

SUMMARY:
The immune system monitors the health of cells and is stimulated by necrosis. Here we examined the receptors and ligands driving this response. In a targeted screen of C-type lectin receptors, a Clec2d reporter responded to lysates from necrotic cells. Biochemical purification identified histones, both free and bound to nucleosomes or neutrophil extracellular traps, as Clec2d ligands. Clec2d recognized poly-basic sequences in histone tails and this recognition was sensitive to post-translational modifications of these sequences. As compared to WT mice, Clec2d−/− mice exhibited reduced proinflammatory responses to injected histones, and less tissue damage and improved survival in a hepatotoxic injury model. In macrophages, Clec2d localized to the plasma membrane and endosomes. Histone binding to Clec2d did not stimulate kinase activation or cytokine production. Rather, histone-bound DNA stimulated endosomal Tlr9-dependent responses in a Clec2d-dependent manner. Thus, Clec2d binds to histones released upon necrotic cell death, with functional consequences to inflammation and tissue damage.
Keywords: C-type lectin, Clec2d, histones, DAMPs, necrosis, macrophages, liver injury, inflammation, histone acetylation, Toll-like receptor, pattern recognition receptor
Graphical Abstract
INTRODUCTION
When cells die by apoptosis in vivo, which is a frequent event in normal physiology, it is usually immunologically silent (Rock and Kono, 2008). Such cells are rapidly cleared by phagocytes and the apoptotic corpses may actively suppress an immune reaction (Peng et al., 2007). In contrast, when cells die in vivo by necrosis, this event is not silent (Rock et al., 2011). Tissue resident sentinel cells, such as macrophages and dendritic cells, recognize the dying cells and initiate responses (Kono et al., 2010b; Rock et al., 2011). The cellular corpses are cleared by phagocytosis and cytokines are elaborated that stimulate inflammation and tissue repair. Dendritic cells also acquire antigens from the dying cells and are stimulated into an activated state capable of initiating adaptive immune responses (Shi and Rock, 2002; Shi et al., 2000). It is thought that the immune system responds in these ways because necrotic death is pathological (Kono and Rock, 2008; Matzinger, 2002). The loss of functional tissue is a threat to the host and an indicator of an injurious process. The immune responses that are mobilized attempt to neutralize or wall off the inciting event, clear debris and ultimately stimulate repair.
The neutrophils and macrophages that are present at a site of inflammation, elaborate potent defense mechanisms, including e.g. reactive oxygen species and proteases. These effector molecules can kill microbes but also damage cells of the host, and this collateral damage can lead to disease. This is illustrated in the setting of an overdose of acetaminophen (APAP), where toxic metabolites of APAP damage hepatocytes, and the necrotic hepatocytes elicit an acute inflammatory response (Krenkel et al., 2015). The ensuing inflammation then causes further tissue damage, which markedly extends the amount of tissue necrosis and hepatic dysfunction (Krenkel et al., 2015). In addition, the production of cytokines stimulated by immune recognition of cell death is thought to drive the development of some cancers (Kuraishy et al., 2011) and to stimulate the growth of malignant cells that survive after cytoablative therapy (Sulciner et al., 2018). Moreover, dendritic cells activated by dying cells can stimulate adaptive immune responses, which further helps mobilize defenses, but is also thought to pose the risk of triggering autoimmunity in some settings (Zelenay and Reis e Sousa, 2013). Because of the protective and pathological roles of cell death-induced immune responses, it is important to understand how the immune system recognizes dead cells and initiates responses.
When a cell undergoes necrosis, its plasma membrane ruptures, which releases intracellular components that stimulate innate immune cells (e.g. macrophages or dendritic cells) (Rock and Kono, 2008). Because these stimulatory endogenous components are not normally present outside of cells, but only exposed after necrotic cell death, their presence in the extracellular space allows the immune system to detect necrosis. Such endogenous “alarm” signals have been termed “damage-associated molecular patterns” (DAMPs) (Land, 2003); this appellation arose by analogy to the term “pathogen-associated molecular patterns” (PAMPs), which are the “alarm” signals that allow the innate immune system to recognize microbes (Janeway, 1989).
So far, a number of DAMPs have been identified and it is almost certain that more exist. Known DAMPs include molecules resident in the cytosol (e.g. ATP, uric acid, and HSPs (heat shock proteins)), granules (e.g. defensins and granulysin), and nucleus (e.g. HMGB1 and histones) (Chen and Nunez, 2010; Xu et al., 2009). Some of the known DAMPs have been shown to play an important role in pathophysiology. For example, depletion of ATP or uric acid, or neutralizing extracellular histones with antibodies has been shown to ameliorate some of the pathological consequences of tissue injury (Kono et al., 2010a; McDonald et al., 2010; Xu et al., 2009). Therefore, it is important to better understand what DAMPs drive responses and how they do so.
Some of the innate pattern recognition receptors (PRRs) that are involved in the recognition of DAMPs have been identified. Some examples include the purinoceptor P2X7 that is stimulated by ATP (Ferrari et al., 2006) and the receptor for advanced glycation end products (RAGE) that is one of the receptors that recognizes HMGB1 (Rauvala and Rouhiainen, 2007). In addition, PAMP receptors have been implicated in responses stimulated by some DAMPs. For instance, TLR4 has been suggested to participate in responses to HSPs, HMGB1, histones, and a number of other DAMPs (Chen and Nunez, 2010; Xu et al., 2011). Nlrp3 has been implicated in responses to monosodium urate crystals, histones and also biglycan (Babelova et al., 2009; Huang et al., 2013; Rock et al., 2010). However, in many cases it is unclear whether the PAMP PRRs recognize DAMPs directly or indirectly, e.g. via some stimulatory molecules bound to or contaminating the DAMPs (Tsan and Gao, 2004, 2007). In any case our current understanding of DAMP receptors and their role is incomplete and it is highly likely that there are additional DAMP receptors yet to be discovered.
A class of PRRs, of which some are involved in recognition of PAMPs, are C-type lectin receptors (CLRs). These are type II transmembrane proteins, and many of them recognize carbohydrate motifs on the PAMPs in a calcium-dependent manner, and some bind protein sequences (Brown et al., 2018). A few CLRs have been found to bind mammalian ligands. For example, Clec9a recognizes F-actin-myosin II complexes released from necrotic cells (Ahrens et al., 2012; Hanc et al., 2015; Sancho et al., 2009; Schulz et al., 2018). Clec9a is expressed only by CD8α+ dendritic cells and does not appear to be involved in death-induced inflammation (Zelenay et al., 2012). Another example is Mincle that recognizes the nuclear protein SAP130 (Yamasaki et al., 2008). Mincle was suggested to be involved in cell death-induced inflammation because this response was inhibited by anti-Mincle antibodies (Yamasaki et al., 2008), however genetic deletion of this receptor did not replicate these results (Kataoka et al., 2014). Other than these few examples, the role of CLRs in recognizing DAMPs and stimulating sterile inflammation is unknown.
Given the precedence of a CLR recognizing injured cells and macrophages being key cells that respond to cell death (Kono et al., 2010b), here we screened a number of macrophage-expressed CLRs to identify a new one, Clec2d, that recognized a ligand that was exposed in necrotic cells. We purified the stimulatory ligands and found that they were histones, which were known DAMPs. Clec2d−/− mice had reduced inflammatory responses to histones and less collateral tissue damage in a liver injury model.
RESULTS
Identifying Clec2d as a Novel DAMP Receptor
To test the hypothesis that a CLR on macrophages was involved in DAMP recognition, we established a cell-based screening system. We constructed chimeric receptors composed of the extracellular domain of a macrophage-expressed C-type lectin (with a C-terminal HA tag) fused to the CD3ζ intracellular domain (Sancho et al., 2009). CD3ζ is a component of the TCR complex that transduces activation signals after TCR stimulation (Alcover et al., 2018). Signaling through CD3ζ activates the transcription factor NFAT, which in turn induces IL-2 gene expression (Huse, 2009). The chimeric receptors were expressed in RF33.70-Luc reporter cells, which are CD8 T cell hybridomas derived from RF33.70 cells (Rock et al., 1990) that were transduced with a luciferase reporter gene under the control of NFAT response elements. When such chimeric receptors bound ligands under the appropriate conditions, luciferase was expressed in the reporter cells (Figure S1A). Because the CLR constructs had C-terminal (extracellular) HA tags, we could use anti-HA antibody to verify expression and also stimulate the chimeric receptors as a positive control. It is well known that signaling through TCR or chimeric CD3ζ fusion proteins generally requires the stimulating ligands be immobilized (e.g. bound to plastic) (Bamezai et al., 1989; Linch et al., 1987) and we confirmed this for our constructs using anti-HA antibody (Figure S1B). Therefore, when using this system to identify potential novel DAMP receptors, we stimulated the CLR reporter cells with supernatants from necrotic cell (EL4) lysates that were coated on microtiter wells.
Using this approach, we found that reporter cells expressing the Clec2d chimeric receptor were stimulated by necrotic EL4 cells. This stimulation required Clec2d because RF33.70-Luc cells expressing Clec1b, Clec4a2, or Dectin-1 chimeric receptors were not stimulated by this necrotic material (Figure 1A & 1B). In contrast, the reporter cells expressing Clec1b and Clec4a2 (as well as Clec2d) were stimulated to express luciferase by anti-HA antibody, and Dectin-1 reporter cells were stimulated by zymosan (a ß-glucan rich particle known to bind Dectin-1), indicating that these CLRs were all expressed and the reporter cells were active (Figure 1A & 1B). To further confirm the participation of the transfected Clec2d in the responses, we used soluble anti-HA antibodies (which bind to the HA tag on the Clec2d- CD3ζ fusion proteins) and found that they blocked Clec2d stimulation by necrotic EL4 cells in the reporter assay (Figure S1C).
Figure 1. Using a Reporter System to Screen for Novel DAMP Receptors.

(A) RF33.70-Luc reporters expressing the indicated CLR-CD3ζ chimeric receptors were stimulated overnight with or without necrotic EL4 cell lysates, and then luciferase activity was assayed. Anti-HA (αHA) was used to stimulate reporter cells as a positive control. Control, reporter cells in media without necrotic lysates. (B) Clec2d or Dectin-1 reporters were left without stimulation (No stim) or stimulated with supernatant from live or necrotic EL4 cultures, and the luciferase activity was measured. Zymosan was used to stimulate Dectin-1 reporters as a positive control. (C, D) Live or necrotic EL4 cells were incubated with soluble Clec2d-Ig or Dectin-1-Ig, followed by staining with secondary antibodies conjugated with fluorophore and analyzed by flow cytometry. (E) Live or necrotic EL4 cells (1x105 or 3x105/well) were fixed with paraformaldehyde and then used to stimulate Clec2d reporters, and the luciferase activity was measured. (F) Reporter cells expressing Clec2d chimeric receptors were stimulated without (control) or with necrotic mouse splenocytes or EL4 cells overnight, and then luciferase activity was measured. (G) Clec2d reporter cells were stimulated overnight without (control) or with necrotic EL4, 293T, or Jurkat cells and then luciferase activity was measured. (H) Six or 22 hr after APAP (300 or 500 mg/KgBW) or PBS injection into WT mice, serum was collected and assayed for ALT activity and stimulation of Clec2d reporter cells. Pearson correlation coefficient analysis of Clec2d reporter activity versus serum ALT activity, with r=0.84, p<0.0001, R2=0.71. See also Figure S1.
We found that supernatant from necrotic but not live EL4 cultures stimulated Clec2d reporters (Figure 1B), indicating that Clec2d indeed recognized a molecular pattern(s) released from damaged cells, i.e. a DAMP(s). Clec2d ligands were also exposed on the surface of necrotic but not live cells as detected by staining with a Clec2d-immunoglobulin fusion protein (Clec2d-Ig). In contrast, Dectin-1-Ig (another CLR-Ig fusion) didn’t bind either necrotic or live EL4 cells. (Figure 1C & 1D), showing specificity. We also found that paraformaldehyde-fixed necrotic, but not live EL4 cells, stimulated reporter cells expressing Clec2d, further indicating that some of the Clec2d ligands are exposed and remain cell-associated on necrotic cells (Figure 1E). We found that Clec2d reporter cells were also stimulated by necrotic lysates of mouse splenocytes (Figure 1F). These findings generalized our initial results and ruled out the possibility that Clec2d was being stimulated by some adventitious microbial contaminant in cultured cells. We also found that Clec2d reporter cells were stimulated by necrotic human cells (293T and Jurkat), indicating that the ligand(s) for Clec2d is broadly expressed and evolutionarily conserved, at least between mouse and human (Figure 1G).
We next investigated whether stimulatory amounts of the Clec2d ligand(s) were released in a situation where there was limited necrosis occurring in vivo. For this purpose, we utilized the well-established APAP overdose mouse model. In mice (and humans) hepatocytes convert APAP to the cytotoxic metabolite, NAPQI (N-acetyl-p-benzoquinone imine), which when present in sufficient amounts causes necrosis in the liver (Ramachandran and Jaeschke, 2017). We found that serum from APAP-treated mice, but not from PBS-treated controls, stimulated Clec2d reporter cells, and this Clec2d-stimulatory effect was significantly and positively correlated to the amount of necrosis as assessed by serum ALT activity (Jaeschke et al., 2014) (Figure 1H). These results again indicated that Clec2d was detecting a bona fide DAMP(s) (a molecular pattern released upon cellular damage), and that this DAMP(s) was released in vivo in sufficient amounts to bind Clec2d. In fact, given that we were measuring this DAMP after it had distributed and been diluted in body fluids, its amount in the damaged liver must have been considerably higher.
Identifying Histones as Clec2d Ligands
We next sought to purify the DAMP(s) that Clec2d recognized. To accomplish this, we first sought to better understand the properties of the Clec2d ligand(s). Proteinase K treatment substantially reduced the lysate’s ability to stimulate Clec2d reporter cells (Figure 2A), suggesting that the Clec2d’s ligand(s) was a protein. We also treated necrotic lysates with urea and/or DTT (Figure 2B), or SDS and/or heat denaturation (Figure 2C) and this did not reduce Clec2d stimulatory activity. In fact, treatment with SDS actually increased the stimulatory activity of the lysates (Figure 2C). These results suggested that Clec2d recognized primary protein sequences rather than the tertiary structure of a DAMP(s), and SDS treatment further exposed the Clec2d stimulatory region.
Figure 2. Characterization of the Necrotic Cell Ligands Recognized by Clec2d.

(A) Necrotic EL4 lysates were treated without or with Proteinase K-agarose beads (0.35 unit/ml) at 37°C for 4 hr. The protease beads were then removed and the lysates were tested for their ability to stimulate Clec2d reporter cells to produce luciferase. ***, p<0.001 with One-way ANOVA. (B) Necrotic EL4 lysates were treated with or without 8 M urea and/or 20 mM DTT at RT for 30 min, and then assayed for their ability to stimulate Clec2d reporter cells to produce luciferase. N.S., no significant difference. (C) Necrotic EL4 lysates were treated with or without 2% SDS and/or heated at 95°C for 10 min. These lysates were then used to stimulated Clec2d reporter cells to produce luciferase. ***, p<0.001 with One-way ANOVA. (D, E) The subcellular fraction enriched with ligand activity (See also Figure S2.) was further fractionated by SDS-PAGE. Part of the gel was used for silver stain to reveal protein bands (E), and the rest of the gel was transferred onto PVDF membrane, and sliced into 23 fractions. Each membrane slice was co-cultured with Clec2d reporter cells, and then luciferase activity was measured (D). Protein bands (arrow heads) in gel fractions with the highest luciferase activity (#3, 5 and 6) were analyzed by Mass Spectrometry, which revealed the presence of histone H4, H2A/H2B, and H3 as the major proteins in each fraction, respectively. (F) Reporter cells with Clec2d or Dectin-1 chimeric receptors were stimulated with purified histones from calf thymus at the indicated concentrations. Luciferase activity was measured in the lysate after overnight stimulation. (G) Zymosan particles (Zym) or agarose beads conjugated with histones (His) or BSA (BSA) were incubated with soluble Clec2d or Dectin-1 receptors expressed as mouse IgG Fc fusion proteins. Proteins that bound these particles were eluted, and analyzed with anti-mouse IgG antibodies by western blots. (H) Reporter cells with Clec2d or Dectin-1 chimera receptors were stimulated overnight with the indicated recombinant human histone proteins or zymosan and then luciferase activity was measured. Control = reporter cells cultured in media without histones or zymosan. See also Figure S2 and S3.
We also used a serial cell extraction kit (Pierce) to fractionate EL4 cells and found that the Clec2d-stimulating activity was present in the last fractions (Figure S2A, see also the materials and methods). Knowing this we took advantage of the resistance of the Clec2d-stimulating activity to protein denaturation to fractionate the DAMP-containing cell fraction by SDS-PAGE and then transferred the fractionated proteins onto a PVDF membrane. This membrane was subsequently sliced into small pieces and three of these fractions (#3, 5, 6) strongly stimulated Clec2d reporter cells (Figure 2D). In a parallel SDS-PAGE gel subjected to a silver stain, the active fractions contained major protein bands (arrow heads in Figure 2E).
We next analyzed the proteins in the 3 gel fractions with the most activity by mass spectrometry and identified the major proteins in fraction #3, 5, and 6 as histone H4, H2A/H2B, and H3, respectively (Figure S3). To verify that histones were indeed stimulating Clec2d, we used histones purified from calf thymus and found that they could strongly stimulate the Clec2d reporter cells (Figure 2F). This stimulation was dependent on Clec2d because control Dectin-1 reporter cells, which were stimulated by the Dectin-1 ligand zymosan (Figure 2H), did not respond to the purified histones (Figure 2F). In addition, the stimulation of Clec2d reporters by histones could be blocked by soluble anti-HA antibodies that bind the HA-tag on Clec2d (Figure S1D). We also found that histone-conjugated agarose beads bound soluble Clec2d-Ig (Figure 2G). This histone-Clec2d binding was specific because soluble Clec2d did not bind BSA-agarose beads or zymosan particles (Figure 2G). In contrast, soluble Dectin-1 was pulled down by zymosan but not histone or BSA-conjugated beads (Figure 2G). Therefore, these data showed that histones could specifically bind to Clec2d, confirming the results of our earlier reporter cell assays.
Recombinant histones H2A, H2B, H3, and H4, including ones purified by SDS-PAGE, could stimulate Clec2d reporter cells in a dose-dependent manner but not the control Dectin-1 reporter (Figure 2H; Figure S2B, S2C and 2D). Given that histone H1 has similarity in sequence and structure to the H2-H4 histones, we tested and found that recombinant H1 also stimulated the Clec2d-expressing but not control reporter cells (Figure 2H; Figure S2B). These results indicated that Clec2d is able to recognize all 5 histone proteins.
Mapping the Clec2d-Binding Regions of Histone Proteins
Since Clec2d recognized all 5 histones, we first looked for candidate regions of identity between these proteins, but no extended region of identity was found. Therefore, to define the stimulatory region, we synthesized and tested overlapping peptides (24-26 a.a. in length) spanning the entire histone sequence of H4. The Clec2d stimulatory activity was located in the N-terminal tail region of H4 (residues 1-30), with the highest activity residing between amino acid residues 2 to 27 (H42-27) (Figure 3A; Figure S4A). We refined this analysis using smaller synthetic peptides and found that H46-21 and H416-27 contained ligand activity while H42-12 did not (Figure 3B; Figure S4C).
Figure 3. Mapping the Clec2d-Binding Regions of Histone H4.

(A) Overlapping synthetic peptides of mouse histone H4 or no peptide (control) were tested for their ability to stimulate Clec2d or Dectin-1 reporter cells to produce luciferase activity. Zymosan and recombinant H4 (Rec. H4) were used as positive controls. The H4 number ranges denote the amino acid positions within the H4 sequence of the synthetic peptides, where 1 is the N-terminal residue). (B) Smaller synthetic peptides located at the N-terminal (H42-12), middle (H46-21), or C-terminal (H416-27) of H42-27 were used to stimulate Clec2d reporters and the luciferase activity was measured. Control = reporter cells cultured without peptides. (C) Similar to (B), synthetic peptides of the tail regions in all 5 histones were used to stimulate the Clec2d reporters. Zymosan was used as a positive control for stimulation of the Dectin-1 reporters. (D) Similar to (B) but the smaller synthetic peptides were within H46-21 containing different numbers of basic residues. Control = reporter cells cultured without peptides. (E) Pearson correlation coefficient analysis of (D) of Clec2d reporter activity versus number of basic residues in the synthetic peptides, with r=0.94, p=0.016, R2=0.89. (F) Similar to (B) except the peptides were H49-21 or its derivative peptides, “K(Ac)” peptide with all lysine residues acetylated, or “K to A” peptide with all lysine residues replaced by alanine. Control = reporter cells cultured without peptides. (G) EL4 cells were treated with or without TSA (50 nM) for 19 hr and then disrupted by freeze and thaw cycles. The resulting lysates were analyzed by western blot and probed for H3K27 acetylation or total H3 histone, or (H) used to stimulate Clec2d reporter cells and after which luciferase activity was measured. ****, p<0.0001, with Student’s t-test. See also Figure S4.
The H4 Clec2d-stimulatory region sequence did not share identity with the other 4 histone proteins, as noted above. However, all of the 5 histones had sequence similarities in their N-terminal (H1-H4) and C-terminal (H1) tail regions. To test if these were the regions recognized by Clec2d, we synthesized peptides from the similar N-terminal tail regions of H1-H4 and the C-terminal tail of H1 and found that they all stimulated the Clec2d reporter cells (Figure 3C; Figure S4B).
Positive Charges on Lysine Residues in Histone Tails are Critical for Clec2d Recognition
A striking feature of the stimulatory histone peptide sequences was that they all contained multiple Lysine (K), Arginine (R), and Histidine (H) (Figure S4B). This led us to analyze whether these basic residues were important for Clec2d recognition. We first synthesized even smaller fragments of the histone H46-21 that contained different numbers of basic residues and found that their stimulatory activity positively correlated with the numbers of basic residues within the fragments (Figure 3D and 3E).
To further test the hypothesis that basic residues contributed to Clec2d recognition, we stimulated the reporter cells with the H49-21 fragment with all four Lysines acetylated (H49-21-K(Ac)), which removed the positive charge from lysines. Another peptide, H49-21-K to A, derived from H49-21 as well but with all four Lysines replaced by Alanines (Figure S4C), was also tested. Both acetylation and Alanine replacement of Lysine residues completely destroyed the stimulatory activity of H49-21, suggesting that the positive charged residues on histone tails are critical for Clec2d recognition (Figure 3F).
Our data showing that the positive charge on lysines was important to Clec2d stimulation, suggested that overall histone acetylation in cells might affect their ability to stimulate Clec2d. To test this hypothesis, we treated EL4 cells with Trichostatin A (TSA), an agent that inhibits histone deacetylase (Yoshida et al., 1990), and confirmed that this treatment increased histone H3 (in this case, H3K27) acetylation in the EL4 cells as expected, while the total H3 amount was unchanged (Figure 3G). We found that necrotic lysates from these TSA-treated cells had reduced Clec2d stimulatory activity (Figure 3H). These results suggested that the state of histone acetylation can indeed affect the activity of histones as DAMPs.
Clec2d-Deficient Mice are More Resistant to the Sequelae of Tissue Injury
While histones are known to be pathogenic DAMPs, how they mediate their effects is not clear and in particular, whether there is a histone receptor involved in this process. Toll-like receptors (TLRs) have been implicated in histone-mediated effects in some studies, but it is unknown if this is a direct histone effect (Allam et al., 2012; Huang et al., 2011; Xu et al., 2011). With our discovery that Clec2d was a histone receptor, we hypothesized that it could be involved in histone-mediated pathophysiological effects. We investigated this issue in the APAP-induced liver injury model wherein DAMP release incites inflammation, which in turn amplifies the extent of tissue damage (Krenkel et al., 2015). Histone release had previously been implicated in amplifying tissue injury and increasing morbidity in this model (Xu et al., 2011).
We found that at an early time point after APAP administration (6 hr), serum ALT activity was comparable between WT and Clec2d−/− mice, indicating that the intrinsic hepatotoxicity of APAP was similar in both groups (Figure 4A). However, at a later time point (12 hr), when the host mounts an inflammatory response that amplifies injury (Chen et al., 2007; Liu et al., 2006; Marques et al., 2012; Zhang et al., 2018), we found that the serum ALT activity had further increased significantly in the WT but not in the Clec2d−/− mice, indicating that WT mice continued to sustain more liver damage over time, while the Clec2d−/− mice did not (Figure 4A). In addition, over time around 30% of WT mice died, while almost all (97%) of Clec2d−/− mice survived and this difference was also significant (Figure 4B). Together these results indicate that Clec2d can sense cell injury in vivo and in response promote subsequent tissue damage.
Figure 4. Clec2d Enhanced Liver Injury in Acetaminophen Overdose.

(A) WT and Clec2d−/− mice were fasted for 24 hr and then injected i.p. with 300 mg/KgBW APAP. Six or 12 hr after APAP injection, serum ALT activity was measured. 6 hr (WT), n=26; 6 hr (Clec2d−/−), n=17; 12 hr (WT), n=54; 12 hr (Clec2d−/−) n=42. Mean ± SEM. *, p<0.05 by One-Way ANOVA. (B) Survival of the mice was monitored for 5 days after injection. WT, n=38; Clec2d−/−, n=32. Survival curve comparison was analyzed by a Log-rank Mantel-Cox test. **, p<0.01. (C) Western blot to detect histone H3 in the serum of WT mice with or without APAP treatment. Mouse serum IgG was also detected on the same blot as a loading control. (D) Histones from the serum of APAP-treated mice were purified by anti-histone (αHis) or isotype control (IsoAb) antibody-conjugated beads. The eluate from the beads were immobilized on the assay plate to stimulate reporter cells expressing Clec2d and luciferase activity was measured after 20 hr. Anti-HA (αHA) antibody was used as a positive control for reporter stimulation. Control=reporter cells in media. (E, F) Histones in the serum of APAP-treated mice (APAP serum) were immunodepleted by anti-histone (αHis) or isotype control (IsoAb) antibody-conjugated beads. Histone H3 in the post-treatment serum or bead-eluates was detected by western blot (E). The APAP serum after treatment was immobilized on a plate and used to stimulate the Clec2d reporter cells (F). Control = reporter cells in media. Mean ± SEM. **, p<0.01 by One-Way ANOVA. (G) WT and Clec2d−/− mice were i.v. injected with purified histones, and 18 hr later cytokine and chemokine concentrations were measured in the serum. WT and Clec2d−/−, n=7; PBS control, n=5. Mean ± SEM. *, p<0.05 by One-Way ANOVA.
The above data are consistent with but do not prove that Clec2d recognition of histones, versus e.g. some other DAMPs or mechanism, is how Clec2d is exacerbating liver injury, and unfortunately, we lack reagents to selectively block histone-Clec2d interaction in vivo to definitively test this point. However, several lines of further evidence suggest that histone – Clec2d interactions are likely to underlie the pathophysiology. Neutralizing histones with antibodies has been shown to similarly limit the late damage in liver injury models, which implicates histones as a major DAMPs inciting further damage, and fits nicely with our findings (Wen et al., 2016; Xu et al., 2011; Yang and Tonnesseen, 2019). This is remarkable because multiple DAMPs contribute to the pathology in this model and function through other receptors (Woolbright and Jaeschke, 2017). In further analyses, we found that histones were detectable in the serum of APAP-treated mice, but not PBS-treated mice (Figure 4C), which was consistent with earlier reports (Allam et al., 2014; Wen et al., 2013; Xu et al., 2011) and confirmed that histones were released from the injured liver. When these APAP-released histones were purified from the serum, they could stimulate Clec2d, indicating that they were biologically active (Figure 4D). Furthermore, we found that Clec2d stimulatory activity in the APAP serum was almost completely removed after immunodepleting histones from the serum (Figure 4E & 4F). These data showed that histones were the major Clec2d ligands in the serum of APAP-induced liver injury model. Moreover, to study the effects of the extracellular histones in vivo, we injected purified histones i.v. into mice and found that inflammatory cytokines were subsequently detectable in the serum of WT mice. This histone-stimulated cytokine response was significantly reduced in Clec2d−/− mice (Figure 4G), indicating that extracellular histones induce inflammatory responses that were at least partially dependent on Clec2d.
Characterization of the Expression of Clec2d in Macrophages
Clec2d was not detectable in the macrophage cell line (iBMDM) (data not shown). In order to study Clec2d expression and function, we transfected iBMDM cells with a Clec2d construct containing a C-terminal HA (extracellular) and an N-terminal mCherry (cytosolic) tags that was under the control of a doxycycline (Dox)-inducible promoter. Upon Dox treatment and staining with anti-HA antibody, expression of Clec2d on the cell surface was detected by flow cytometry (Figure 5A). Using confocal microscopy to visualize mCherry, we found Clec2d was present both on the plasma membrane and in intracellular vesicles. The expression in vesicles colocalized with Rab7 (a late endosomal protein) and Calreticulin (an ER protein) but not Eea1 (an early endosomal protein) (Figure 5B). We have not been able to similarly visualize the subcellular distribution of endogenous Clec2d due to the lack of suitable antibodies; however, we could detect endogenous Clec2d in phagosomes purified from peritoneal macrophages by western blot (Figure S5A), indicating that endogenous Clec2d also traffics to peripheral vesicles.
Figure 5. Characterization of Clec2d in Macrophages.

(A) iBMDM cells transduced with a Dox-inducible Clec2d-HA construct were cultured with or without Dox and then analyzed for surface HA staining by flow cytometry. (B) iBMDM cells transfected with a Dox-inducible Clec2d-mCherry construct (Red) were cultured with Dox, permeabilized, stained (green) for Eea1, Rab7, or Calreticulin, and analyzed by confocal microscopy. Colocalization of Clec2d with Rab7 or calreticulin is shown in yellow in the merged images and highlighted with arrows. Bars=10 μm (C) Western blot data showing that Bafilomycin (Baf) rescues total Clec2d amounts when peritoneal macrophages were treated with Cycloheximide (CHX). N/S = non-specific band on the same αClec2d blot, which served as a loading control. (D) iBMDM cells transduced with a Dox-inducible Clec2d construct were treated with or without Dox and then stimulated for overnight with or without histones (30 μg/ml) and/or CpG (1 μg/ml). TNFα and IL-6 were measured in the culture supernatant by ELISA. The data was pooled from 5 experiments. Mean ± SEM. ***, p<0.001; ****, p<0.0001 by Two-Way ANOVA. (E) Primary peritoneal macrophages were stimulated for overnight with 30 μg/ml histones and/or 0.1 μg/ml CpG. IL-6 and IP-10 were measured in the supernatant. Mean ± SEM, *, p<0.05; **, p<0.01 by Two-Way ANOVA. (F) Peritoneal macrophages from WT and Clec2d−/− mice were plated and stimulated without (None) or with 30 μg/ml histones (His) or 40 μg/ml zymosan (Zym) for 30 min. The phosphorylation of MAPKs, NFκB (p65), and Syk were detected in the cell lysates by western blot. (G) Peritoneal macrophages from WT, Clec2d−/− and/or Tlr9−/− mice were stimulated for overnight with or without histones (30 μg/ml) and/or CpG (0.1 μg/ml). RANTES was measured in the supernatant. Mean ± SEM, ****, p<0.0001 by Two-Way ANOVA. ns, not significant. See also Figure S5.
We also examined the turnover of Clec2d. When primary peritoneal macrophages, (which express high amounts of endogenous Clec2d), were treated for 5 hr with cycloheximide to inhibit protein synthesis and then evaluated by western blot, cellular amounts of Clec2d decreased to ~20%. However, if these cells were also treated with bafilomycin, an agent that inhibits endolysosomal proteolysis, then Clec2d amounts were largely maintained (Figure 5C). These data further indicated that endogenous Clec2d trafficked to endosomes, where it was degraded
Role of Clec2d in Histone-Stimulated Cytokine Responses
Since histones stimulate inflammation (Allam et al., 2014; Huang et al., 2013; Xu et al., 2011; Xu et al., 2009), and we had observed Clec2d-dependent histone stimulation of proinflammatory cytokines in vivo, we investigated the role of Clec2d in histone stimulation of macrophages to produce cytokines. In gain-of-function experiments, we tested whether purified histones stimulated cytokine responses from iBMDM cells in the absence (−Dox) or presence (+Dox) of Clec2d. In reciprocal loss-of-function experiments we tested histone-stimulated responses of Clec2d-deficient versus Clec2d-sufficient (WT) peritoneal macrophages. In both of these systems, purified histones stimulated no-to-weak production of cytokines, which when present, was not consistently dependent on Clec2d (Figure 5D and 5E).
The fact that stimulation of Clec2d with histones did not induce cytokine responses and Clec2d lacks ITAMs suggested that Clec2d was not a signaling receptor. To further explore this issue, we investigated whether histone stimulation of Clec2d on macrophages resulted in any detectable kinase activation. We found that histone stimulation of Clec2d-expressing macrophages did not activate Syk, the downstream signaling kinase of activating CLRs. We also found no detectable Clec2d-dependent activation of NFκB and other MAPKs, including p38, Erk, and Jnk upon histone treatment of the macrophages (Figure 5F). Together these data suggest that Clec2d is not a signaling receptor.
Since histones bind immunostimulatory ligands, such as DNA, and Clec2d traffics to endosomes, we hypothesized that this receptor might deliver such histone-complexes to endosomal TLRs. To test this hypothesis, we used histones and CpG DNA. We first verified that CpG oligonucleotides were not directly recognized by Clec2d, using our Clec2d-RF33.70-Luc reporter cells (Figure S5B). We also verified that CpG bound histones (Figure S5C). We then treated our Dox-inducible Clec2d cell lines with media versus Dox, stimulated them with histones and/or limiting concentrations of CpG DNA and then measured the production of TNFα and IL-6. Histones augmented the stimulation of the macrophages by CpG and this augmentation was significantly increased when the expression of Clec2d was induced (Figure 5D). We also examined this issue using peritoneal macrophages from WT and Clec2d−/− mice. Histones augmented the cytokine responses of WT macrophages to CpG (both with native phosphodiester and phosphorothioate backbones), but in Clec2d−/− cells, these responses were reduced significantly (Figure 5E and S5D). The reduced responses to histone + CpG in Clec2d−/− macrophages was not due to any change in the Tlr9 expression (Figure S5E). In the absence of Tlr9, the production of cytokines and chemokines to histone + CpG was almost completely lost (Figure 5G). Together, these results indicate that Clec2d can help deliver histone-TLR agonists to TLRs in endosomes in ways that simulate responses.
Role of Clec2d in Response to Physiological Ligands
Since much of the histones released from necrotic cells are complexed to DNA in nucleosomes, we tested whether nucleosomes stimulated microphages in a Clec2d-dependent manner. We found that nucleosomes activated Clec2d reporter cells, although with less potency than purified histones, presumably due to a lower density of binding epitopes (Figure 6A). We further found that nucleosomes by themselves could stimulate cytokine and chemokine expression in WT macrophages, and this response was significantly reduced in Clec2d−/− cells (Figure 6B). Importantly, when nucleosomes were treated with DNase I to remove DNA, they lost the ability to stimulate cytokine production in macrophages (Figure 6C). These data again support our hypothesis that it was the DNA within histones-DNA complex (nucleosomes) that stimulated macrophage responses.
Figure 6. Clec2d Recognizes Physiological Ligands.

(A) Purified nucleosomes or histones were coated on a plate, cultured with reporter cells expressing Clec2d, and luciferase activity was measured. (B) Peritoneal macrophages from WT or Clec2d−/− mice were stimulated with purified nucleosomes for 18 hr and the RANTES concentration in the supernatant was measured by ELISA. (C) Purified nucleosomes were pre-treated with or without DNase I at 37°C for 1 hr and used to stimulate peritoneal macrophages from WT and Clec2d−/− mice. RANTES concentration was measured by ELISA in the supernatant after 18 hr stimulation. DNase I alone serves as a control. Mean ± SEM , ****, P<0.001 by Two-Way ANOVA. (D) Neutrophils isolated from WT mice were treated with 10 μM Ionomycin for 4 hr to induce NETs formation in a 96-well plate. Reporter cells expressing Clec2d or Dectin-1 were then cultured in the plate and luciferase activity was measured after 20 hr stimulation. Immobilized histones (for Clec2d reporters) and zymosan (for Dectin-1 reporters) serve as positive controls for reporter stimulation. (E) Purified neutrophils were treated with 10 μM Ionomycin for 4 hr to induce NETs formation in the chamber slide. After treatment with 2% paraformaldehyde for 5 min, the NETs were stained with Clec2d-Ig or Dectin-1-Ig followed by secondary antibodies conjugated with fluorophore. The slides were then mounted with mounting oil containing DAPI. Bars=20 μm.
Another situation where extracellular histones are found is in neutrophil extracellular traps (NETs) (Sollberger et al., 2018). To test whether Clec2d recognized NETs, we stimulated purified primary neutrophils with ionomycin and found that the resulting NETs that were produced stimulated the RF33.70 Clec2d cells to produce luciferase (Figure 6D). Moreover, we found that soluble Clec2d-Ig but not Dectin-1-Ig stained NETs (Figure 6E). These data show that Clec2d recognized NETs.
DISCUSSION
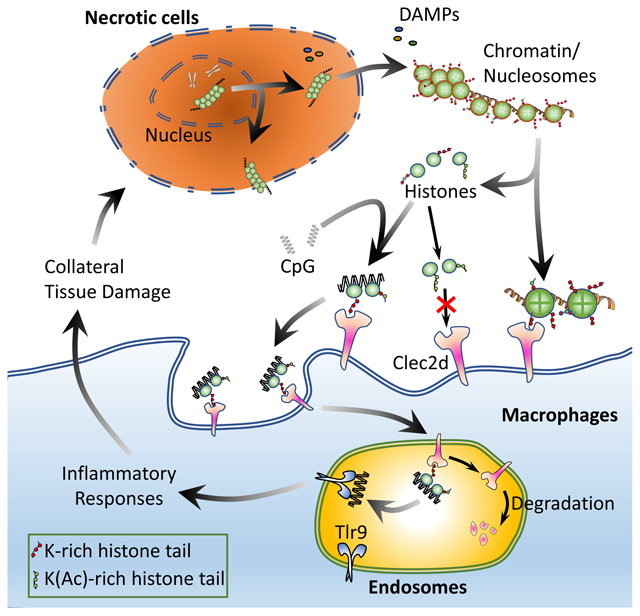
Histones are known DAMPs, but how they are detected by the immune system was poorly understood (Silk et al., 2017). The major findings in this report are that: Clec2d is a receptor that bound unmodified poly-basic regions on histones. Upon such recognition, Clec2d delivered immune-stimulatory histone complexes to TLRs in endosomes and stimulated cytokine responses. In the in vivo setting, Clec2d participated in histone-stimulated pro-inflammatory cytokine responses, and promoted collateral tissue damage. Since histones are ubiquitously expressed in all nucleated cells, Clec2d is a universal sensor of cell death. Moreover, since histones are exposed and released when a dying cell’s plasma membrane loses integrity, it is likely that Clec2d will sense cells dying by necrosis, pyroptosis, necroptosis and even by apoptosis (upon secondary necrosis). In addition, extracellular traps formed by activated innate immune cells, which contain histones and DNA (Okubo et al., 2018; Silk et al., 2017; Sollberger et al., 2018), were recognized by Clec2d.
Clec2d binds some carbohydrates, including sulfated glycosaminoglycans such as chondroitin, fucoidan and λ-carrageenan (Gange et al., 2004). Histones can undergo O-linked glycosylation and conceivably this might contribute to binding to Clec2d (Fujiki et al., 2011; Mir and Moinuddin, 2015). However, since Clec2d recognized H1-H4 recombinant proteins (expressed in bacteria) as well as synthetic peptides, all of which lack carbohydrate, this CLR can directly bind to histone protein sequences.
Clec2d recognized histones via sequences in the N-terminal tail of all histones and also in the C-terminal tail of H1. Given the conservation of these sequences through evolution, it will be interesting to explore in future studies whether Clec2d can also recognize histones of lower eukaryotes and thereby sense infections with pathogens, such as parasites. The stimulatory sequences in the 5 different histones are not identical to one another, they all are rich in basic residues and where examined, the positively-charged residues were required for recognition by Clec2d. Therefore, Clec2d is sensing a degenerative poly-basic motif. A number of other DAMPs, such as HMGB1 and some defensins (Bauer et al., 2001; Yang et al., 2013), have polybasic regions and it will be of interest to explore whether they might be Clec2d ligands.
The side chains of basic residues in histone tails are post-translationally modified as part of chromatin and gene regulation (Chen et al., 2017), and some of these modifications can extinguish the residue’s positive charge (Bannister and Kouzarides, 2011). We found that lysine acetylation blocked recognition by Clec2d. Moreover, increasing histone acetylation by inhibiting HDAC, reduced the ability of injured cells to stimulate Clec2d. Interestingly, HDAC inhibition in vivo reduces inflammation and is protective in a liver injury model (Kim et al., 2016). The ability of Clec2d to sense histone modifications is also of interest because cancers can have globally reduced amounts of histone modification (Chervona and Costa, 2012; Khan et al., 2015). Therefore, our findings raise the possibility that Clec2d might be particularly attuned to sensing such abnormal cells.
DAMPs in general, and histones in particular, can contribute to tissue damage by amplifying the extent of injury (Rock and Kono, 2008; Rock et al., 2011). This effect is at least in part a consequence of the inflammation incited by DAMPs, which causes collateral damage in adjacent tissue. The discovery that Clec2d is a histone receptor is of particular interest because it is well established that histones are pathogenic DAMPs (Allam et al., 2014). We further showed that the histones released after APAP liver injury were the major DAMP recognized by Clec2d. Moreover, we found that extracellular histones in vivo stimulated Cled2d-dependent proinflammatory cytokines. Loss of Clec2d reduced the late liver damage observed in APAP-treated mice, which is caused by cytokine-driven inflammation (Chen et al., 2007; Krenkel et al., 2015; Marques et al., 2012; Zhang et al., 2018). Similarly, administration of anti-histone antibodies reduced the late cell injury and mortality in APAP and other liver injury models (Allam et al., 2014). While we can’t prove that Clec2d is mediating its effects in the APAP model through interactions with histones, the above points make this mechanism of action likely. In future studies, it will of interest to further define the underlying cellular and molecular mechanisms of Clec2d’s effects in tissue injury.
Clec2d lacks ITAM motifs or known signaling adaptors used by other stimulatory CLRs and we found that histone stimulation of cytokine production by cultured macrophages was weak and inconsistent. We also could not detect any Clec2d-dependent stimulation of multiple MAP kinases, Syk and NFκB by purified histones. Therefore, Clec2d is likely not itself an activating receptor. However, histones are naturally complexed with DNA (and can bind other bioactive molecules) and therefore might be able to deliver their bound cargo to pattern recognition receptors in cells, such as Tlr9 in endosomes. Consistent with this mechanism of action, we found that Clec2d trafficked to late endosomes and augmented the responses of macrophages to histones + CpG DNA. This response was completely dependent on Tlr9. How histone-DNA complexes bound to Clec2d interact with Tlr9 and whether Clec2d affects Tlr9 signaling in other ways remains to be determined. While injection of purified histones into mice elicited Clec2d-dependent cytokine responses, histone-bound immunostimulatory adducts could still be involved in such responses because TLR ligands such as DNA and LPS are normal constituents in the plasma and could bind to the injected histones (Augusto et al., 2003). Our findings fit with previous reports that endosomal TLRs are involved in histone-induced pathology (Allam et al., 2012; Huang et al., 2011; Xu et al., 2011). We also observed a Clec2d-independent component of histone + CpG responses, which could come from non-specific (e.g. absorptive) endocytosis of the complexes or perhaps another as of yet unrecognized histone receptor.
Indirect crosstalk has been observed between CLRs and TLRs, wherein Syk-pathway signaling from CLRs synergizes with the signaling through TLRs (Ostrop and Lang, 2017). This is very unlikely to occur with Clec2d because it does not stimulate Syk. Therefore, it is much more likely that the Clec2d augments Tlr9-dependent responses directly, by delivering the immunostimulatory ligand complex. Importantly, the Clec2d augmentation of responses was not just seen with synthetic CpG oligonucleotides, but also with nucleosomes, which are released under pathophysiological conditions; the DNA in such complexes was essential for Clec2d-dependent cytokine responses. This carrier function of Clec2d is similar to that of CD14, a receptor that interacts with LPS as well as the DAMP oxPAPC (Zanoni et al., 2017). Another analogous example of CpG chaperoning is observed with RAGE, although it is through a distinct mechanism (Sirois et al., 2013). TLR stimulation markedly upregulates Clec2d RNA and protein expression (BioGPS database and data not shown) and this kind of crosstalk would likely create a positive feedback loop in responses to cell injury.
Clec2d is expressed on macrophages and at about 2-fold higher on Kupffer cells (ImmGen RNAseq database). Upon liver injury, overall amounts of Clec2d do not change in the liver, but drop about 50% in Kupffer cells (GEO dataset #GDS4928 & ImmGen RNAseq database). In addition, Clec2d is expressed on a number of cell types, including other leukocytes, epithelial and mesenchymal cells (BioGPS & ImmGen RNAseq database) (Zhou et al., 2002). Clec2d is expressed on osteoclasts and osteoblasts and thought to influence bone homeostasis because the Clec2d−/− mouse has mild osteoporosis (Kartsogiannis et al., 2008); how Clec2d mediates this effect is unknown. B cells make anti-histone autoantibodies in autoimmunity (van der Vlag and Berden, 2011). Since B cells express Clec2d, it will be of Interest in future studies to investigate whether Clec2d and its ligands play a role in the generation of such autoantibodies or affect the physiology of other Clec2d-expressing cells in the setting of tissue injury.
Clec2d is a ligand of the NK inhibitory receptors, NKR-P1B and NKR-P1D (Carlyle et al., 2004). These Clec2d-NK receptor interactions are unlikely to be involved in the phenomena studied here because Clec2d recognized dead cells that lack NKR-P1B, NKR-P1D. Moreover, NK cell depletion doesn’t affect the pathophysiologic sequelae in the liver injury model (Masson et al., 2008).
Given the role of Clec2d in the pathophysiology of tissue injury, Clec2d could be a potential and attractive therapeutic target because a single agent, such as a blocking biologic, could reduce response to multiple ligands, such as the 5 histones. If effective, this approach could help limit damage in ischemic diseases, such as stroke and myocardial infarction, trauma and other settings of tissue injury.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kenneth L. Rock (Kenneth.Rock@umassmed.edu). The mouse lines and reagents obtained from other laboratories are described below and may require a Material Transfer Agreement (MTA) with the providing scientists.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Clec2d-deficient mice (Kartsogiannis et al., 2008) were a gift from Dr. Matthew Gillespie (Monash University, Australia) and provided by Dr. James Carlyle (Sunnybrook Research Institute, Canada); after receiving the mice, we further backcrossed this line to C57BL/6J mice at least 6 more generations and 384 SNPs genotyping (Charles River, Wilmington, MA) was performed to confirm the mice were 100% C57BL/6J background. C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). Tlr9-deficient mice (Hemmi et al., 2000) were made available by Shizuo Akira (Osaka University) and obtained from Dr. Douglas Golenbock (UMass Medical School) and further bred to generate Clec2d and Tlr9 double deficient mice. Male mice, age 8-14 weeks old, were used for the study. All mice were housed in specific-pathogen-free animal facility at UMASS Medical School, and all the animal experiments were approved by the UMASS IACUC committee.
METHODS DETAILS
Constructs for chimeric receptors and tagged-Clec2d
The MSCV-Clec9a retroviral vector was used to construct CLR extracellular domains fused to the NKG2D transmembrane domain and the CD3ζ chain intracellular domain. Clec9a extracellular sequence was replaced by other CLR extracellular domain. At the C-terminal of CLR extracellular domains, an HA tag was added. In addition, an IRES-EGFP was placed behind the CLR chimera sequence to facilitate identification and sorting of transduced cells expressing the construct. The pCMV6-AN-mFc-S vector was used to construct CLR-Ig fusion proteins consisting of a CLR extracellular domain fused at the N-terminus with a mouse IgG1 Fc domain. The pTRIPZ lentiviral vector was used to construct full length mouse Clec2d with N-terminal mCherry and C-terminal HA tags.
Reporter assay
RF33.70 cells containing an NFAT-Luc reporter gene (RF33.70-Luc) were transduced with CLR chimeric constructs. Necrotic cell lysates, histones, histone peptides, anti-HA antibodies or nothing (for background control) were immobilized on 96-well EIA plates (Corning, #3361), and then RF33/CLR reporter cells (5x104/well) were cultured in these wells for overnight. The cells were then lysed with passive lysis buffer (20 μl/well, Promega, #E1941) at RT for 40 min after which 100 μl/well of Luciferase Assay Reagent (Promega, #E1501) was added to the lysates and luciferase activity was quantified using an Envision plate reader (PerkinElmer). Cell necrosis was induced by UV irradiation, freeze/thaw cycles, or heat shock (45°C, 10 min) followed by incubation at 37°C for 8 hr. The necrotic lysates were clarified by centrifugation at 10,000 rpm for 5 min, and the supernatant was collected for subsequent reporter assays. In some experiments, necrotic EL4 lysates were treated with Proteinase K-agarose beads, urea, DTT, and/or SDS before being immobilized and washed on EIA plates. For immobilizing synthetic histone peptides, 96-well EIA plates were first coated with streptavidin (5 μg/ml) at 4°C overnight. After washing with PBS, histone peptides (2 μg/ml) conjugated with N-terminal biotin were immobilized in the wells, 4°C for 2 hr. Under the conditions we used, the reporter cells did not themselves undergo necrosis after stimulation with necrotic lysates or histones (data not shown).
Acetaminophen-induced liver injury
Wild type (WT) or Clec2d−/− male mice aged 10-14 weeks old were fasted for 24 hr, food was restored for 1 hr and the mice were then injected intra-peritoneally (i.p.) with APAP (300 mg/KgBW) in PBS. Six or 12 hr after APAP injection, serum was collected to measure ALT activity. To detect Clec2d ligand activity in the serum, 6 or 22 hr after APAP (300 or 500 mg/KgBW) injection, serum was collected from WT mice and immobilized on the EIA plate for Clec2d reporter assay. Serum ALT activity was measured by the UMASS Memorial Hospital Lab service using a Beckman AU5800 System.
Immunodepletion of histones from the serum of APAP-treated mice
Serum from WT male mice treated with APAP for 12 hr were diluted 4 folds with PBS and incubated with Dynabeads conjugated with anti-histone (PL2-3) (Losman et al., 1992) or isotype control antibodies to deplete histones. After histone depletion, western blot for histone H3 was used to confirm the depletion efficiency and the serum was immobilized in 96-well assay plate to stimulate Clec2d reporter cells. Histones pulled down from the APAP serum were also eluted from the beads and used to stimulate Clec2d reporter cells.
In vivo histone stimulation of inflammatory mediators
Male WT and Clec2d−/− mice were i.v. injected with 750 μg/mouse of purified histones, and 18 hr later serum was collected and assay for the concentration of inflammatory mediators by Luminex.
SDS-PAGE fractionation of EL4 lysate for reporter assay
EL4 cells were first subjected to a Subcellular Protein Fractionation Kit (Pierce, #78840) according to the manufacturer’s protocol. This procedure uses serial extractions into crude cytosolic, membrane-bound, nuclear soluble, chromatin-bound, and a final crude “cytoskeletal fraction”, which is what remains or spills over from previous extractions. As described in the results section, we observed a lot of histone in the final cytoskeletal fraction, which presumably was histone bound to cytoskeleton, incompletely solubilized, and/or had spilled over into this fraction. This fraction containing enriched Clec2d ligand activity was used for further fractionation by reducing SDS-PAGE (10% Bis-Tris gel, MOPS buffer). After electrophoresis, proteins in the gel were transferred onto PVDF membrane (Millipore, #IPVH00010), which was then blocked with 5% FBS/PBS overnight at 4°C. The membrane was then sliced from high to low molecular weight into 23 small fragments and the membrane fragments were put into a 96-well plate. Clec2d reporter cells were then cultured on top of these membrane slices overnight, and luciferase activity was measured. In a parallel SDS-PAGE, the gel strip was subjected to silver stain to reveal the protein bands in the gel.
Histone beads pull-down assay
Two mg of NHS-activated agarose beads (Pierce, #26196) were saturated with histones or BSA proteins. These beads (2 mg) or zymosan particles (0.5 mg) were incubated with 293T culture supernatant containing soluble Clec2d or Dectin-1 mouse Fc fusion proteins, and incubated at 4°C for overnight. After washing with PBS, the bound proteins were eluted from the beads or particles with LDS sample buffer with DTT, run on Western blot, which was then probed to detect the Fc-fusion CLRs.
In vitro iBMDM stimulation
An immortalized macrophage cell line (iBMDM) was transduced with pTRIPZ-mCherry-Clec2d-HA and sorted for mCherry+ cells. The expression of Clec2d was induced by 1 μg/ml doxycycline (Dox) for at least 1 day. In 96-well plate, 8x104/well iBMDM were stimulated with 30 μg/ml histones and/or 1 μg/ml CpG1826 for overnight, and the culture supernatant was analyzed by ELISA (eBioscience) to quantify IL-6 and TNFα.
iBMDM confocal microscopy
iBMDM cells transduced with pTRIPZ-Clec2d-mCherry-HA were treated with Dox (1 μg/ml), seeded in chamber slides (Thermo Fisher Scientific, #155411) and cultured overnight for attachment. The bound cells were then washed with PBS and fixed with 1% glutaraldehyde/PBS at RT for 10 min. Glutaraldehyde was then removed and the cells were immediately treated with 1% sodium borohydride/PBS for 10 min to quench autofluorescence. The cells were then washed with PBS and permeabilized with 0.2% Triton-X-100/PBS for 4 min, followed by washing in PBS and blocking with 5% FBS/PBS at RT for 2 hr. The cells were then stained with antibodies against Eea1 (Santa Cruz, #sc-6414), Calreticulin (Abcam, #ab2907), or Rab7 (Cell Signaling, #D95F2), at RT for 2 hr, followed by PBS wash and incubate with proper secondary Abs conjugated with AF488. The cells were then analyzed with a Leica TCS SP8 confocal microscope system.
Phagosome isolation from peritoneal macrophage
The protocol for isolating phagosomes was adapted from a previous publication (Desjardins et al., 1994). Briefly, 3x107 peritoneal macrophages from WT male mice were resuspended in 1 ml of complete medium containing 100 nM Bafilomycin and with 1.2x109 BSA-blocked latex beads (Polyscience, #07310, 1 μm) in a 1.5 ml eppendorf tube. After a 2 hr incubation rotating (30 rpm) at 37°C, the cells were pelleted at 400g x 5 min at 4°C. The resulting cell pellet was resuspended in 1 ml of ice-cold homogenization buffer (250 mM sucrose, 3 mM Imidazole, pH 7.4, protease inhibitors) and homogenized on ice in a Dounce homogenizer with a tight-fitting pestle. The homogenization was carried out until about 80% of cells were disrupted (trypan blue+) without substantial nuclear disruption, as monitored by light microscopy. Intact cells and nuclei were pelleted by centrifugation at 400g x 5 min at 4°C. The resulting supernatant (containing phagosomes) was mixed with an equal volume of 62% sucrose/3 mM Imidazole to bring the final sucrose concentration to 40%, layered on top of 1 ml of 62% Sucrose in an ultracentrifuge tube (Beckman Coulter, #331372), and then overlaid sequentially with 2 ml of 35% sucrose, 2 ml of 25% sucrose, and 2 ml of 10% sucrose solutions. This discontinuous density gradient was centrifuged in a swinging bucket rotor (SW41Ti; Beckman Instruments, Palo Alto, CA) for 1 hr at 100,000g at 4°C then allowed to stop without a brake engaged. The phagosome-containing fraction was collected between the interface of the 10 and 25% sucrose solutions and resuspended in 12 ml of cold PBS. The beads were finally pelleted by a 15 min centrifugation at 40,000g in an SW41Ti rotor at 4°C. The proteins were then solubilized and subjected to western blotting to detect Clec2d, Rab7(phagosomes), Calreticulin (ER), and Erk (cytosol).
In vitro primary peritoneal macrophage stimulation
Peritoneal exudate cells (PECs) from naïve male mice were collected by lavage with 8 ml/mouse RPMI-2% FBS and then plated on culture dishes with complete RPMI. After incubating overnight, the dishes were washed 4 times with DPBS to remove non-adherent cells and the adherent cells were incubated with DPBS-5 mM EDTA at 37°C for 7 min. The macrophages were then pipetted off the dish with complete RPMI. The purity of macrophages (CD11bhi, F4/80hi) was greater than 97% (data not shown). In 96-well plates, 1x105/well macrophages were stimulated with 30 μg/ml histones and/or 0.1 μg/ml CpG1826 ODN for overnight. The culture supernatant was then harvested and assayed for cytokines by Luminex (Eve Technologies, Calgary, AB, Canada) to detect inflammatory mediators.
In vitro peritoneal macrophage stimulation to detect kinase phosphorylation
Thioglycollate media (Remel, #R07178) was injected i.p. into male WT and Clec2d−/− mice for 3-5 days to elicit PECs. PECs were collected by lavage of the peritoneum with 8 ml/mouse RPMI-2%FBS, washed once with HCM, and then allowed to attach to tissue culture dishes for 30 min at 37°C. After washing off non-adherent cells, the dishes were incubated with 5 mM EDTA/PBS for 15 min to loosen the adherent macrophages. The cells were then pipetted off the dish with complete RPMI. The purity of macrophages (CD11b+, F4/80+) was greater than 95% (data not shown). The macrophages were added to 12-well plates at 2x106/well and incubated for 4h before stimulating them without or with 30 μg/ml purified histones or 40 μg/ml Zymosan for 30 min. The cells were then washed with PBS and lysed with 120 μl of RIPA buffer (Boston Bioproducts, #BP-115) containing protease inhibitors (Roche, #11697498001) and phosphatase inhibitors (Sigma, #P5726), followed by mixing with 4X LDS sample buffer (Invitrogen, #NP0008) with reducing agent (Invitrogen, #NP0009) and heating at 95°C for 10 min. Western blot was carried out to detect phospho-MAPKs (p38, ERK, JNK), phospho-Syk, and phospho-NFκB (p65), as well as total MAPKs and Syk.
Endogenous Clec2d stability analysis
Peritoneal macrophages from naïve male mice were collected/enriched from PECs. The cells were then cultured in complete RPMI for 5 hr without or with 10 μM cycloheximide (CHX) and/or 100 nM Bafilomycin (Baf). After the treatment, macrophages were harvested, lysed with RIPA buffer, and lysates analyzed for endogenous Clec2d amounts by western blot.
Nucleosomes stimulation of reporter cells and macrophages
Purified poly-nucleosomes from HeLa cells (EpiCypher, #16-0003) were added to coat microtiter wells, RF33.70 Clec2d reporter cells added and after 20 hr of incubation at 37°C luciferase activity was measured. For macrophage stimulation, macrophages were purified from thioglycollate-elicited PECs of WT and Clec2d−/− male mice as described above. In 96-well plates, 1x105/well macrophages were stimulated overnight at 37°C with 15 μg/ml of nucleosomes. The culture supernatant was then harvested and assayed for RANTES by ELISA (R&D System) according to the manufacturer’s instructions. For some experiments, the nucleosomes were pre-treated with DNase I (4000 units/ml) at 37°C for 1 hr before adding to macrophages.
Neutrophil isolation and NETs induction
WT male mice were i.p. injected with 1 ml thioglycollate media (Remel,#R07178) and after ~18 hr PEC were collected with 8 ml/mouse RPMI-2%FBS, followed by staining with αLy6G-PE (BD#561104). The Ly6G+ population (neutrophils) were isolated by sorting with a flow cytometer, cultured in RPMI-SF and treated with 10 μM lonomycin for 4 hr to induce NETosis. For reporter assays, the NETs were induced in the 96-well plate and washed 3 times with PBS before adding the reporter cells. The luciferase activity was then measured after 20 hr of stimulation. For CLR-Ig staining, NETs were induced on chamber slides (Lab-Tek, #154534) for 4 hr and then fixed with 2% paraformaldehyde for 5 min. After washing with PBS, soluble CLR-Ig was incubated with the NETs overnight at 4°C, followed by staining with secondary antibodies conjugated with fluorophores at RT for 1 hr. After washing with PBS, the slides were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen, #P36935) and examined with a fluorescence microscope (Zeiss Zen Pro System).
Databases used to check Clec2d expression
ImmGen (The Immunological Genome Project), http://www.immgen.org BioGPS, http://biogps.org GEO datasets (Gene Expression Omnibus), https://www.ncbi.nlm.nih.gov/gds/
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was done by GraphPad Prism 7 or 8 (GraphPad Software). Comparison between multiple groups was analyzed by Student’s t-test, Mann-Whitney test, one-way ANOVA, or two-way ANOVA. Correlation between two parameters was analyzed by Pearson correlation coefficient analysis. Survival curve was analyzed by Log-rank Mantel-Cox test. Differences with p values <0.05 were considered significant. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p<0.0001. n represents number of biological replicates.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| goat anti-mouse Clec2d | R&D System | Cat#AF3376 |
| rabbit anti-histone H3 | Abcam | Cat#ab1791 |
| rabbit anti-histone H3 (acetyl-K27) | Abcam | Cat#ab4729 |
| mouse anti-HA | Sigma-Aldrich | Cat#H3663; Clone: HA-7 |
| goat anti-Eea1 | Santa Cruz Biotechnology | Cat#sc-6414 |
| rabbit anti-Calreticulin | Abcam | Cat#ab2907 |
| rabbit anti-Rab7 | Cell Signaling Technology | Cat#9367; Clone: D95F2 |
| goat anti-mouse IgG (H+L)-HRP | Thermo Fisher Scientific | Cat#31430 |
| goat anti-mouse IgG (H+L)-Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11001 |
| goat anti-Rabbit IgG (H+L)-Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-11034 |
| bovine anti-goat IgG (H+L)-HRP | Jackson ImmunoResearch | Cat#805-035-180 |
| bovine anti-goat IgG (H+L)-Alexa Fluor 647 | Jackson ImmunoResearch | Cat#805-605-180 |
| mouse anti-histones | Ann Marshak-Rothstein (Losman et al., 1992) | Clone PL2-3 |
| rabbit anti-p38 | Cell Signaling Technology | Cat#8690 |
| rabbit anti-p-p38 | Cell Signaling Technology | Cat#4511 |
| rabbit anti-Jnk | Cell Signaling Technology | Cat#9295 |
| rabbit anti-pJnk | Cell Signaling Technology | Cat#4668 |
| rabbit anti-Erk | Cell Signaling Technology | Cat#4605 |
| rabbit anti-pErk | Cell Signaling Technology | Cat#4370 |
| rabbit anti-p-p65 | Cell Signaling Technology | Cat#3033 |
| rabbit anti-Syk | Cell Signaling Technology | Cat#2712 |
| rabbit anti-Syk (Y317/323) | Thermo Fisher Scientific | Cat#44-234G |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline | Sigma-Aldrich | Cat#D3072-1ML |
| glutaraldehyde 25% | Sigma-Aldrich | Cat#G5882 |
| Sodium borohydride | Sigma-Aldrich | Cat#213462-25G |
| Paraformaldehyde | Sigma-Aldrich | Cat#P6148 |
| EDTA | Thermo Fisher Scientific | Cat#21857 |
| BSA | Roche | Cat#03-116-964-001 |
| Trypan Blue | Thermo Fisher Scientific | Cat#15250061 |
| Urea | Sigma-Aldrich | Cat#U5378 |
| DTT | Sigma-Aldrich | Cat#D0632 |
| SDS (Sodium dodecyl sulfate) | Sigma-Aldrich | Cat# 436143 |
| Cycloheximide, 100mg/ml | Sigma-Aldrich | Cat#C4859-1ML |
| Bafilomycin A1 | Sigma-Aldrich | Cat#B1793 |
| Zymosan A | Sigma-Aldrich | Cat#Z4250 |
| Trichostatin A | Cell Signaling Technology | Cat#9950 |
| DNase I | Sigma-Aldrich | Cat#DN25 |
| Recombinant H1 | NEB | Cat#M2501 |
| Recombinant H2A | NEB | Cat#M2502 |
| Recombinant H2B | NEB | Cat#M2505 |
| Recombinant H3 | NEB | Cat#M2503 |
| Recombinant H4 | NEB | Cat#M2504 |
| Histones from calf thymus | Sigma-Aldrich | Cat#H9250 |
| HeLa Polynucleosomes, Purified | Epicypher | Cat#16-0003 |
| Thioglycollate media | Remel | Cat#R07178 |
| H4 (2-27) peptide | LifeTien | N/A |
| H4 (17-40) peptide | LifeTien | N/A |
| H4 (28-53) peptide | LifeTien | N/A |
| H4 (42-65) peptide | LifeTien | N/A |
| H4 (54-78) peptide | LifeTien | N/A |
| H4 (67-90) peptide | LifeTien | N/A |
| H4 (79-103) peptide | LifeTien | N/A |
| H4 (2-17) peptide | LifeTien | N/A |
| H4 (2-12) peptide | LifeTien | N/A |
| H4 (16-27) peptide | LifeTien | N/A |
| H4 (6-21) peptide | LifeTien | N/A |
| H4 (13-21) peptide | LifeTien | N/A |
| H4 (6-17) peptide | LifeTien | N/A |
| H4 (6-13) peptide | LifeTien | N/A |
| H4 (9-17) peptide | LifeTien | N/A |
| H4 (9-21) WT peptide | LifeTien | N/A |
| H4 (9-21) K(Ac) peptide | LifeTien | N/A |
| H4 (9-21) K to A peptide | LifeTien | N/A |
| H1 (12-23) peptide | Abclonal | N/A |
| H1 (180-194) peptide | Abclonal | N/A |
| H2A (4-19) peptide | Abclonal | N/A |
| H2B (12-29) peptide | Abclonal | N/A |
| H3 (9-28) peptide | Abclonal | N/A |
| Critical Commercial Assays | ||
| Pierce Subcellular Protein Fractionation Kit for Cultured Cells | Thermo Fisher Scientific | Cat#78840 |
| Luciferase Assay System | Promega | Cat#E1501 |
| Passive Lysis 5X Buffer | Promega | Cat#E1941 |
| Mouse Cytokine Array / Chemokine Array 31-Plex | Eve Technologies | Cat#MD-31 |
| Mass Spectrometry | UMASS Proteomics Core | N/A |
| ALT assay | UMASS Memorial Hospital Lab | Beckman AU5800 |
| eBioscience™ Mouse TNFα ELISA Ready-SET-Go!™ Kit | Thermo Fisher Scientific | Cat#50-173-31 |
| eBioscience™ Mouse IL-6 ELISA Ready-SET-Go!™ Kit | Thermo Fisher Scientific | Cat#50-112-8696 |
| Mouse CCL5/RANTES Duoset ELISA | R&D System | Cat#DY-478 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| Experimental Models: Cell Lines | ||
| IMMP | (Hornung et al., 2008) | N/A |
| RF33.70 | (Rock et al., 1990) | N/A |
| HEK 293T | ATCC | Cat#CRL-3216 |
| EL4 | ATCC | Cat#TIB-39 |
| Jurkat | ATCC | Cat#TIB-152 |
| Experimental Models: Organisms/Strains | ||
| Mouse: WT or Clec2d+/+: C57BL/6J | Jackson Laboratory | Stock# 000664 |
| Mouse: Clec2d−/− | Matthew Gillespie (Kartsogiannis et al., 2008) | N/A |
| Mouse: Tlr9−/− | Shizuo Akira (Hemmi et al., 2000) | N/A |
| Oligonucleotides | ||
| CpG1826-PTO (tccatgacgttcctgacgtt) | IDT | N/A |
| CpG1826-PD (tccatgacgttcctgacgtt) | IDT | N/A |
| CpG1826-Biotin | Invivogen | Cat#tlrl-1826b |
| CpG1826-FITC | Invivogen | Cat#tlrl-1826f |
| Recombinant DNA | ||
| MSCV-clec9a | Caetano Reis e Sousa (Sancho et al., 2009) | N/A |
| MSCV-dectin1 | Caetano Reis e Sousa (Sancho et al., 2009) | N/A |
| pTRIPZ-N-mCherry | Open Biosystem (Dharmacon) | N/A |
| pCMV6-AN-mFc-S | ORIGENE | Cat#PS100056 |
| Software and Algorithms | ||
| GraphPad Prism | GraphPad Software Inc. | Version 7.04 |
| Leica Application Suite X | Leica Microsystems | Version 3.3.0 |
| Zeiss Zen Pro | ZEISS | |
| FlowJo | FlowJo LLC | Version 9 |
| Other | ||
| Immobilon-P Transfer membrane (PVDF) | Merck Millipore | Cat#IPVH00010 |
| Pierce NHS-Activated Agarose, Dry | Thermo Fisher Scientific | Cat#26196 |
| Proteinase K–Agarose from Tritirachium album | Sigma-Aldrich | Cat#P9290-10UN |
| Dynabeads-Pan Mouse IgG | Invitrogen | Cat#110.41 |
| Latex beads | Polysciences | Cat#07310 |
ACKNOWLEDGEMENTS
We would like to thank: Dr. Caetano Reis e Sousa for providing the MSCV-Clec9a/CD3 and MSCV-Dectin-1/CD3 chimeric plasmids; Dr. Matthew Gillespie and Dr. James Carlyle for providing Clec2d−/− mice; Dr. Shizuo Akira for making available Tlr9−/− mice; Dr. Ann Marshak-Rothstein for Anti-histone antibodies (PL2-3); Dr. Kate Fitzgerald for critical review of the manuscript; Dr. Diego J. Farfán-Arribas, Dr. Jeff Colbert, and Dr. Vicky Kartsogiannis for helpful discussion and technical assistance; and Yu-Hui Chen for help with statistical analysis. This work was supported by the Grant AI129966 from the NIH.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahrens S, Zelenay S, Sancho D, Hanc P, Kjaer S, Feest C, Fletcher G, Durkin C, Postigo A, Skehel M, et al. (2012). F-actin is an evolutionarily conserved damage-associated molecular pattern recognized by DNGR-1, a receptor for dead cells. Immunity 36, 635–645. [DOI] [PubMed] [Google Scholar]
- Alcover A, Alarcon B, and Di Bartolo V (2018). Cell Biology of T Cell Receptor Expression and Regulation. Annu Rev Immunol 36, 103–125. [DOI] [PubMed] [Google Scholar]
- Allam R, Kumar SV, Darisipudi MN, and Anders HJ (2014). Extracellular histones in tissue injury and inflammation. J Mol Med (Berl) 92, 465–472. [DOI] [PubMed] [Google Scholar]
- Allam R, Scherbaum CR, Darisipudi MN, Mulay SR, Hagele H, Lichtnekert J, Hagemann JH, Rupanagudi KV, Ryu M, Schwarzenberger C et al. (2012). Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J Am Soc Nephrol 23, 1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augusto LA, Decottignies P, Synguelakis M, Nicaise M, Le Marechal P, and Chaby R (2003). Histones: a novel class of lipopolysaccharide-binding molecules. Biochemistry 42, 3929–3938. [DOI] [PubMed] [Google Scholar]
- Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, Bruckner P, Pfeilschifter J, Schaefer RM, Grone HJ, and Schaefer L (2009). Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors. J Biol Chem 284, 24035–24048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamezai A, Goldmacher V, Reiser H, and Rock KL (1989). Internalization of phosphatidylinositol-anchored lymphocyte proteins. I. Documentation and potential significance for T cell stimulation. J Immunol 143, 3107–3116. [PubMed] [Google Scholar]
- Bannister AJ, and Kouzarides T (2011). Regulation of chromatin by histone modifications. Cell Res 21, 381–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer F, Schweimer K, Kluver E, Conejo-Garcia JR, Forssmann WG, Rosch P, Adermann K, and Sticht H (2001). Structure determination of human and murine beta-defensins reveals structural conservation in the absence of significant sequence similarity. Protein Sci 10, 2470–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Willment JA, and Whitehead L (2018). C-type lectins in immunity and homeostasis. Nat Rev Immunol 18, 374–389. [DOI] [PubMed] [Google Scholar]
- Carlyle JR, Jamieson AM, Gasser S, Clingan CS, Arase H, and Raulet DH (2004). Missing self-recognition of Ocil/Clr-b by inhibitory NKR-P1 natural killer cell receptors. Proc Natl Acad Sci U S A 101, 3527–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Kono H, Golenbock D, Reed G, Akira S, and Rock KL (2007). Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 13, 851–856. [DOI] [PubMed] [Google Scholar]
- Chen GY, and Nunez G (2010). Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10, 826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Li S, Subramaniam S, Shyy JY, and Chien S (2017). Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu Rev Biomed Eng 19, 195–219. [DOI] [PubMed] [Google Scholar]
- Chervona Y, and Costa M (2012). Histone modifications and cancer: biomarkers of prognosis? Am J Cancer Res 2, 589–597. [PMC free article] [PubMed] [Google Scholar]
- Desjardins M, Huber LA, Parton RG, and Griffiths G (1994). Biogenesis of phagolysosomes proceeds through a sequential series of interactions with the endocytic apparatus. J Cell Biol 124, 677–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, and Di Virgilio F (2006). The P2X7 receptor: a key player in IL-1 processing and release. J Immunol 176, 3877–3883. [DOI] [PubMed] [Google Scholar]
- Fujiki R, Hashiba W, Sekine H, Yokoyama A, Chikanishi T, Ito S, Imai Y, Kim J, He HH, Igarashi K et al. (2011). GlcNAcylation of histone H2B facilitates its monoubiquitination. Nature 480, 557–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gange CT, Quinn JM, Zhou H, Kartsogiannis V, Gillespie MT, and Ng KW (2004). Characterization of sugar binding by osteoclast inhibitory lectin. J Biol Chem 279, 29043–29049. [DOI] [PubMed] [Google Scholar]
- Hanc P, Fujii T, Iborra S, Yamada Y, Huotari J, Schulz O, Ahrens S, Kjaer S, Way M, Sancho D et al. (2015). Structure of the Complex of F-Actin and DNGR-1, a C-Type Lectin Receptor Involved in Dendritic Cell Cross-Presentation of Dead Cell-Associated Antigens. Immunity 42, 839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, and Akira S (2000). A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745. [DOI] [PubMed] [Google Scholar]
- Huang H, Chen HW, Evankovich J, Yan W, Rosborough BR, Nace GW, Ding Q, Loughran P, Beer-Stolz D, Billiar TR, et al. (2013). Histones Activate the NLRP3 Inflammasome in Kupffer Cells during Sterile Inflammatory Liver Injury. J Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Evankovich J, Yan W, Nace G, Zhang L, Ross M, Liao X, Billiar T, Xu J, Esmon CT, and Tsung A (2011). Endogenous histones function as alarmins in sterile inflammatory liver injury through Toll-like receptor 9 in mice. Hepatology 54, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M (2009). The T-cell-receptor signaling network. J Cell Sci 122, 1269–1273. [DOI] [PubMed] [Google Scholar]
- Jaeschke H, Xie Y, and McGill MR (2014). Acetaminophen-induced Liver Injury: from Animal Models to Humans. J Clin Transl Hepatol 2, 153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA Jr. (1989). Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 54 Pt 1, 1–13. [DOI] [PubMed] [Google Scholar]
- Kartsogiannis V, Sims NA, Quinn JM, Ly C, Cipetic M, Poulton IJ, Walker EC, Saleh H, McGregor NE, Wallace ME, et al. (2008). Osteoclast inhibitory lectin, an immune cell product that is required for normal bone physiology in vivo. J Biol Chem 283, 30850–30860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka H, Kono H, Patel Z, Kimura Y, and Rock KL (2014). Evaluation of the contribution of multiple DAMPs and DAMP receptors in cell death-induced sterile inflammatory responses. PLoS One 9, e104741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA, Reddy D, and Gupta S (2015). Global histone post-translational modifications and cancer: Biomarkers for diagnosis, prognosis and treatment? World J Biol Chem 6, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Park JS, Lee DW, and Lee SM (2016). Trichostatin A Protects Liver against Septic Injury through Inhibiting Toll-Like Receptor Signaling. Biomol Ther (Seoul) 24, 387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Chen CJ, Ontiveros F, and Rock KL (2010a). Uric acid promotes an acute inflammatory response to sterile cell death in mice. J Clin Invest 120, 1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Karmarkar D, Iwakura Y, and Rock KL (2010b). Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J Immunol 184, 4470–4478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, and Rock KL (2008). How dying cells alert the immune system to danger. Nat Rev Immunol 8, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenkel O, Mossanen JC, and Tacke F (2015). Immune mechanisms in acetaminophen-induced acute liver failure. Hepatobiliary Surg Nutr 3, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraishy A, Karin M, and Grivennikov SI (2011). Tumor promotion via injury- and death-induced inflammation. Immunity 35, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land W (2003). Allograft injury mediated by reactive oxygen species: from conserved proteins of Drosophila to acute and chronic rejection of human transplants. Part III: interaction of (oxidative) stress-induced heat shock proteins with toll-like receptor-bearing cells of innate immunity and its consequences for the development of acute and chronic allograft rejection. Transplantation Reviews 17, 67–86. [Google Scholar]
- Linch DC, Wallace DL, and O'Flynn K (1987). Signal transduction in human T lymphocytes. Immunol Rev 95, 137–159. [DOI] [PubMed] [Google Scholar]
- Liu ZX, Han D, Gunawan B, and Kaplowitz N (2006). Neutrophil depletion protects against murine acetaminophen hepatotoxicity. Hepatology 43, 1220–1230. [DOI] [PubMed] [Google Scholar]
- Losman MJ, Fasy TM, Novick KE, and Monestier M (1992). Monoclonal autoantibodies to subnucleosomes from a MRL/Mp(−)+/+ mouse. Oligoclonality of the antibody response and recognition of a determinant composed of histones H2A, H2B, and DNA. J Immunol 148, 1561–1569. [PubMed] [Google Scholar]
- Marques PE, Amaral SS, Pires DA, Nogueira LL, Soriani FM, Lima BH, Lopes GA, Russo RC, Avila TV, Melgaco JG, et al. (2012). Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56, 1971–1982. [DOI] [PubMed] [Google Scholar]
- Masson MJ, Carpenter LD, Graf ML, and Pohl LR (2008). Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology 48, 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger P (2002). The danger model: a renewed sense of self. Science 296, 301–305. [DOI] [PubMed] [Google Scholar]
- McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, Beck PL, Muruve DA, and Kubes P (2010). Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330, 362–366. [DOI] [PubMed] [Google Scholar]
- Mir AR, and Moinuddin (2015). Glycoxidation of histone proteins in autoimmune disorders. Clin Chim Acta 450, 25–30. [DOI] [PubMed] [Google Scholar]
- Okubo K, Kurosawa M, Kamiya M, Urano Y, Suzuki A, Yamamoto K, Hase K, Homma K, Sasaki J, Miyauchi H, et al. (2018). Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med 24, 232–238. [DOI] [PubMed] [Google Scholar]
- Ostrop J, and Lang R (2017). Contact, Collaboration, and Conflict: Signal Integration of Syk-Coupled C-Type Lectin Receptors. J Immunol 198, 1403–1414. [DOI] [PubMed] [Google Scholar]
- Peng Y, Martin DA, Kenkel J, Zhang K, Ogden CA, and Elkon KB (2007). Innate and adaptive immune response to apoptotic cells. J Autoimmun 29, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran A, and Jaeschke H (2017). Mechanisms of acetaminophen hepatotoxicity and their translation to the human pathophysiology. J Clin Transl Res 3, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauvala H, and Rouhiainen A (2007). RAGE as a receptor of HMGB1 (Amphoterin): roles in health and disease. Curr Mol Med 7, 725–734. [DOI] [PubMed] [Google Scholar]
- Rock KL, and Kono H (2008). The inflammatory response to cell death. Annu Rev Pathol 3, 99–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Lai JJ, and Kono H (2011). Innate and adaptive immune responses to cell death. Immunol Rev 243, 191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Latz E, Ontiveros F, and Kono H (2010). The sterile inflammatory response. Annu Rev Immunol 28, 321–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, Rothstein L, and Gamble S (1990). Generation of class I MHC-restricted T-T hybridomas. J Immunol 145, 804–811. [PubMed] [Google Scholar]
- Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P, Rosewell I, and Reis e Sousa C (2009). Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature 458, 899–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Hanc P, Bottcher JP, Hoogeboom R, Diebold SS, Tolar P, and Reis ESC (2018). Myosin II Synergizes with F-Actin to Promote DNGR-1-Dependent Cross-Presentation of Dead Cell-Associated Antigens. Cell Rep 24, 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, and Rock KL (2002). Cell death releases endogenous adjuvants that selectively enhance immune surveillance of particulate antigens. Eur J Immunol 32, 155–162. [DOI] [PubMed] [Google Scholar]
- Shi Y, Zheng W, and Rock KL (2000). Cell injury releases endogenous adjuvants that stimulate cytotoxic T cell responses. Proc Natl Acad Sci U S A 97, 14590–14595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silk E, Zhao H, Weng H, and Ma D (2017). The role of extracellular histone in organ injury. Cell Death Dis 8, e2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirois CM, Jin T, Miller AL, Bertheloot D, Nakamura H, Horvath GL, Mian A, Jiang J, Schrum J, Bossaller L, et al. (2013). RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J Exp Med 210, 2447–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollberger G, Tilley DO, and Zychlinsky A (2018). Neutrophil Extracellular Traps: The Biology of Chromatin Externalization. Dev Cell 44, 542–553. [DOI] [PubMed] [Google Scholar]
- Sulciner ML, Serhan CN, Gilligan MM, Mudge DK, Chang J, Gartung A, Lehner KA, Bielenberg DR, Schmidt B, Dalli J, et al. (2018). Resolvins suppress tumor growth and enhance cancer therapy. J Exp Med 215, 115–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsan MF, and Gao B (2004). Endogenous ligands of Toll-like receptors. J Leukoc Biol 76, 514–519. [DOI] [PubMed] [Google Scholar]
- Tsan MF, and Gao B (2007). Pathogen-associated molecular pattern contamination as putative endogenous ligands of Toll-like receptors. J Endotoxin Res 13, 6–14. [DOI] [PubMed] [Google Scholar]
- van der Vlag J, and Berden JH (2011). Lupus nephritis: role of antinucleosome autoantibodies. Semin Nephrol 31, 376–389. [DOI] [PubMed] [Google Scholar]
- Wen Z, Lei Z, Yao L, Jiang P, Gu T, Ren F, Liu Y, Gou C, Li X, and Wen T (2016). Circulating histones are major mediators of systemic inflammation and cellular injury in patients with acute liver failure. Cell Death Dis 7, e2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Liu Y, Li F, Ren F, Chen D, Li X, and Wen T (2013). Circulating histones exacerbate inflammation in mice with acute liver failure. J Cell Biochem 114, 2384–2391. [DOI] [PubMed] [Google Scholar]
- Woolbright BL, and Jaeschke H (2017). Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J Hepatol 66, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhang X, Monestier M, Esmon NL, and Esmon CT (2011). Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J Immunol 187, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, Taylor FB, Esmon NL, Lupu F, and Esmon CT (2009). Extracellular histones are major mediators of death in sepsis. Nat Med 15, 1318–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, and Saito T (2008). Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol 9, 1179–1188. [DOI] [PubMed] [Google Scholar]
- Yang D, Wei F, Tewary P, Howard OM, and Oppenheim JJ (2013). Alarmin-induced cell migration. Eur J Immunol 43, 1412–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, and Tonnesseen TI (2019). DAMPs and sterile inflammation in drug hepatotoxicity. Hepatol Int 13, 42–50. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Kijima M, Akita M, and Beppu T (1990). Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem 265, 17174–17179. [PubMed] [Google Scholar]
- Zanoni I, Tan Y, Di Gioia M, Springstead JR, and Kagan JC (2017). By Capturing Inflammatory Lipids Released from Dying Cells, the Receptor CD14 Induces Inflammasome-Dependent Phagocyte Hyperactivation. Immunity 47, 697–709 e693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenay S, Keller AM, Whitney PG, Schraml BU, Deddouche S, Rogers NC, Schulz O, Sancho D, and Reis e Sousa C (2012). The dendritic cell receptor DNGR-1 controls endocytic handling of necrotic cell antigens to favor cross-priming of CTLs in virus-infected mice. J Clin Invest 122, 1615–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenay S, and Reis e Sousa C (2013). Adaptive immunity after cell death. Trends Immunol 34, 329–335. [DOI] [PubMed] [Google Scholar]
- Zhang C, Feng J, Du J, Zhuo Z, Yang S, Zhang W, Wang W, Zhang S, Iwakura Y, Meng G et al. (2018). Macrophage-derived IL-1alpha promotes sterile inflammation in a mouse model of acetaminophen hepatotoxicity. Cell Mol Immunol 15, 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Kartsogiannis V, Quinn JM, Ly C, Gange C, Elliott J, Ng KW, and Gillespie MT (2002). Osteoclast inhibitory lectin, a family of new osteoclast inhibitors. J Biol Chem 277, 48808–48815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.