

Abstract
Rhesus theta defensin-1 (RTD-1), a macrocyclic immunomodulatory host defense peptide from Old World monkeys, is therapeutic in pristane-induced arthritis (PIA) in rats, a model of rheumatoid arthritis (RA). RNA-sequence (RNA-Seq) analysis was used to interrogate the changes in gene expression in PIA rats, which identified 617 differentially expressed genes (DEGs) in PIA synovial tissue of diseased rats. Upstream regulator analysis showed upregulation of gene expression pathways regulated by TNF, IL1B, IL6, proinflammatory cytokines, and matrix metalloproteases (MMPs) involved in RA. In contrast, ligand-dependent nuclear receptors like the liver X-receptors NR1H2 and NR1H3 and peroxisome proliferator-activated receptor gamma (PPARG) were downregulated in arthritic synovia. Daily RTD-1 treatment of PIA rats for 1–5 days following disease presentation modulated 340 of the 617 disease genes, and synovial gene expression in PIA rats treated 5 days with RTD-1 closely resembled the gene signature of naive synovium. Systemic RTD-1 inhibited proinflammatory upstream regulators such as TNF, IL1, and IL6 and activated antiarthritic ligand-dependent nuclear receptor pathways, including PPARG, NR1H2, and NR1H3, that were suppressed in untreated PIA rats. RTD-1 also inhibited proinflammatory responses in IL-1β-stimulated human RA fibroblast-like synoviocytes (FLS) in vitro and diminished expression of human orthologs of disease genes that are induced in rat PIA synovium. Thus, the antiarthritic mechanisms of systemic RTD-1 include homeostatic regulation of arthritogenic gene networks in a manner that correlates temporally with clinical resolution of rat PIA.
Keywords: defensin, pristane induced arthritis, rheumatoid arthritis, RNA-Seq
INTRODUCTION
Rheumatoid arthritis (RA) is an autoimmune disease that afflicts ~0.5–1% of the population worldwide (29). Pathogenesis of RA involves chronic inflammation of the synovium, joint erosion, infiltration of immune cells, and the dysregulation of immune signaling and cytokine gene expression pathways (29). Fibroblast-like synoviocytes (FLS) play a central role in disease pathogenesis and synovial tissue infiltration by leukocytes stimulates FLS to produce inflammatory cytokines, chemokines and matrix metalloproteinases (MMPs) which mediate invasion and degradation of cartilage and bone. Several animal models have been used to study RA pathogenesis (22, 31). Dark Agouti (DA) rats injected with pristane develop chronic inflammation of joints which is pathologically similar to human RA (49, 50). In a recent study, systemic administration of rhesus θ-defensin-1 (RTD-1) was effective in arresting and reversing joint damage in evolving and severe rat pristane-induced arthritis (PIA) (39).
θ-Defensins are 18-amino acid macrocyclic peptides produced by head-to-tail ligation of nonapeptides excised from two precursor polypeptides (Fig. 1). θ-Defensins are expressed in Old World monkeys, e.g., rhesus macaques and baboons, but not in humans or other primates (15, 32, 41, 44). Initially, θ-defensins were characterized on the basis of the in vitro antimicrobial activities (44, 48). However, similar to a number of other host defense peptides (HDPs) (18), θ-defensins are now recognized as pleiotropic mediators of innate immunity, because they have potent microbicidal activities in vitro (4, 6, 15, 44, 45, 47, 48) and also possess remarkable immune modulating properties both in vitro and in vivo (5, 19, 38, 46, 51). For example, RTD-1 reduced mortality in a mouse model of severe acute respiratory syndrome coronavirus infection by reducing inflammation in the lungs of infected animals (51), promoted survival in murine polymicrobial sepsis with concomitant suppression of inflammatory cytokines (38), and reduced inflammatory cytokine secretion from human blood leukocytes stimulated with diverse Toll-like receptor (TLR) agonists (38). RTD-1 inhibits NF-κB and MAP kinase pathways in LPS-stimulated THP-1 cells and human primary monocytes by activating the PI3K/Akt pathway, a negative regulator of those pathways (46). RTD-1 and related natural θ-defensin isoforms also reduce the secretion of TNF-α by inhibiting tumor necrosis factor alpha converting enzyme (TACE/ADAM17) (37).
Fig. 1.

Macrocyclic structure of θ-defensins. Top: biosynthesis of theta defensins involves ligation of 2 nonamer peptides (highlighted in yellow and blue) from two polypeptide precursors to form the macrocyclic peptide. Bottom: a schematic representation of rhesus theta-defensin-1 (RTD-1) showing the sequence and cyclic structure of the peptide, the disulfide linkages, and the shaded sequences indicate the nonamer peptides from two precursor polypeptides. Adapted from (39).
In the rat PIA model, parenteral RTD-1 treatment of rats with evolving or established PIA rapidly arrested joint disease progression, restored limb mobility, and preserved normal joint architecture (39). RTD-1 also reduced the levels of joint IL-1β compared with saline-treated PIA controls, suppressed secretion of IL-6 by FLS cells stimulated with TNF-α and IL-1β, and inhibited FLS invasiveness (39). To further characterize the antiarthritic mechanisms of RTD-1, we analyzed the temporal effects of RTD-1 on arthritis gene signatures in rat PIA synovial tissue.
METHODS
Animals.
All animal experiments were approved by Institutional Animal Care and Use Committee (approval #20378 and 11355) at University of Southern California.
PIA model.
Arthritis was induced in female DA/OlaHsd rats (6–8 wk; Harlan; Indianapolis, IN) by injection of 250 µl pristane (Millipore Sigma, St. Louis, MO) administered by three or four intradermal injections into the tail via a 26-gauge needle (39). In the PIA cohort, arthritis was observed 10–14 days after pristane injection, and the disease was allowed to progress to an arthritis severity score (AS) (7) of at least 3 in each of the rear limbs of individual animals, and the following day was defined as time zero (t = 0). Rats were treated by subcutaneous administration of 5 mg/kg RTD-1 formulated in normal saline or vehicle alone (normal saline) in the loose interscapular skin as previously described (39). Rats (n = 6–11 per cohort) were euthanized 1, 3, or 5 days after initiation of treatment. Studies were nonblinded, and treatment was randomized beginning at t = 0. Rats with established PIA that received no treatment, euthanized at t = 0 represent the control disease cohort in all experiments (denoted throughout as PIA). Euthanasia was performed by cardiac puncture following intraperitoneal injection with 500 µl of ketamine/xylazine solution (20%/10% in saline).
Tissue retrieval and processing.
Synovial tissue was dissected from ankle joints of euthanized rats, transferred to tubes prechilled with dry ice, and stored at −80°C. Synovial tissue from both ankle joints of untreated naive or RTD-1-treated (for 3 or 5 days) healthy rats was pooled, whereas synovial tissue from ankle joints of PIA rats was collected and individually analyzed. RNA for sequence analysis (RNA-Seq) was treated with DNase using Quick RNA Miniprep purification kits (Zymoresearch, Irvine, CA). RNA samples (n = 4–8 per cohort) that had RNA integrity number values ≥ 6.5 were selected for RNA-Seq analysis.
RNA-Seq.
Strand-specific library preparation was carried out using Clonetech SMARTer Stranded Total RNA-Seq (Pico) Kit. Libraries were generated from total RNA for sequencing. Sequencing was performed on Illumina Nextseq500 with 1× 75 single end reads. Library preparation and sequencing were performed by the UCLA Sequencing Core Facility.
Bioinformatics analyses of RNA-Seq.
Image analysis and base-calling was carried out using RTA 1.13.48.0. Final file formatting, and FASTQ generation and demultiplexing were performed using CASAVA v 1.8.2. The quality of raw sequencing reads was checked using in-house RNA-Seq workflow, consisting of several open source tools. Briefly, sequencing reads were checked in FastQC. Reads were trimmed on both ends based on quality scores of >20 for each sequence using FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) (23), and adaptor sequences were removed. High-quality reads were aligned to the RGSC6 genome using TopHat-2, which uses Bowtie 2 at different steps, allowing one mismatch and in conjunction with the gene model from Ensemble release 95. Reads were quantified as number of reads across exons. Differentially expressed genes were identified by combining two approaches that use different algorithms. A gene was called differentially expressed if it passed the false discovery rate (FDR) adjusted P < 0.05 in DEseq2 (2) and employing the null model hypothesis (21). DESeq2 tests the differential expression based on negative binomial generalized linear model (27), while the null model hypothesis takes the average expression of groups into consideration. The gene list was further ranked by fold-change criteria. To investigate potential functional enrichment of various biological pathways in differentially expressed genes in RNA-Seq, a ranked P value was computed for each pathway from the Fisher exact test based on the binomial distribution and independence for probability of any gene belonging to any enriched set (14). Unless specified, hierarchical clustering, principal component analysis, and statistical analysis were performed in R (http://www.r-project.org). Data were visualized in Integrated Genomics Viewer (http://software.broadinstitute.org/software/igv/).
Ingenuity Pathway Analysis.
Ingenuity Pathway Analysis (IPA) core analyses were used to determine the molecular activities of the differentially expressed genes. Upstream regulator analysis was used to predict the activation or inhibition of upstream regulators (24). The P value of the enrichment score was used to evaluate the significance of the overlap between observed and predicted gene sets, while the activation Z score was used to assess the match between observed and predicted patterns of activation (Z ≥ 2) and inhibition (Z ≤ −2). Upstream regulators are not italicized and are denoted as per IPA output.
Quantitative real-time PCR analysis.
Samples of RNA prepared for RNA-Seq were analyzed by quantitative real-time PCR (qRT-PCR). RNA from synovial tissues of animals from indicated cohorts was used to produce first-strand cDNA with the RT2 First strand synthesis kit (Qiagen). Real-time PCR was performed in duplicate with RT2 SYBR green qPCR master mix and validated primers (Qiagen) to quantify expression of a subset of genes identified as differentially expressed by RNA-Seq analysis (Supplementary Table S1; all supplementary material is available at https://doi.org/10.6084/m9.figshare.8855588). The cycling parameters for PCR were 1 cycle for 10 min at 95°C, 40 cycles of 15 s at 95°C, and 60°C for 1 min on a C1000 Thermal Cycler equipped with CFX96 real-time system (Bio-Rad, Hercules, CA). A reverse-transcriptase control was included to confirm comparable efficiency of reverse transcription in all samples, and melting curve analysis was performed to confirm formation of a single PCR product. Gene expression was normalized to the ACTB gene, and fold changes in samples from RTD-1-treated versus saline-treated rats were calculated with the Geneglobe Data Analysis center (Qiagen) by the 2−ΔΔCT method.
FLS cells.
Two primary human RA-FLS cell lines (25) were employed. FLS cells were plated in 24-well plates at a density of 5 × 104 cells per well. After 1 day in culture, the medium was removed and replaced with serum-free DMEM medium. Cells were stimulated with 0.1 ng/ml recombinant human IL-1β (R & D Systems, Minneapolis, MN) for 6 h with 0–10 µg/ml of RTD-1. RNA was isolated from the cells using Quick RNA Miniprep isolation kit, and gene expression analysis was performed with qRT-PCR, performed as above (Supplementary Table S1 for list of primers for human genes from Qiagen). Human ACTB was used as the reference gene, and the difference in CT values (ΔCT) was calculated as ΔCT = CTACTB − CTGene of Interest and analyzed by t test (paired, two tailed) (34). Experiments were performed twice for each cell line (total 4 independent experiments). The medium from the cells was centrifuged for 8 min at 250 g and then at 8,000 g for 5 min and used for cytokine analysis using the Milliplex MAP kit (EMD Millipore, Billerica, MA) using a Bio-Rad Bioplex HTF Luminex reader.
Statistical analysis.
Pearson’s correlation coefficient and Student’s t test were performed as indicated in the legends to the figures with GraphPad Prism or Microsoft Excel.
RESULTS
Identification of differentially expressed genes in synovial tissues of PIA and naive rats.
To evaluate the effects of RTD-1 on synovial tissue gene expression in PIA, we first analyzed gene expression alterations in synovial tissue in PIA rats compared with naive animals. The synovial tissue from PIA animals was harvested 1 day following the onset of disease with AS ≥ 3 in each rear limb (the total arthritic score from all four limbs was between 8 and 13 in PIA rats; animal maximum AS = 16). RNA-Seq analysis of joint synovial tissues from the two cohorts identified 617 differentially expressed genes (DEGs) with an FDR <0.05, hereafter referred to as disease genes. Principal component analysis (PCA) of the DEGs allowed for identification of two gene clusters that segregated with naive or PIA rats (Fig. 2A). Two-way hierarchical cluster analysis using Euclidean distance and average linkage also identified distinct gene groups in PIA and naive animals (Fig. 2B). RNA-Seq results were validated by qRT-PCR of up- and downregulated DEGs in PIA rat synovial tissue RNAs compared with that from naive rats (Supplementary Fig. S1).
Fig. 2.

Differential expression of disease genes in synovium of pristane-induced arthritic (PIA) and naive rats. A: unsupervised principal component analysis (PCA) plots of the 617 differentially expressed genes (DEGs) shows two distinct clusters expressed in synovial tissues of PIA and naive animals. Each circle represents an individual tissue sample. B: hierarchical clustering of differentially expressed genes [false discovery rate (FDR) P < 0.05] using Euclidean distance and average linkage indicates altered expression profiles in PIA and naive synovium. C: functional categories of disease genes identified in PIA tissue determined by Ingenuity Pathway Analysis (IPA). D: gene set enrichment analysis of DEGs identifies multiple arthritis-related pathways. Data sets used to extract the pathway terms are indicated in parentheses. A ranked P value was computed for each pathway from the Fisher exact test based on the binomial distribution along with Benjamini-Hochberg correction (P < 0.05).
The 617 disease genes were associated with numerous cellular and functional groups (Fig. 2C and Supplementary Table S2). Of note, genes implicated in RA were also expressed in PIA synovium at elevated levels; these include inflammatory cytokines and chemokines (IL6, IL1B, CXCL6, CCL20, and CCL9 ≥ 30-fold), proteases (MMPs 3, 8, 9, 10, and 13 ≥ 10-fold), transmembrane receptors (IL21R, TLR1, and CLEC7A ≥ 10-fold), enzymes (HAS2 and CHI3L1 ≥ 10-fold), and others (PTX3, S100A9, SLPI > 50-fold). Expression of many genes encoding ion channels and ligand-dependent nuclear receptors were suppressed in arthritic joints: GRIA2, CLIC5, and KCNN3 < −30-fold and PPARG and NR3C2 < −10-fold, respectively, relative to naive synovial tissue. Gene set enrichment analysis of the disease genes disclosed their association with RA and/or pathways associated with progression of RA such as TNF signaling, cytokine signaling, and inflammatory responses (Fig. 2D). In addition to known arthritis-related pathways, response to fatty acid and abnormal liver triglyceride genes were also enriched in PIA synovial tissues. These results demonstrate that diverse sets of genes are regulated during PIA, many of which have previously been implicated in RA.
Upstream regulator analysis of disease genes identified several highly activated regulators, the highest being TNF, IL6, and IL1B (Z scores > 5), well-characterized mediators of RA (Supplementary Table S3). In addition, IFNG, TLR3, TLR9, TGFB1, IL-17, NF-κB, and MAP kinase pathways, all implicated in RA, were all markedly upregulated in PIA synovium (Supplementary Table S3). Of note, several anti-inflammatory ligand-dependent nuclear receptors were downregulated in PIA, with PPARG being the most affected upstream regulator (Z score < −5). These findings are consistent with the finding that proinflammatory pathways are activated and anti-inflammatory or protective pathways are downregulated in PIA and RA.
Concordance of rat PIA RNA-Seq results with published arthritis-specific gene signatures.
Disease genes identified by RNA-Seq analysis were compared with DEGs implicated in arthritis susceptibility or resistance by Brenner et al. (8), who compared synovial tissue gene expression of PIA-susceptible DA rats with the expression pattern in arthritis-resistant congenic rat strains. DEGs identified by our RNA-Seq analysis included 193 genes that were the same as DEGs identified by transcriptomic comparisons of congenic strains (8), of which 77 genes were upregulated and 116 downregulated in both data sets (Pearson correlation, r = 0.8409, P < 0.0001; Fig. 3 and Supplementary Table S4). Moreover, both data sets predicted many of the same activated or inhibited upstream regulators, including previously mentioned arthritogenic cytokines (TNF, IL6, IL1B, IFNG) and signaling pathways (TLR, NF-κB, MAP kinase). Similarly, several ligand-dependent nuclear receptor genes (PPARA, NR1H3, NR1H2, NR3C1, PPARD, and PPARG), which are protective, anti-inflammatory upstream regulators, were suppressed during disease in both studies (data not shown).
Fig. 3.
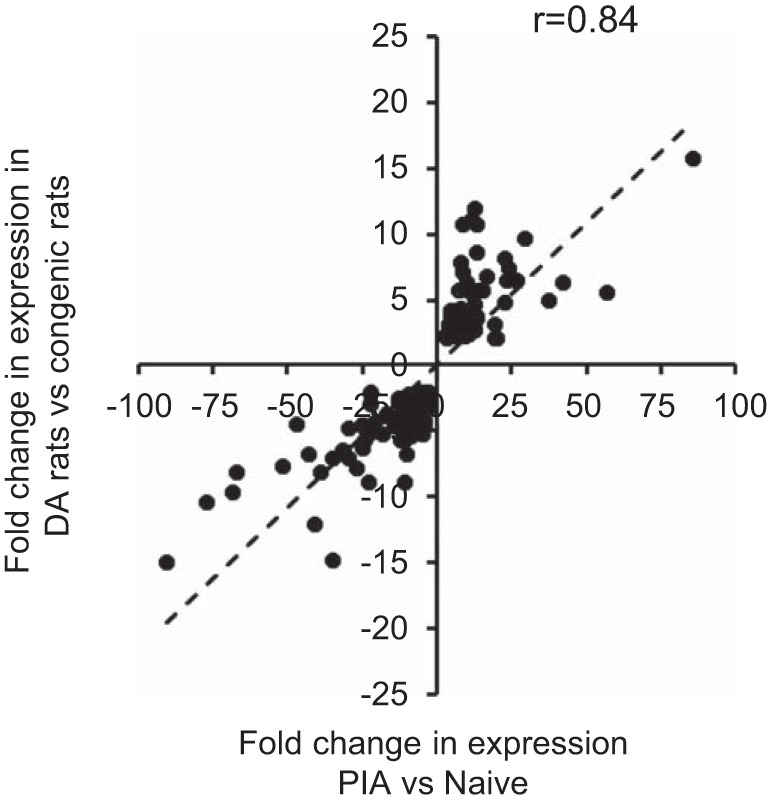
Concordance of synovial gene expression in PIA determined by RNA-Seq analysis and arthritis genes identified by microarray analysis. Linear regression analysis correlates 193 PIA-altered genes common to microarray analysis [Brenner et al. (8)] and RNA-Seq in this study (Supplementary Table S4). Log-transformed fold-change values were used to generate the scatter plot. Pearson correlation coefficient r = 0.84; P < 0.0001.
RTD-1 mediated changes in PIA synovial tissue gene expression.
Previously, we reported that subcutaneous administration of RTD-1 to rats with established PIA arrested disease within 3 days of treatment (39). To evaluate the temporal effect of RTD-1 on synovial gene expression in this model, RNA-Seq analysis was performed on synovial tissue from PIA rats that were administered saline or 5 mg/kg RTD-1 for 1, 3, or 5 days (Supplementary Fig. S2). Over this period, RTD-1 modified expression levels of 340 of the 617 PIA disease genes (Fig. 4). After a single RTD-1 treatment, 32 disease genes (FDR P < 0.05) were differentially expressed compared with saline-treated controls (Fig. 4, A and D). However, PCA and hierarchical analyses showed that PIA, saline-treated, and cohorts dosed 1 day with RTD-1 clustered together, evidence that RTD-1 had little effect on disease gene expression and consistent with a single dose of peptide lacking clinical effect (Supplementary Fig. S2). However, after 3 days of RTD-1 administration, a new cluster of 258 disease genes emerged that were responsive to peptide treatment (Fig. 4, B and D and Supplementary Table S5). This cluster was distinct from genes expressed in joints of naive animals and of those expressed in synovium of PIA rats and saline controls, which clustered together. Among these RTD-1-responsive disease genes, 89 were upregulated and 169 downregulated (Supplementary Table S5).
Fig. 4.

RTD-1 alters disease gene expression. Gene expression was analyzed by PCA (top panels) and unsupervised hierarchical two-way clustering analysis (bottom panels) of disease genes after RTD-1 treatment for 1 day (32 genes) (A), 3 days (258 genes) (B), and 5 days (107 genes) (C). D: Venn diagram showing number of disease genes regulated by RTD-1 treatment for 1, 3, or 5 days.
Daily administration of RTD-1 to PIA rats homeostatically restored normal disease gene expression (Fig. 4C; Supplementary Figs. S3, S4). Five days of RTD-1 treatment reduced severity of clinical arthritis (Supplementary Fig. S2) and further altered differential expression of 107 disease genes compared with saline controls (Fig. 4, C and D; Supplementary Table S6). PCA and cluster analyses showed that disease gene expression in rats receiving 5 days of RTD-1 administration overlapped with that of naive animals but was distinct from PIA and saline control gene expression profiles, which remained clustered together (Fig. 4C).
The temporal normalization of disease gene expression was further evident in volcano plots of global transcriptional changes after 3 and 5 days of RTD-1 treatment (Supplementary Fig. S3). For example, disease genes such as CCL7, CCL20, IL6, and FCNB that were markedly elevated in PIA were all highly suppressed after 5 days of treatment with RTD-1, but normalization was only partially restored in PIA rats treated 3 days with peptide (Supplementary Fig. S3). Conversely, several genes that were downregulated in PIA synovium, e.g., SCD1, ASCL1, CCL24, PHEX, were markedly upregulated after 5 days of RTD-1 administration (Supplementary Fig. S3). The temporal effects of systemic RTD-1 on disease gene expression were analyzed further and validated by qRT-PCR (Supplementary Fig. S4, A and B), the results of which correlated well (r = 0.96, P < 0.0001) with RNA-Seq data. In addition, a representative genomic snapshot showed that expression of the stearoyl-CoA desaturase-1 (SCD1) gene, which was markedly suppressed in PIA (Supplementary Table S2), was restored to normal levels by treatment with RTD-1 for 5 days (Supplementary Fig. S4C).
To further evaluate gene expression changes associated with disease resolution, two-way hierarchical clustering analysis of the 107 disease genes modulated by 5 days of RTD-1 administration was performed for each of the treated cohorts (Fig. 5A). The expression profile of rats that received RTD-1 for 5 days most closely resembled that of tissue from naive rats (Fig. 5A). In contrast, synovial gene expression profiles obtained from the 1-day and 3-day RTD-1 treatment groups were similar to those of untreated PIA rats (PIA, t = 0), and the effect of RTD-1 on the 107 gene cluster differed greatly from expression in cohorts receiving saline vehicle for 3 and 5 days (Fig. 5A), clearly differentiating peptide-mediated resolution from that mediated by vehicle administration alone.
Fig. 5.

RTD-1-responsive PIA disease genes. A: two-way hierarchical clustering of the 107 genes regulated by 5-day treatment with RTD-1 across all treatment cohorts shows similar patterns of genes in synovium of 5-day peptide treatment group and that of naive animals. B: gene enrichment of differentially expressed genes in PIA after 3 days and 5 days of RTD-1 treatment. Each enriched pathway is ranked based on the P value computed from the Fisher exact test based on the binomial distribution and independence for probability of any gene belonging to any enriched set. Source databases are shown in parentheses. C: plots of Z scores of common upstream regulators predicted by IPA in untreated PIA and in PIA following 5-day RTD-1 treatment show the homeostatic effects of peptide treatment on arthritis associated pathways.
The transcriptomic responses to systemic RTD-1 were interrogated further by gene set enrichment analyses. RTD-1-regulated DEGs, analyzed after 3 and 5 days of peptide treatment, are involved in RA, stress, immune system regulation, receptor binding, regulation of cytokine activity, cell adhesion, leukocyte migration, and response to fatty acids. Genes responsive to rosiglitazone, a PPAR-γ agonist, and 1-methyl-3-isobutylxanthine, a cAMP regulator, also were enriched in the RTD-1 regulated gene set (Fig. 5B).
Upstream regulator analysis showed that PIA rats treated for 5 days with RTD-1 responded by inhibition of proinflammatory upstream regulators, including IL6, IL1, TGFB1, TNF, and NF-κB, all of which are implicated in RA (Fig. 5C). Additionally, the ligand-dependent nuclear receptors RARA, RXRA, NR1H3, NR1H2, and PPARG, known anti-inflammatory effectors, were activated in synovial tissues of 5-day RTD-1-treated cohorts (Fig. 5C, Supplementary Table S7). Of note, PPARG, NR1H2, and NR1H3 were markedly repressed in untreated PIA. We conclude from these findings that RTD-1 administration reverses the proinflammatory transcriptional program that is induced in diseased PIA synovium.
Effect of RTD-1 on gene expression in naive synovial tissue in healthy animals.
RTD-1 treatment of healthy rats for 3 or 5 days induced 88 and 91 synovial DEGs, respectively. Those DEGs were dissimilar from the disease genes modulated by RTD-1 in PIA rats (Supplementary Fig. S5, A and B). For example, in healthy rats treated for 3 days with RTD-1, only two of the 258 disease genes modulated by RTD-1 in PIA rats were differentially expressed, and only four of the 107 disease genes modulated by 5 days of RTD-1 treatment in PIA rats were expressed differentially when healthy animals were treated similarly (Supplementary Fig. S5A). Furthermore, 68 DEGs were common to healthy rats that received systemic RTD-1 for 3 and 5 days (Supplementary Fig. S5C), but those DEGs involved pathways that are not related to arthritis, which included translation, senescence, and RNA metabolism (data not shown). These results show that, although RTD-1 modulates the expression of certain genes in naive synovium, the induced expression is distinct from disease gene expression in PIA synovium, and gene regulation in PIA joints is highly selective and associated with disease resolution.
RTD-1 inhibits proinflammatory responses in RA-FLS cells.
In a previous study, we showed that IL-1β protein is elevated in joints of PIA rats and that synovial IL-1β levels are suppressed following RTD-1 treatment (39). Because IL-1β increases IL6 and CXCL8 expression by human RA-FLS cells (16), we stimulated RA-FLS cells with IL-1β and showed that IL6 and CXCL8 mRNA expression was repressed by RTD-1 in a concentration-dependent manner (Fig. 6, A and B). Also, RTD-1 markedly reduced levels of IL-6 and IL-8 protein in culture media (Fig. 6, C and D) and inhibited release of additional inflammatory chemokines and cytokines, e.g., CCL3, CCL4, CCL5, CCL7, IFN-γ and TNF-α, from IL-1β-stimulated RA-FLS cells (Supplementary Fig. S6). Finally, we measured whether expression of human orthologs of rat disease genes that are induced in PIA is affected by RTD-1 treatment of RA-FLS cells exposed to IL-1β. IL-1β stimulation induced increased levels of RA-FLS cell IL1B, CCL20, CCL7, and PTGS2 mRNAs (Fig. 7), each of which shows elevated expression in rat PIA synovium. RTD-1 inhibited expression of each of these mRNAs, while GAPDH, which was unaltered during PIA, was unaffected by IL-1β stimulation or by treatment with RTD-1 (Fig. 7). Thus, consistent with the effects of RTD-1 in PIA, RTD-1 inhibited proinflammatory responses in human RA-FLS cells.
Fig. 6.

RTD-1 suppresses IL-6 and IL-8 expression in IL-1β-stimulated rheumatoid arthritis-fibroblast-like synoviocyte (RA-FLS) cells. RA-FLS cells were stimulated with IL-1β ± RTD-1 for 6 h and expression of IL6 mRNA (A) and CXCL8 mRNA (B) was analyzed by quantitative (q)RT-PCR and normalized to ACTB gene expression. Culture medium from A and B was analyzed for secretion of IL-6 (C) and IL-8 (D) using the Milliplex MAP kit. Data show means ± SD (t test, two tailed: *P < 0.05, **P < 0.01; comparison of IL-1β + RTD-1 with IL-1β treatment).
Fig. 7.

Effect of RTD-1 on gene expression by IL-1β-stimulated RA-FLS cells. RA-FLS cells were stimulated with IL-1β ± RTD-1 for 6 h, and the RNA was used to analyze the expression of human genes by qRT-PCR and normalized to ACTB gene expression. Data show means ± SD (t test, two tailed: *P < 0.05, **P < 0.01; comparison of IL-1β + RTD-1 with IL-1β treatment). With the exception of GAPDH, addition of RTD-1 significantly suppressed expression of each gene.
DISCUSSION
RTD-1 possesses immune-modulating properties in both in vitro and in animal models of infection and sepsis (5, 19, 38, 39, 46, 51). In each of the animal models investigated, the therapeutic effect of RTD-1 was the moderation of proinflammatory responses by suppression of proinflammatory cytokine levels (5, 19, 51). In vitro, RTD-1 blocks proinflammatory gene expression in stimulated blood leukocytes (38) and THP-1 macrophages and suppresses NF-κB and MAP kinase pathways (46). RTD-1 also has antiprotease properties, inhibiting MMPs and cathepsins, and reversibly inhibiting TACE/ADAM17, the sheddase that releases soluble TNF-α (37, 39). Consistent with the effects of RTD-1 modulation of dysregulated inflammation in infectious disease models, RTD-1 arrested and induced resolution of autoimmune arthritis in the rat (38), demonstrating the inflammation resolving properties in vivo in the absence of infection.
In previous studies, when RTD-1 treatment was initiated at early PIA (total AS of 3 for all 4 limbs), we observed rapid inhibition of arthritis, and RTD-1 efficacy was similarly observed in animals with severe, established arthritis (39). However, to identify the genes and networks involved in the pathogenesis of arthritis and those regulated by RTD-1, synovial tissues were harvested from animals with PIA that had AS of at least 3 in each rear limb. RTD-1 treatment at this intermediate time point during evolving arthritis led to reduction in AS after 5 days (average AS ± SD after 5 d treatment 9.55 ± 4.57 vs. 12.2 ± 2.66, P = 0.0625 in RTD-1- and saline-treated animals, respectively). Systemic RTD-1 administration alters the gene expression program of synovial tissue in the rat PIA disease model (Fig. 4). The therapeutic response was consistent with a previous study demonstrating that RTD-1 arrested arthritis progression and promoted resolution of severe joint disease with concomitant suppression of synovial IL-1β levels (39). DEGs encoding proinflammatory cytokines and MMPs were among the prominent arthritis-relevant genes that were upregulated in synovial tissues of diseased animals (Fig. 2 and Supplementary Table S2).
Consistent with the chronic inflammation of RA, gene enrichment analysis in PIA indicated changes in rheumatoid and osteoarthritis genes and genes associated with immune responses and immune signaling pathways. In fact, nearly 200 of the disease genes in PIA also were identified by Brenner et al. (8) as protective or pathogenic based on their study of susceptible (DA) and resistant congenic rat strains (Supplementary Table S4). Upstream regulator analyses for both studies implicated alterations in proinflammatory cytokines (TNF, IL1B, IL6, CCL2), inflammatory signaling pathways (NF-κB, MAP kinase), and protective ligand-dependent nuclear receptors (liver X receptors, PPARG, PPARA, PPARD) in PIA. In RA, chronic elevation of TNF-α (28), IL-6 (11), and IL-1β (10) levels is common and implicated in disease progression, consistent with our finding that IL1B and IL6 gene expression was markedly induced in PIA synovial tissue. Interestingly, TNF gene expression was not elevated in PIA, consistent with other studies (8), but activation of TNF-responsive pathways was clearly revealed in synovium from PIA rats. Moreover, we (39) and others (49) have shown that anti-TNF biologics reduce disease severity in rat PIA, confirming a role for TNF-α in pathogenesis of rat PIA.
Identification of gene subsets that were temporally affected over the 5-day course of RTD-1 treatment showed that peptide administration homeostatically restores the disease gene expression profile in PIA rat joints to one that resembles joints of healthy animals (Figs. 4 and 5A). Consistent with PIA and RA pathogenesis and resolution, DEGs modulated in joints of PIA rats after 5 days of systemic RTD-1 are enriched for genes implicated in RA, immune and cytokine responses, leukocyte migration and cell adhesion, regulation of stress responses, and extracellular matrix-associated proteins (Fig. 5B). Induction of PIA is associated with infiltration of cells in the joint tissue (50), and reduction in joint cellular infiltration was observed after RTD-1 treatment (39). Therefore, changes in the cellular composition of the joint may be at least partially responsible for the observed differential gene expression in synovial tissues of RTD-1-treated and untreated animals. RTD-1 administration to healthy rats did not affect synovial disease gene expression as it did in dysregulated PIA synovium, demonstrating that the peptide targets disease pathways selectively.
The antiarthritic mechanisms of RTD-1 include temporal attenuation of proinflammatory arthritis gene signatures in rat PIA synovial tissue. Expression of arthritogenic chemokine mRNAs that were upregulated in PIA and suppressed by RTD-1 treatment included CCL7, a monocyte chemoattractant that is elevated in the synovium and sera of RA patients (35), as well as CCL9 and CCL20. In addition to suppressing CCL7 expression in diseased PIA synovia, RTD-1 inhibited CCL7 expression by IL-1β-stimulated RA-FLS cells (Fig. 7). CCL9 is implicated in RA, because it stimulates osteoclast spreading and migration (26), thereby promoting bone resorption in RA (40). CCL20 is elevated in RA synovium and contributes to synovial hyperplasia by promoting chemotaxis of monocytes and lymphocytes (43). Systemic RTD-1 also suppressed expression of arthritogenic MMPs that are implicated in PIA and RA pathogenesis (9). For example, MMP-9 (1), MMP-10 (3), and MMP-13 (30) were induced in RA synovium but were suppressed in PIA synovia when rats were treated with RTD-1 for 5 days.
RTD-1 administration to PIA rats markedly changed the activation states of upstream regulators compared with PIA animals (Fig. 5C, Supplementary Table S7). The upstream regulators TNF, IL1B, and IL6, which had been activated in diseased joint tissue, were inhibited by RTD-1. In contrast, ligand-dependent nuclear receptors whose expression was repressed in PIA became activated following RTD-1 treatment. Interestingly, arthritis-relevant upstream regulators that include the proinflammatory cytokines IL6, IL1, TNF, IL17A, IL18, IL6R, and IL17F and signaling/transcription factors STAT3, PI3K, NFkB, ERK 1/2, HMGB1, AP1, and RELA were activated in PIA. Also, the protective upstream regulators PPARG, DUSP1, and Nr1h were repressed in PIA as they are in early and established RA, consistent with the similarity of the rat PIA model and human RA (17). Furthermore, 99 of the 107 genes selectively regulated by 5 days of RTD-1 administration have human orthologs, suggesting that RTD-1 may modify similar dysregulated pathways in human disease.
We have reported previously that IL-1β levels, which increase in joints of diseased rats, are reduced by RTD-1 treatment, and RTD-1 also inhibited secretion of IL-6 from rat FLS cells that were stimulated with IL-1β and TNF-α (39). Here, we have shown that RTD-1 suppressed both expression of IL6 and CXCL8 mRNAs as well as the secretion of these cytokines into the medium of IL-1β-induced human RA-FLS cells. RTD-1 also inhibited secretion of other inflammatory cytokines/chemokines including CCL7, CCL3, CCL4, CCL5, IFN-γ and TNF-α by RA-FLS cells. Finally, RTD-1 also inhibited the upregulation of IL1B, CCL20, CCL7, and PTGS2 mRNAs in IL-1β-stimulated RA-FLS, all of which are elevated in PIA rat synovial tissue.
The effect of RTD-1 on gene expression of ligand-dependent nuclear receptors such as PPAR-γ and liver X receptors was notable because of the protective role of these genes in PIA and RA (8, 13, 25). Among the nuclear receptors suppressed in PIA, PPARG expression was the most inhibited upstream regulator (~14-fold). After 5 days of RTD-1 treatment, expression levels of the common PPARG target genes ACSL1, CES1, CXCL6, DGAT2, FABP4, GPD1, IL6, LPL, MMP9, PC, SCD1, and SERPINE1 were reversed relative to their levels in PIA tissue, reflecting the activation of PPARG upstream regulator function (Supplementary Fig S7). This effect is consistent with previous studies wherein stimulation of PPAR-γ activity inhibited expression of MMP-1 (13), PPAR-γ agonist troglitazone inhibited secretion of TNF-α, IL-6, IL-8, and MMPs by human RA FLS cells (52), and PPAR-γ agonist rosiglitazone reduced plasma TNF-α, IL-1β, and IL-6 levels and diminished joint bone erosion in mice with collagen-induced arthritis (12). Several PPAR-γ agonists have been studied in RA and remain viable as potential therapeutic agents for RA (42).
As noted above, RTD-1 treatment of PIA rats induced activation of liver X receptors NR1H2 and NR1H3 (Fig. 5C). Liver X receptors have anti-inflammatory roles by inhibiting NF-κB activation and downregulating expression of proinflammatory genes, including PTGS2, IL1B, and IL6 in LPS-stimulated macrophages (20). Consistent with that role, systemic administration of RTD-1 suppressed the expression of PTGS2, IL1B, and IL6 in PIA synovial tissues. In studies of rat FLS cells, activation of liver X receptors inhibited invasiveness of IL-1β- and IL-6-stimulated rat FLS cells and expression of IL-6, IL-1β and MMP-3 (25). Similarly, expression of SCD1, a regulator of lipid metabolism and inflammation (36), was downregulated in PIA synovial tissue, consistent with the decreased expression of its known transcriptional inducers PPAR-γ, liver X receptors, and SREBP1/SREBF1 (33). Treatment of PIA rats systemically with RTD-1 for 5 days led to increased SCD1 expression as well as activation of PPARG and additional liver X receptor upstream regulators noted above. These results suggest that the antiarthritic effects of RTD-1 are mediated by stimulation of downstream effectors of ligand-dependent nuclear receptors in diseased synovial tissue.
The therapeutic effects of RTD-1 are associated with regulation of gene expression and signaling pathway networks in synovial tissue of PIA rats. To our knowledge, RTD-1 is the first HDP demonstrated to regulate arthritogenic gene expression pathways. Peptide treatment results in homeostatic resolution of pro- and anti-inflammatory pathways, many of which are similarly dysregulated in both PIA and RA. The therapeutic effect of RTD-1 on PIA synovial gene expression was also confirmed in studies of peptide effects on human RA-FLS cells, directly underscoring the potential translational relevance of these observations to human disease. Previously, RTD-1 was shown to be effective in arresting PIA and inducing resolution of clinical disease when administered subcutaneously as infrequently as once every 5 days (39). The cyclic conformation of RTD-1 confers remarkable stability and lack of immunogenicity (38, 39), and preclinical safety studies of RTD-1 have disclosed no systemic toxicities in vertebrates (data not shown). Based on these studies, a novel formulation of RTD-1 is being evaluated for its suitability as an RA therapeutic.
GRANTS
Funded by National Institutes of Health (NIH) Grants RO1-AI22931 (M. E. Selsted) and R44AR-068833-01 (D. Q. Tran), Arthritis Foundation Grant 5997 (M. E. Selsted), Southern California Clinical and Translational Science Institute Grant ULTR000130 (M. E. Selsted), the Seth MacFarlane Foundation (M. E. Selsted), the University of Southern California Norris Comprehensive Cancer Center, and NIH Grant P30 CA-014089.
DISCLOSURES
Oryn Therapeutics, LLC provided salary support to D. Q. Tran but did not have additional role in design, data collection, analysis, manuscript preparation or publication.
M. E. Selsted is a cofounder of Oryn Therapeutics, LLC, which has licensed theta defensin technology for therapeutic development in the field of immunology and infectious disease. Dr. Selsted is an equity holder in Oryn Therapeutics but receives no direct or indirect compensation from the company. Coauthor D. Q. Tran is employed by Oryn Therapeutics as scientific director. Besides Dr. Tran, Oryn played no role in the design, execution, analysis of the experiments or in the writing of the manuscript. Moreover, the data in the manuscript were reviewed by a USC faculty member assigned to monitor experimental work conducted in my laboratory as an element of the Conflict of Interest management plan that has been approved by the university.
AUTHOR CONTRIBUTIONS
P.T. and M.E.S., conceived study; P.T., V.P., D.Q.T., J.B.S., A.J.O. and M.E.S., planned experiments; P.T., A.S., D.Q.T., K.K.T. and J.B.S., performed experiments; V.P. performed bioinformatics analyses; T.L., provided and characterized cell lines; P.G., provided reagents; P.T., V.P., A.S., J.B.S., A.J.O., P.G. and M.E.S., analyzed data; P.T., V.P., P.G., A.J.O. and M.E.S., wrote manuscript; all authors approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Patti Tran for invaluable management and coordination of laboratory operations.
REFERENCES
- 1.Ahrens D, Koch AE, Pope RM, Stein-Picarella M, Niedbala MJ. Expression of matrix metalloproteinase 9 (96-kd gelatinase B) in human rheumatoid arthritis. Arthritis Rheum 39: 1576–1587, 1996. doi: 10.1002/art.1780390919. [DOI] [PubMed] [Google Scholar]
- 2.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 11: R106, 2010. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barksby HE, Milner JM, Patterson AM, Peake NJ, Hui W, Robson T, Lakey R, Middleton J, Cawston TE, Richards CD, Rowan AD. Matrix metalloproteinase 10 promotion of collagenolysis via procollagenase activation: implications for cartilage degradation in arthritis. Arthritis Rheum 54: 3244–3253, 2006. doi: 10.1002/art.22167. [DOI] [PubMed] [Google Scholar]
- 4.Basso V, Garcia A, Tran DQ, Schaal JB, Tran P, Ngole D, Aqeel Y, Tongaonkar P, Ouellette AJ, Selsted ME. Fungicidal Potency and Mechanisms of θ-Defensins against Multidrug-Resistant Candida Species. Antimicrob Agents Chemother 62: e00111-18, 2018. doi: 10.1128/AAC.00111-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bensman TJ, Jayne JG, Sun M, Kimura E, Meinert J, Wang JC, Schaal JB, Tran D, Rao AP, Akbari O, Selsted ME, Beringer PM. Efficacy of Rhesus Theta-Defensin-1 in Experimental Models of Pseudomonas aeruginosa Lung Infection and Inflammation. Antimicrob Agents Chemother 61: e00154-17, 2017. doi: 10.1128/AAC.00154-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beringer PM, Bensman TJ, Ho H, Agnello M, Denovel N, Nguyen A, Wong-Beringer A, She R, Tran DQ, Moskowitz SM, Selsted ME. Rhesus θ-defensin-1 (RTD-1) exhibits in vitro and in vivo activity against cystic fibrosis strains of Pseudomonas aeruginosa. J Antimicrob Chemother 71: 181–188, 2016. doi: 10.1093/jac/dkv301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brand DD, Kang AH, Rosloniec EF. The mouse model of collagen-induced arthritis. Methods Mol Med 102: 295–312, 2004. doi: 10.1385/1-59259-805-6:295. [DOI] [PubMed] [Google Scholar]
- 8.Brenner M, Laragione T, Gulko PS. Analyses of synovial tissues from arthritic and protected congenic rat strains reveal a new core set of genes associated with disease severity. Physiol Genomics 45: 1109–1122, 2013. doi: 10.1152/physiolgenomics.00108.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Front Biosci 11: 529–543, 2006. doi: 10.2741/1817. [DOI] [PubMed] [Google Scholar]
- 10.Chikanza IC, Kingsley G, Panayi GS. Peripheral blood and synovial fluid monocyte expression of interleukin 1 alpha and 1 beta during active rheumatoid arthritis. J Rheumatol 22: 600–606, 1995. [PubMed] [Google Scholar]
- 11.Choy E. Interleukin 6 receptor as a target for the treatment of rheumatoid arthritis. Ann Rheum Dis 62, Suppl 2: ii68–ii69, 2003. doi: 10.1136/ard.62.suppl_2.ii68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cuzzocrea S, Mazzon E, Dugo L, Patel NS, Serraino I, Di Paola R, Genovese T, Britti D, De Maio M, Caputi AP, Thiemermann C. Reduction in the evolution of murine type II collagen-induced arthritis by treatment with rosiglitazone, a ligand of the peroxisome proliferator-activated receptor gamma. Arthritis Rheum 48: 3544–3556, 2003. doi: 10.1002/art.11351. [DOI] [PubMed] [Google Scholar]
- 13.Fahmi H, Pelletier JP, Di Battista JA, Cheung HS, Fernandes JC, Martel-Pelletier J. Peroxisome proliferator-activated receptor gamma activators inhibit MMP-1 production in human synovial fibroblasts likely by reducing the binding of the activator protein 1. Osteoarthritis Cartilage 10: 100–108, 2002. doi: 10.1053/joca.2001.0485. [DOI] [PubMed] [Google Scholar]
- 14.Feng J, Jing J, Li J, Zhao H, Punj V, Zhang T, Xu J, Chai Y. BMP signaling orchestrates a transcriptional network to control the fate of mesenchymal stem cells in mice. Development 144: 2560–2569, 2017. doi: 10.1242/dev.150136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garcia AE, Osapay G, Tran PA, Yuan J, Selsted ME. Isolation, synthesis, and antimicrobial activities of naturally occurring theta-defensin isoforms from baboon leukocytes. Infect Immun 76: 5883–5891, 2008. doi: 10.1128/IAI.01100-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Georganas C, Liu H, Perlman H, Hoffmann A, Thimmapaya B, Pope RM. Regulation of IL-6 and IL-8 expression in rheumatoid arthritis synovial fibroblasts: the dominant role for NF-kappa B but not C/EBP beta or c-Jun. J Immunol 165: 7199–7206, 2000. doi: 10.4049/jimmunol.165.12.7199. [DOI] [PubMed] [Google Scholar]
- 17.Guo Y, Walsh AM, Fearon U, Smith MD, Wechalekar MD, Yin X, Cole S, Orr C, McGarry T, Canavan M, Kelly S, Lin TA, Liu X, Proudman SM, Veale DJ, Pitzalis C, Nagpal S. CD40L-Dependent Pathway Is Active at Various Stages of Rheumatoid Arthritis Disease Progression. J Immunol 198: 4490–4501, 2017. doi: 10.4049/jimmunol.1601988. [DOI] [PubMed] [Google Scholar]
- 18.Haney EF, Mansour SC, Hancock RE. Antimicrobial Peptides: An Introduction. Methods Mol Biol 1548: 3–22, 2017. doi: 10.1007/978-1-4939-6737-7_1. [DOI] [PubMed] [Google Scholar]
- 19.Jayne JG, Bensman TJ, Schaal JB, Park AYJ, Kimura E, Tran D, Selsted ME, Beringer PM. Rhesus θ-Defensin-1 Attenuates Endotoxin-induced Acute Lung Injury by Inhibiting Proinflammatory Cytokines and Neutrophil Recruitment. Am J Respir Cell Mol Biol 58: 310–319, 2018. doi: 10.1165/rcmb.2016-0428OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat Med 9: 213–219, 2003. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 21.Kani K, Garri C, Tiemann K, Malihi PD, Punj V, Nguyen AL, Lee J, Hughes LD, Alvarez RM, Wood DM, Joo AY, Katz JE, Agus DB, Mallick P. JUN-Mediated Downregulation of EGFR Signaling Is Associated with Resistance to Gefitinib in EGFR-mutant NSCLC Cell Lines. Mol Cancer Ther 16: 1645–1657, 2017. doi: 10.1158/1535-7163.MCT-16-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kannan K, Ortmann RA, Kimpel D. Animal models of rheumatoid arthritis and their relevance to human disease. Pathophysiology 12: 167–181, 2005. doi: 10.1016/j.pathophys.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 23.Kim K, Punj V, Kim JM, Lee S, Ulmer TS, Lu W, Rice JC, An W. MMP-9 facilitates selective proteolysis of the histone H3 tail at genes necessary for proficient osteoclastogenesis. Genes Dev 30: 208–219, 2016. doi: 10.1101/gad.268714.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krämer A, Green J, Pollard J Jr, Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30: 523–530, 2014. doi: 10.1093/bioinformatics/btt703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laragione T, Gulko PS. Liver X receptor regulates rheumatoid arthritis fibroblast-like synoviocyte invasiveness, matrix metalloproteinase 2 activation, interleukin-6 and CXCL10. Mol Med 18: 1009–1017, 2012. doi: 10.2119/molmed.2012.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lean JM, Murphy C, Fuller K, Chambers TJ. CCL9/MIP-1gamma and its receptor CCR1 are the major chemokine ligand/receptor species expressed by osteoclasts. J Cell Biochem 87: 386–393, 2002. doi: 10.1002/jcb.10319. [DOI] [PubMed] [Google Scholar]
- 27.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maini RN, Elliott M, Brennan FM, Williams RO, Feldmann M. TNF blockade in rheumatoid arthritis: implications for therapy and pathogenesis. APMIS 105: 257–263, 1997. doi: 10.1111/j.1699-0463.1997.tb00567.x. [DOI] [PubMed] [Google Scholar]
- 29.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med 365: 2205–2219, 2011. doi: 10.1056/NEJMra1004965. [DOI] [PubMed] [Google Scholar]
- 30.Moore BA, Aznavoorian S, Engler JA, Windsor LJ. Induction of collagenase-3 (MMP-13) in rheumatoid arthritis synovial fibroblasts. Biochim Biophys Acta 1502: 307–318, 2000. doi: 10.1016/S0925-4439(00)00056-9. [DOI] [PubMed] [Google Scholar]
- 31.Moudgil KD, Kim P, Brahn E. Advances in rheumatoid arthritis animal models. Curr Rheumatol Rep 13: 456–463, 2011. doi: 10.1007/s11926-011-0200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen TX, Cole AM, Lehrer RI. Evolution of primate theta-defensins: a serpentine path to a sweet tooth. Peptides 24: 1647–1654, 2003. doi: 10.1016/j.peptides.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 33.Paton CM, Ntambi JM. Biochemical and physiological function of stearoyl-CoA desaturase. Am J Physiol Endocrinol Metab 297: E28–E37, 2009. doi: 10.1152/ajpendo.90897.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reimann S, Fink L, Wilhelm J, Hoffmann J, Bednorz M, Seimetz M, Dessureault I, Troesser R, Ghanim B, Klepetko W, Seeger W, Weissmann N, Kwapiszewska G. Increased S100A4 expression in the vasculature of human COPD lungs and murine model of smoke-induced emphysema. Respir Res 16: 127, 2015. doi: 10.1186/s12931-015-0284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rump L, Mattey DL, Kehoe O, Middleton J. An initial investigation into endothelial CC chemokine expression in the human rheumatoid synovium. Cytokine 97: 133–140, 2017. doi: 10.1016/j.cyto.2017.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sampath H, Ntambi JM. The role of stearoyl-CoA desaturase in obesity, insulin resistance, and inflammation. Ann N Y Acad Sci 1243: 47–53, 2011. doi: 10.1111/j.1749-6632.2011.06303.x. [DOI] [PubMed] [Google Scholar]
- 37.Schaal JB, Maretzky T, Tran DQ, Tran PA, Tongaonkar P, Blobel CP, Ouellette AJ, Selsted ME. Macrocyclic θ-defensins suppress tumor necrosis factor-α (TNF-α) shedding by inhibition of TNF-α-converting enzyme. J Biol Chem 293: 2725–2734, 2018. doi: 10.1074/jbc.RA117.000793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schaal JB, Tran D, Tran P, Ösapay G, Trinh K, Roberts KD, Brasky KM, Tongaonkar P, Ouellette AJ, Selsted ME. Rhesus macaque theta defensins suppress inflammatory cytokines and enhance survival in mouse models of bacteremic sepsis. PLoS One 7: e51337, 2012. doi: 10.1371/journal.pone.0051337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaal JB, Tran DQ, Subramanian A, Patel R, Laragione T, Roberts KD, Trinh K, Tongaonkar P, Tran PA, Minond D, Fields GB, Beringer P, Ouellette AJ, Gulko PS, Selsted ME. Suppression and resolution of autoimmune arthritis by rhesus θ-defensin-1, an immunomodulatory macrocyclic peptide. PLoS One 12: e0187868, 2017. doi: 10.1371/journal.pone.0187868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schett G. Cells of the synovium in rheumatoid arthritis. Osteoclasts. Arthritis Res Ther 9: 203, 2007. doi: 10.1186/ar2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol 6: 551–557, 2005. doi: 10.1038/ni1206. [DOI] [PubMed] [Google Scholar]
- 42.Shahin D, Toraby EE, Abdel-Malek H, Boshra V, Elsamanoudy AZ, Shaheen D. Effect of peroxisome proliferator-activated receptor gamma agonist (pioglitazone) and methotrexate on disease activity in rheumatoid arthritis (experimental and clinical study). Clin Med Insights Arthritis Musculoskelet Disord 4: 1–10, 2011. doi: 10.4137/CMAMD.S5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Szekanecz Z, Vegvari A, Szabo Z, Koch AE. Chemokines and chemokine receptors in arthritis. Front Biosci (Schol Ed) 2: 153–167, 2010. doi: 10.2741/s53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang YQ, Yuan J, Osapay G, Osapay K, Tran D, Miller CJ, Ouellette AJ, Selsted ME. A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated alpha-defensins. Science 286: 498–502, 1999. doi: 10.1126/science.286.5439.498. [DOI] [PubMed] [Google Scholar]
- 45.Tongaonkar P, Tran P, Roberts K, Schaal J, Osapay G, Tran D, Ouellette AJ, Selsted ME. Rhesus macaque θ-defensin isoforms: expression, antimicrobial activities, and demonstration of a prominent role in neutrophil granule microbicidal activities. J Leukoc Biol 89: 283–290, 2011. doi: 10.1189/jlb.0910535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tongaonkar P, Trinh KK, Schaal JB, Tran D, Gulko PS, Ouellette AJ, Selsted ME. Rhesus macaque θ-defensin RTD-1 inhibits proinflammatory cytokine secretion and gene expression by inhibiting the activation of NF-κB and MAPK pathways. J Leukoc Biol 98: 1061–1070, 2015. doi: 10.1189/jlb.3A0315-102R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tran D, Tran P, Roberts K, Osapay G, Schaal J, Ouellette A, Selsted ME. Microbicidal properties and cytocidal selectivity of rhesus macaque theta defensins. Antimicrob Agents Chemother 52: 944–953, 2008. doi: 10.1128/AAC.01090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tran D, Tran PA, Tang YQ, Yuan J, Cole T, Selsted ME. Homodimeric theta-defensins from rhesus macaque leukocytes: isolation, synthesis, antimicrobial activities, and bacterial binding properties of the cyclic peptides. J Biol Chem 277: 3079–3084, 2002. doi: 10.1074/jbc.M109117200. [DOI] [PubMed] [Google Scholar]
- 49.Tuncel J, Haag S, Hoffmann MH, Yau AC, Hultqvist M, Olofsson P, Bäcklund J, Nandakumar KS, Weidner D, Fischer A, Leichsenring A, Lange F, Haase C, Lu S, Gulko PS, Steiner G, Holmdahl R. Animal Models of Rheumatoid Arthritis (I): Pristane-Induced Arthritis in the Rat. PLoS One 11: e0155936, 2016. doi: 10.1371/journal.pone.0155936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vingsbo C, Sahlstrand P, Brun JG, Jonsson R, Saxne T, Holmdahl R. Pristane-induced arthritis in rats: a new model for rheumatoid arthritis with a chronic disease course influenced by both major histocompatibility complex and non-major histocompatibility complex genes. Am J Pathol 149: 1675–1683, 1996. [PMC free article] [PubMed] [Google Scholar]
- 51.Wohlford-Lenane CL, Meyerholz DK, Perlman S, Zhou H, Tran D, Selsted ME, McCray PB Jr. Rhesus theta-defensin prevents death in a mouse model of severe acute respiratory syndrome coronavirus pulmonary disease. J Virol 83: 11385–11390, 2009. doi: 10.1128/JVI.01363-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamasaki S, Nakashima T, Kawakami A, Miyashita T, Ida H, Migita K, Nakata K, Eguchi K. Functional changes in rheumatoid fibroblast-like synovial cells through activation of peroxisome proliferator-activated receptor gamma-mediated signalling pathway. Clin Exp Immunol 129: 379–384, 2002. doi: 10.1046/j.1365-2249.2002.01876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]