagonism (summarized in Figure 11). Specifically, we propose that the AIP macrocycle exhibits a hydrophobic face at the C-terminus that facilitates receptor binding, and that the positioning of that hydrophobic face is best facilitated by a type II’ β-turn macrocycle that is not present in native AIP-I. Instead, we found AIP-I has a dispensable stabilizing interaction in its macrocycle that we predict impairs adoption of the optimal conformation for binding its own cognate receptor. Additionally, a bulky hydrophobic group on the tail (e.g., Val3), oriented properly by the macrocycle, is the essential factor for AgrC-I receptor activation. Removal of this hydrophobic group, while maintaining the β-turn macrocycle, results in a highly potent AgrC-I antagonist (i.e., AAA). Removal of the tail altogether, as in t-AIP-I, yields a moderate antagonist with a macrocycle conformation analogous to the native AIP-I. We found the native AIP-II and AIP-III to adopt structures quite different from AIP-I, with each having highly different tail structures. Interestingly, both AIP-II and AIP-III are capable of antagonizing AgrC-I,30, 50 and have macrocycle conformations more similar to the β-turn motif of antagonist AAA, again suggesting that the most important features for binding to AgrC-I are hydrophobic residues in the macrocycle and a β-turn type macrocycle, while additional contacts made by the exocyclic tail may improve the AIP-I:AgrC-I binding interaction but are secondary to the macrocycle interactions. To further underscore the importance of the macrocyclic β-turn motif for peptide:AgrC-I binding, we found that analogs of AIP-I D1AS6A and AAA that reinforced the β-turn motif were also highly potent, with the antagonist AAdA having a subnanomolar IC50 value and constituting, to our knowledge, the most potent AgrC antagonist in S. epidermidis to be reported. In turn, analogs in which β-turn formation was perturbed exhibited dramatic losses in potency, along with large perturbations of key NMR chemical shifts not present for other β-turn containing peptides, emphasizing the key role that the β-turn plays for AgrC-I binding.
Figure 11.

Model for AgrC-I activation by AIP-I. AIP-I is shown with a Connolly surface indicating relative hydrophobicity of residues, with yellow being most hydrophobic and green being most hydrophilic.
The proposed models of AgrC-I agonism and antagonism are in agreement with the relative potencies of the AIP analogs observed in the cell-based reporter assay, strongly suggesting that the NMR structures and associated data reported herein are biologically relevant. We note that while the presence of two hydrophobic side chains near the C-terminus in conjunction with a type II’ β-turn was sufficient for subnanomolar antagonism in S. epidermidis, such levels of antagonistic activity have been reserved for AIP analogs with at least three major hydrophobic groups and highly variable macrocyclic structures in the related bacterial species, S. aureus.52 These structural differences may contribute to the relatively weak cross-inhibition observed for the native AIP signals and related analogs between S. aureus and S. epidermidis.50
These structures and biological data for the native S. epidermidis AIPs and non-native AIP-I analogs are significant as they provide the first insights into the mechanism of AgrC receptor activation in in this emerging pathogen and shape our understanding of agr-type QS. These data and insights should also guide the design of new chemical probes to study QS in S. epidermidis, as we showcase through the synthesis and study of β-turn modifying analogs herein. Indeed, the discovery of AAdA as a highly potent AgrC-I antagonist serves to validate structure-function studies such as this one. Looking to the future, because the primary mechanism by which S. epidermidis causes infection is biofilm formation and the activation of the agr system can reduce biofilm accumulation,12, 25, 27, 29 designing AIP analogs that can agonize all three AgrC receptors in S. epidermidis would be of great interest. Our structural studies suggest that, while the branched hydrophobic side chain of residue Val3 is crucial for receptor activation, the proper orientation of this side chain may not be dictated by any specific interactions between the side chain and the macrocycle. Consequently, we hypothesize that a Val residue, or another moiety with an adequately bulky hydrophobic group, could be introduced onto a type II’ β-turn macrocycle mimetic, the AIP-II macrocycle, or AIP-III macrocycle, turning the resulting peptide into a highly potent activator for multiple AgrC receptors. Further simplified AgrC-I antagonists and agonists also could be identified using a method similar to that previously described for S. aureus AgrC receptors, where a number of different hydrophobic residues were substituted at the C-terminal region, and the other residues in the macrocycle were replaced with a single amino acid linker.43, 48 Studies toward these and related goals are ongoing and will be reported in due course.
Supplementary Material
ACKNOWLEDGEMENT
Financial support for this work was provided by the Office of Naval Research (N00014-16-1-2185), the NSF (CHE-1708714), and the Wisconsin Alumni Research Foundation (UW 2020 Program). J.K.V. was supported in part by an NSF Graduate Research Fellowship (DGE-1747503). J.K.V., K.H.J.W., and T.J.P. were supported in part by the UW–Madison NIH Chemistry–Biology Interface Training Program (T32 GM008505), the UW–Madison Office of the Vice Chancellor for Research and Graduate Education (with funding from the Wisconsin Alumni Research Foundation). T.Y. was supported in part by the UW–Madison NIH Biotechnology Training Program (T32 GM008349). G.C. and M.T. were supported by NIH grant P41 GM103399. This study made use of the National Magnetic Resonance Facility at Madison, which is supported by NIH grants P41 GM103399 and P41 GM66326. Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA.
ABBREVIATIONS
- QS
quorum sensing
- AIP
autoinducing peptide
- agr
accessory gene regulator
- SAR
structure-activity relationship
- AgrC
accessory gene regulator signal receptor protein C
- NMR
nuclear magnetic resonance
- RMS
root-mean-square
Footnotes
Experimental procedures, characterization data for new peptides, biological assay data, NMR spectra and associated analyses, Xplor constraint file statistics, images of structure ensembles, and .pdb files for the representative structures of each peptide are available as Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest. This article is dedicated to Prof. Ronald T. Raines and Prof. Samuel H. Gellman on the occasion of their 60th birthdays.
REFERENCES
- 1.Vuong C; Otto M, Staphylococcus epidermidis infections. Microb. Infect 2002, 4 (4), 481–489. [DOI] [PubMed] [Google Scholar]
- 2.von Eiff C; Peters G; Heilmann C, Pathogenesis of infections due to coagulase-negative staphylococci. The Lancet Infectious Diseases 2002, 2 (11), 677–685. [DOI] [PubMed] [Google Scholar]
- 3.Otto M, Staphylococcus epidermidis — the ‘accidental’ pathogen. Nature Reviews Microbiology 2009, 7, 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mack D; Davies AP; Harris LG; Rohde H; Horstkotte MA; Knobloch JK-M, Microbial interactions in Staphylococcus epidermidis biofilms. Anal. Bioanal. Chem 2007, 387 (2), 399–408. [DOI] [PubMed] [Google Scholar]
- 5.McCann MT; Gilmore BF; Gorman SP, Staphylococcus epidermidis device-related infections: pathogenesis and clinical management. J. Pharm. Pharmacol 2008, 60 (12), 1551–1571. [DOI] [PubMed] [Google Scholar]
- 6.Rogers KL; Fey PD; Rupp ME, Coagulase-Negative Staphylococcal Infections. Infect. Dis. Clin. North Am 2009, 23 (1), 73–98. [DOI] [PubMed] [Google Scholar]
- 7.Mertens A; Ghebremedhin B, Genetic determinants and biofilm formation of clinical Staphylococcus epidermidis isolates from blood cultures and indwelling devises. Eur. J. Microbiol. Immunol. (Bp) 2013, 3 (2), 111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris LG; Dudley E; Rohde H; Frommelt L; Siemssen N; Wilkinson TS; Mack D, Limitations in the use of PSMγ, agr, RNAIII, and biofilm formation as biomarkers to define invasive Staphylococcus epidermidis from chronic biomedical device-associated infections. Int. J. Med. Microbiol 2017, 307 (7), 382–387. [DOI] [PubMed] [Google Scholar]
- 9.Costerton JW; Stewart PS; Greenberg EP, Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284 (5418), 1318–1322. [DOI] [PubMed] [Google Scholar]
- 10.Claessens J; Roriz M; Merckx R; Baatsen P; Van Mellaert L; Van Eldere J, Inefficacy of vancomycin and teicoplanin in eradicating and killing Staphylococcus epidermidis biofilms in vitro. Int. J. Antimicrob. Agents 2015, 45 (4), 368–375. [DOI] [PubMed] [Google Scholar]
- 11.Sabaté Brescó M; Harris LG; Thompson K; Stanic B; Morgenstern M; O’Mahony L; Richards RG; Moriarty TF, Pathogenic Mechanisms and Host Interactions in Staphylococcus epidermidis Device-Related Infection. Front. Microbiol 2017, 8 (1401). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vuong C; Dürr M; Carmody AB; Peschel A; Klebanoff SJ; Otto M, Regulated expression of pathogen-associated molecular pattern molecules in Staphylococcus epidermidis: quorum-sensing determines pro-inflammatory capacity and production of phenol-soluble modulins. Cell. Microbiol 2004, 6 (8), 753–759. [DOI] [PubMed] [Google Scholar]
- 13.Kong K-F; Vuong C; Otto M, Staphylococcus quorum sensing in biofilm formation and infection. Int. J. Med. Microbiol 2006, 296 (2), 133–139. [DOI] [PubMed] [Google Scholar]
- 14.Yao Y; Vuong C; Kocianova S; Villaruz AE; Lai Y; Sturdevant DE; Otto M, Characterization of the Staphylococcus epidermidis Accessory-Gene Regulator Response: Quorum-Sensing Regulation of Resistance to Human Innate Host Defense. J. Infect. Dis 2006, 193 (6), 841–848. [DOI] [PubMed] [Google Scholar]
- 15.Wang C; Li M; Dong D; Wang J; Ren J; Otto M; Gao Q, Role of ClpP in biofilm formation and virulence of Staphylococcus epidermidis. Microb. Infect 2007, 9 (11), 1376–1383. [DOI] [PubMed] [Google Scholar]
- 16.Cheung GYC; Joo H-S; Chatterjee SS; Otto M, Phenol-soluble modulins – critical determinants of staphylococcal virulence. FEMS Microbiol. Rev 2014, 38 (4), 698–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otto M, Phenol-soluble modulins. Int. J. Med. Microbiol 2014, 304 (2), 164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Camilli A; Bassler BL, Bacterial Small-Molecule Signaling Pathways. Science 2006, 311 (5764), 1113–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rutherford ST; Bassler BL, Bacterial Quorum Sensing: Its Role in Virulence and Possibilities for Its Control. Cold Spring Harb. Perspect. Med 2012, 2:a012427 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyon GJ; Novick RP, Peptide signaling in Staphylococcus aureus and other Gram-positive bacteria. Peptides 2004, 25 (9), 1389–1403. [DOI] [PubMed] [Google Scholar]
- 21.George EA; Muir TW, Molecular Mechanisms of agr Quorum Sensing in Virulent Staphylococci. ChemBioChem 2007, 8 (8), 847–855. [DOI] [PubMed] [Google Scholar]
- 22.Novick RP; Geisinger E, Quorum Sensing in Staphylococci. Annu. Rev. Genet 2008, 42, 541–64. [DOI] [PubMed] [Google Scholar]
- 23.Thoendel M; Kavanaugh JS; Flack CE; Horswill AR, Peptide Signaling in the Staphylococci. Chem. Rev 2011, 111 (1), 117–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Otto M, Virulence factors of the coagulase-negative staphylococci. Front. Biosci 2004, 9, 841–863. [DOI] [PubMed] [Google Scholar]
- 25.Wang R; Khan BA; Cheung GYC; Bach T-HL; Jameson-Lee M; Kong K-F; Queck SY; Otto M, Staphylococcus epidermidis surfactant peptides promote biofilm maturation and dissemination of biofilm-associated infection in mice. J. Clin. Invest 2011, 121 (1), 238–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Otto M, Staphylococcal Infections: Mechanisms of Biofilm Maturation and Detachment as Critical Determinants of Pathogenicity. Annu. Rev. Med 2013, 64 (1), 175–188. [DOI] [PubMed] [Google Scholar]
- 27.Le KY; Dastgheyb S; Ho TV; Otto M, Molecular determinants of staphylococcal biofilm dispersal and structuring. Frontiers Cell. Infect. Microbiol 2014, 4, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung GYC; Rigby K; Wang R; Queck SY; Braughton KR; Whitney AR; Teintze M; DeLeo FR; Otto M, Staphylococcus epidermidis Strategies to Avoid Killing by Human Neutrophils. PLoS Path 2010, 6 (10), e1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boles BR; Horswill AR, agr-Mediated Dispersal of Staphylococcus aureus Biofilms. PLoS Path 2008, 4 (4), e1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olson ME; Todd DA; Schaeffer CR; Paharik AE; Van Dyke MJ; Büttner H; Dunman PM; Rohde H; Cech NB; Fey PD; Horswill AR, Staphylococcus epidermidis agr Quorum-Sensing System: Signal Identification, Cross Talk, and Importance in Colonization. J. Bacteriol 2014, 196 (19), 3482–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qin L; Da F; Fisher EL; Tan DCS; Nguyen TH; Fu C-L; Tan VY; McCausland JW; Sturdevant DE; Joo H-S; Queck SY; Cheung GYC; Otto M, Toxin Mediates Sepsis Caused by Methicillin-Resistant Staphylococcus epidermidis. PLoS Path 2017, 13 (2), e1006153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le KY; Otto M, Quorum-sensing regulation in staphylococci-an overview. Front. Microbiol 2015, 6, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayville P; Ji G; Beavis R; Yang H; Goger M; Novick RP; Muir TW, Structure-activity analysis of synthetic autoinducing thiolactone peptides from Staphylococcus aureus responsible for virulence. Proc. Natl. Acad. Sci. U.S.A 1999, 96 (4), 1218–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lyon GJ; Mayville P; Muir TW; Novick RP, Rational design of a global inhibitor of the virulence response in Staphylococcus aureus, based in part on localization of the site of inhibition to the receptor-histidine kinase, AgrC. Proc. Natl. Acad. Sci. U. S. A 2000, 97 (24), 13330–13335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDowell P; Affas Z; Reynolds C; Holden MT; Wood SJ; Saint S; Cockayne A; Hill PJ; Dodd CE; Bycroft BW; Chan WC; Williams P, Structure, activity and evolution of the group I thiolactone peptide quorum-sensing system of Staphylococcus aureus. Mol. Microbiol 2001, 41 (2), 503–12. [DOI] [PubMed] [Google Scholar]
- 36.Lyon GJ; Wright JS; Muir TW; Novick RP, Key Determinants of Receptor Activation in the agr Autoinducing Peptides of Staphylococcus aureus. Biochemistry 2002, 41 (31), 10095–10104. [DOI] [PubMed] [Google Scholar]
- 37.Scott RJ; Lian L-Y; Muharram SH; Cockayne A; Wood SJ; Bycroft BW; Williams P; Chan WC, Side-chain-to-tail thiolactone peptide inhibitors of the staphylococcal quorum-sensing system. Bioorg. Med. Chem. Lett 2003, 13 (15), 2449–2453. [DOI] [PubMed] [Google Scholar]
- 38.Fowler SA; Stacy DM; Blackwell HE, Design and Synthesis of Macrocyclic Peptomers as Mimics of a Quorum Sensing Signal from Staphylococcus aureus. Org. Lett 2008, 10 (12), 2329–2332. [DOI] [PubMed] [Google Scholar]
- 39.George EA; Novick RP; Muir TW, Cyclic Peptide Inhibitors of Staphylococcal Virulence Prepared by Fmoc-Based Thiolactone Peptide Synthesis. J. Am. Chem. Soc 2008, 130 (14), 4914–4924. [DOI] [PubMed] [Google Scholar]
- 40.Tal-Gan Y; Stacy DM; Foegen MK; Koenig DW; Blackwell HE, Highly Potent Inhibitors of Quorum Sensing in Staphylococcus aureus Revealed Through a Systematic Synthetic Study of the Group-III Autoinducing Peptide. J. Am. Chem. Soc 2013, 135 (21), 7869–7882. [DOI] [PubMed] [Google Scholar]
- 41.Tal-Gan Y; Stacy DM; Blackwell HE, N-Methyl and peptoid scans of an autoinducing peptide reveal new structural features required for inhibition and activation of AgrC quorum sensing receptors in Staphylococcus aureus. Chem. Commun 2014, 50 (23), 3000–3003. [DOI] [PubMed] [Google Scholar]
- 42.Johnson JG; Wang B; Debelouchina GT; Novick RP; Muir TW, Increasing AIP Macrocycle Size Reveals Key Features of agr Activation in Staphylococcus aureus. ChemBioChem 2015, 16 (7), 1093–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasquez JK; Tal-Gan Y; Cornilescu G; Tyler KA; Blackwell HE, Simplified AIP-II Peptidomimetics Are Potent Inhibitors of Staphylococcus aureus AgrC Quorum Sensing Receptors. ChemBioChem 2017, 18 (4), 413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kirchdoerfer RN; Garner AL; Flack CE; Mee JM; Horswill AR; Janda KD; Kaufmann GF; Wilson IA, Structural Basis for Ligand Recognition and Discrimination of a Quorum-quenching Antibody. J. Biol. Chem 2011, 286 (19), 17351–17358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen A; Mansson M; Bojer MS; Gram L; Larsen TO; Novick RP; Frees D; Frokiaer H; Ingmer H, Solonamide B inhibits quorum sensing and reduces Staphylococcus aureus mediated killing of human neutrophils. PLoS One 2014, 9 (1), e84992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sully EK; Malachowa N; Elmore BO; Alexander SM; Femling JK; Gray BM; DeLeo FR; Otto M; Cheung AL; Edwards BS; Sklar LA; Horswill AR; Hall PR; Gresham HD, Selective Chemical Inhibition of agr Quorum Sensing in Staphylococcus aureus Promotes Host Defense with Minimal Impact on Resistance. PLoS Path 2014, 10 (6), e1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tal-Gan Y; Ivancic M; Cornilescu G; Yang T; Blackwell HE, Highly Stable, Amide-Bridged Autoinducing Peptide Analogues that Strongly Inhibit the AgrC Quorum Sensing Receptor in Staphylococcus aureus. Angew. Chem. Int. Ed 2016, 55 (31), 8913–8917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vasquez JK; Blackwell HE, Simplified Autoinducing Peptide Mimetics with Single-Nanomolar Activity Against the Staphylococcus aureus AgrC Quorum Sensing Receptor. ACS Infect. Dis 2019, 5 (4), 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dufour P; Jarraud S; Vandenesch F; Greenland T; Novick RP; Bes M; Etienne J; Lina G, High Genetic Variability of the agr Locus in Staphylococcus Species. J. Bacteriol 2002, 184 (4), 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang T; Tal-Gan Y; Paharik AE; Horswill AR; Blackwell HE, Structure–Function Analyses of a Staphylococcus epidermidis Autoinducing Peptide Reveals Motifs Critical for AgrC-type Receptor Modulation. ACS Chem. Biol 2016, 11 (7), 1982–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tal-Gan Y; Ivancic M; Cornilescu G; Blackwell HE, Characterization of structural elements in native autoinducing peptides and non-native analogues that permit the differential modulation of AgrC-type quorum sensing receptors in Staphylococcus aureus. Org. Biomol. Chem 2016, 14 (1), 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tal-Gan Y; Ivancic M; Cornilescu G; Cornilescu CC; Blackwell HE, Structural Characterization of Native Autoinducing Peptides and Abiotic Analogues Reveals Key Features Essential for Activation and Inhibition of an AgrC Quorum Sensing Receptor in Staphylococcus aureus. J. Am. Chem. Soc 2013, 135 (49), 18436–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwieters CD; Kuszewski JJ; Tjandra N; Clore GM, The Xplor-NIH NMR Molecular Structure Determination Package. J. Magn. Reson 2003, 160, 66–74. [DOI] [PubMed] [Google Scholar]
- 54.Schwieters C; Kuszewski J; Mariusclore G, Using Xplor–NIH for NMR molecular structure determination. Prog. Nucl. Magn. Reson. Spectrosc 2006, 48 (1), 47–62. [Google Scholar]
- 55.The PyMOL Molecular Graphics System, Version 1.7, Schrödinger, LLC: 2015. [Google Scholar]
- 56.Vijayakumar M; Qian H; Zhou HX, Hydrogen bonds between short polar side chains and peptide backbone: Prevalence in proteins and effects on helix forming propensities. Proteins: Structure, Function, and Bioinformatics 1999, 34 (4), 497–507. [PubMed] [Google Scholar]
- 57.Flöckner H; Braxenthaler M; Lackner P; Jaritz M; Ortner M; Sippl MJ, Progress in fold -recognition. Proteins: Structure, Function, and Bioinformatics 1995, 23 (3), 376–386. [DOI] [PubMed] [Google Scholar]
- 58.Khan BA; Yeh AJ; Cheung GY; Otto M, Investigational therapies targeting quorum-sensing for the treatment of Staphylococcus aureus infections. Exp. Opin. Invest. Drugs 2015, 24 (5), 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wilmot CM; Thornton JM, β-Turns and their distortions: a proposed new nomenclature. Protein Eng 1990, 3 (6), 479–493. [DOI] [PubMed] [Google Scholar]
- 60.Davis IW; Leaver-Fay A; Chen VB; Block JN; Kapral GJ; Wang X; Murray LW; Arendall WB 3rd; Snoeyink J; Richardson JS; Richardson DC, MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 2007, 35 (suppl_2), W375–W383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen VB; Arendall III WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst., Section D: Struct. Biol 2010, 66 (1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stanger HE; Gellman SH, Rules for Antiparallel β-Sheet Design: d-Pro-Gly Is Superior to l-Asn-Gly for β-Hairpin Nucleation. J. Am. Chem. Soc 1998, 120 (17), 4236–4237. [Google Scholar]
- 63.Mitchell JBO; Smith J, D-amino acid residues in peptides and proteins. Proteins: Structure, Function, and Bioinformatics 2003, 50 (4), 563–571. [DOI] [PubMed] [Google Scholar]
- 64.Mielke SP; Krishnan VV, Characterization of protein secondary structure from NMR chemical shifts. Prog. Nucl. Magn. Reson. Spectrosc 2009, 54 (3–4), 141–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Saitô H, Conformation-dependent 13C chemical shifts: A new means of conformational characterization as obtained by high-resolution solid-state 13C NMR. Magn. Reson. Chem 1986, 24 (10), 835–852. [Google Scholar]
- 66.Szilagyi L, Chemical shifts in proteins come of age. Prog. Nucl. Magn. Reson. Spectrosc 1995, 27, 325–443. [Google Scholar]
- 67.Wishart DS; Sykes BD, The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 1994, 4 (2), 171–80. [DOI] [PubMed] [Google Scholar]
- 68.Shen Y; Bax A, Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 2013, 56 (3), 227–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Williamson MP, Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc 2013, 73, 1–16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.