

Abstract
Pulmonary emphysema is characterized by alveolar type II (ATII) cell death, destruction of alveolar wall septa, and irreversible airflow limitation. Cigarette smoke induces oxidative stress and is the main risk factor for this disease development. ATII cells isolated from nonsmokers, smokers, and patients with emphysema were used for this study. ATII cell apoptosis in individuals with this disease was detected. DJ-1 and S100A8 have cytoprotective functions against oxidative stress-induced cell injury. Reduced DJ-1 and S100A8 interaction was found in ATII cells in patients with emphysema. The molecular function of S100A8 was determined by an analysis of the oxidation status of its cysteine residues using chemoselective probes. Decreased S100A8 sulfination was observed in emphysema patients. In addition, its lower levels correlated with higher cell apoptosis induced by cigarette smoke extract in vitro. Cysteine at position 106 within DJ-1 is a central redox-sensitive residue. DJ-1 C106A mutant construct abolished the cytoprotective activity of DJ-1 against cell injury induced by cigarette smoke extract. Furthermore, a molecular and complementary relationship between DJ-1 and S100A8 was detected using gain- and loss-of-function studies. DJ-1 knockdown sensitized cells to apoptosis induced by cigarette smoke extract, and S100A8 overexpression provided cytoprotection in the absence of DJ-1. DJ-1 knockout mice were more susceptible to ATII cell apoptosis induced by cigarette smoke compared with wild-type mice. Our results indicate that the impairment of DJ-1 and S100A8 function may contribute to cigarette smoke-induced ATII cell injury and emphysema pathogenesis.
Keywords: alveolar type II cells, cigarette smoke, DJ-1, emphysema, S100A8
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is characterized by irreversible airflow obstruction and encompasses a spectrum of diseases, including emphysema (20). Pulmonary emphysema is characterized by airspace enlargements and alveolar wall destruction. Alveolar type II (ATII) cells produce and secrete pulmonary surfactant, maintain alveolar fluid homeostasis, and restore the epithelium after damage to the more sensitive alveolar type I cells (29). Cigarette smoke is a major risk factor for emphysema development (49). It induces oxidative stress and protease and anti-protease imbalance, which can result in extracellular matrix destruction and alveolar epithelial cell injury and death. Effective treatment against this disease is very limited (45).
S100A8 (also known as calgranulin-A or MRP-8) belongs to S100 calcium-binding protein family (17). It is expressed in a wide variety of cell types including human primary ATII cells as we previously reported (27). S100A8 preferentially forms a heterocomplex with S100A9 which mediates most cell functions; however, it can also function independently and in association with other proteins (36). It has been shown that S100A8 protects against acute lung injury (16). We previously demonstrated that S100A8 has a cytoprotective activity (27); however, the mechanism is not well known. S100A8 has a conservative cysteine residue at position 42 (C42), which regulates the function of this protein (40). Oxidative stress induces S100A8 expression and the protein activity is abrogated by substitution of C42 with alanine (39). Oxidized C42 within S100A8 was identified in sputum obtained from asthma patients, which indicates the importance of S100A8 in the lung (14).
DJ-1 is a homodimer and a redox-sensitive molecular chaperone (10, 46). It is ubiquitously expressed in mammalian tissue, including the lung (19, 42), and human primary ATII cells as we previously reported (4). It plays an important role in the antioxidant defense system as we described (3, 4). DJ-1 deficiency is implicated in various pathological processes and diseases (5, 12, 18, 43). Cysteine residue at position 106 (C106) within DJ-1 is a sensor of redox status. Loss of DJ-1 function increased oxidative stress-induced cell death caused by hydrogen peroxide (41). We and others reported that DJ-1 overexpression and activation protected cells against injury (4, 10). In addition, we have recently demonstrated murine ATII cell injury in DJ-1 knockout (KO) mice exposed to cigarette smoke (42).
In the present study, we used primary human ATII cells obtained from control organ donors and emphysema patients, gain- and loss-of-function approaches in vitro and DJ-1 KO mice. Our data indicate a complementary cytoprotective role of DJ-1 and S100A8 against injury induced by cigarette smoke in ATII cells.
MATERIALS AND METHODS
Isolation of human primary ATII cells.
Lung tissue was obtained from deidentified nonsmoker and smoker organ donors (n = 3–8 per group, 45–69 yr old, women and men), whose lungs were unsuitable for transplantation and were donated for medical research from the Gift of Life Foundation (Philadelphia, PA). Nonsmokers were individuals who never smoked and smokers were smoking 10–20 cigarettes every day for at least 3 yr. We selected donors with a reasonable lung function: a / ratio of >250, limited time on a ventilator, a clinical history, and X-ray that did not reveal any infection. Lungs were obtained from emphysema patients (GOLD 4) who underwent lung transplants through Temple Biobank (Temple University, Philadelphia, PA). The study was approved by the Institutional Review Board (IRB) at Partners Healthcare and Temple University and performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from patients.
ATII cells were isolated as we previously described (25). Briefly, after instillation of 12.9 U/mL elastase (Worthington, Lakewood, NJ) the lung was minced followed by centrifugation. The cell suspension was filtrated and enriched by density gradient centrifugation made of Optiprep (Accurate Chemical Scientific, Westbury, NY). ATII cells were purified using Ep-CAM-positive selection (Miltenyi Biotec, Germany). The purity of freshly isolated ATII cells was 90% as we analyzed by staining for prosurfactant protein C (proSP-C) (25, 50). Freshly isolated ATII cells were used for all experiments.
Western blotting and immunoprecipitation.
Cells were lysed, and human and murine lung tissue was homogenized in lysis buffer with protease and phosphatase inhibitor cocktail (Gold Biotechnology, Olivette, MO). The lysates were centrifuged at 14,000 rpm for 20 min to remove cell debris followed by an analysis of protein expression by Western blotting. The following antibodies were used: β-actin (Sigma, St. Louis, MO), active caspase-3 and anti-mouse S100A8 (both from Abcam, Cambridge, MA), anti-human S100A8, and DJ-1 (both from Santa Cruz Biotechnology, Dallas, TX), His tag (Bio-Rad, Hercules, CA), and anti-cysteine with oxidized thiol groups (SOH, SO2H, SO3H; Enzo Life Sciences, Farmingdale, NY). We applied horseradish peroxidase (HRP)-conjugated AffiniPure donkey anti-rabbit IgG or HRP-conjugated AffiniPure donkey anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA). The blots were developed using Luminata Forte Western HRP Substrate (Millipore, Billerica, MA) for chemiluminescent detection of protein expression. To perform immunoprecipitation, lysates were incubated with S100A8, DJ-1, or control IgG antibodies for 18 h followed by adding G Mag Sepharose beads (GE Healthcare, Chicago, IL) for 2 h at 4°C. After washes, the protein complex was eluted in sample buffer and used for Western blotting as described above. ImageJ (NIH, Bethesda, MD) was applied for densitometric analysis of protein expression normalized to β-actin or immunoprecipitated S100A8 or DJ-1 and presented as a ratio to nonsmoker controls.
Detection of S100A8 sulfenylation and sulfinylation.
The levels of S100A8 sulfenylation and sulfinylation were analyzed using DCP-Bio and NO-Bio chemoselective probes (Kerafast, Boston, MA), respectively, per manufacturer’s recommendations. Briefly, S100A8 in ATII cell or lung tissue lysates was immunoprecipitated for 18 h, followed by incubation with protein G Mag Sepharose beads for 2 h. For detection of S100A8 sulfenylation, precipitated proteins were incubated with 2.5 mM DCP-Bio probe for 1 h. Biotin-labeled samples were washed with PBS followed by Western blotting analysis using streptavidin (Cell Signaling Technology, Danvers, MA) and S100A8 antibodies. For detection of S100A8 sulfinylation, free thiols in the precipitated proteins were trapped in the reaction with 2 mM 2,2′-dipyridyl disulfide (DPS) for 30 min. After washing with PBS, protein sulfinylation was assayed by incubation with 250 µM NO-bio sulfinic acid probe for 30 min, and biotin-labeled samples were analyzed by Western blotting using streptavidin and S100A8 antibodies. The levels of S100A8 sulfenylation or sulfination were normalized to S100A8 expression and were presented as a ratio to nonsmoker controls using Image J (NIH).
Gene knockdown and overexpression.
The human alveolar A549 cell line was maintained in DMEM medium (GE Healthcare) supplemented with 10% FBS (GE Healthcare), 125 U/mL penicillin and 125 U/mL streptomycin (GE Healthcare). For gene knockdown, cells were transfected with 60 nM of S100A8-targeting small interfering (si)RNA (Santa Cruz Biotechnology) or 100 nM of DJ-1 siRNA: 5′-GGAGCAGGAAAACCGGAAGtt-3′ and 5′-CUUCCGGUUUUCCUGCUCCtt-3′ (7) obtained from Invitrogen (Carlsbad, CA). We used Lipofectamine RNAiMax reagent (Invitrogen) for cell transfection for 48 h or 24 h to knockdown S100A8 or DJ-1 gene, respectively, before treatment with cigarette smoke extract. The nontargeting (NT) siRNA was used as a control as we reported (23). For transient gene expression, the following plasmids were used: 2.5 µg empty vector pcDNA3.1, wild-type DJ-1-His tag, DJ-1 with a substitution of cysteine at position 106 with alanine (C106A)-His tag or S100A8 (Addgene, Cambridge, MA). A549 cells were transfected with plasmids using polyethylenimine (PEI) 25K (Polysciences, Warrington, PA) (26) at a 3:1 ratio of PEI:DNA for 48 h per manufacturer’s instructions. Knockdown and overexpression of the target genes were confirmed by Western blotting as described above.
Cigarette smoke extract.
Cigarette smoke extract was prepared as we previously described (4). Briefly, the smoke of one 3R4F cigarette (Kentucky Tobacco Research & Development Center, Lexington, KY) without a filter was extracted in 12.5 mL DMEM medium using a peristaltic pump (Mannostat, 72-310-000; Thermo Fisher Scientific, Waltham, MA) and filtered through a 0.22 μm filter. Obtained 100% cigarette smoke extract was diluted in DMEM to a desired concentration and used immediately for cell treatment for the indicated time.
Acridine orange and ethidium bromide double staining.
S100A8 knockdown or overexpression in A549 cells was performed as described above followed by exposure to 20% cigarette smoke extract for 24 h. Double staining using acridine orange and ethidium bromide (both from Sigma) was used as we reported previously (21). Briefly, cells were stained with 100 µg/mL acridine orange and 100 µg/mL ethidium bromide and immediately analyzed using fluorescence microscopy (Zeiss). The following stages were identified: live cells (green nucleus), early apoptotic cells (cell membrane still continuous but chromatin condensation with an irregular green nucleus is visible), late apoptosis (orange nuclei with fragmentation or condensation of chromatin), and necrotic cells (uniform red nuclei).
Flow cytometry analysis.
Alexa Fluor 488 Annexin V/Dead Cell Apoptosis kit (Thermo Fisher Scientific) was used to detect A549 cell apoptosis induced by cigarette smoke extract per manufacturer’s instructions. Briefly, 5 μL Annexin V conjugated to Alexa Fluor 488 and 1 μg/mL propidium iodide (PI) were diluted in binding buffer and cells were stained for 5 min. LSR-II flow cytometer (BD Biosciences, San Jose, CA) and FlowJo (TreeStar, Ashland, OR) were used for data acquisition and analysis.
Mouse exposure to cigarette smoke.
Wild-type C57BL/6 and DJ-1 KO mice were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained at the Animal Facility of Temple University. Animals were age- and sex-matched. We used 4-wk-old mice for all experiments. Mice were exposed to cigarette smoke generated from Kentucky reference cigarette 3R4F for 2 h/day every day for 3 wk using a Teague TE-10 smoking system (Teague Enterprises, Woodland, CA). The total suspended particles (TSP) was 150 mg/m3, and CO concentrations were less than 300 ppm. All experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Temple University.
Real-time PCR.
RT-PCR was performed as we previously described (27). Briefly, RNA was obtained from murine lung tissue using TRIzol reagent (Thermo Fisher Scientific) and then reverse-transcribed into cDNA by using Superscript II Reverse Transcript kit (Thermo Fisher Scientific). PCR amplification was performed using a SYBR Green Master Mix kit (Thermo Fisher Scientific) and StepOnePlus Real-Time PCR System (Applied Biosystems, Foster City, CA). Murine gene-specific primers were retrieved from PrimerBank (https://pga.mgh.harvard.edu/primerbank/) and ordered from Invitrogen. We used the following primers for mouse S100A8: forward 5′-AAATCACCATGCCCTCTACAAG-3′ and reverse 5′-CCCACTTTTATCACCATCGCAA-3′; and mouse GAPDH: forward 5′-CATGGCCTTCCGTGTTCCT-3′ and reverse 5′-CCTGCTTCACCACCTTCTT-3′. Gene expression levels were normalized to GAPDH and presented as a ratio to control. The ΔΔCt method was used for data analysis.
Immunohistofluorescence.
Paraffin-embedded human and murine lung tissue sections were incubated with proSP-C (Millipore), SP-C (Santa Cruz Biotechnology), active caspase-3, S100A8, or DJ-1 antibodies for 18 h. The sections were stained with secondary antibodies Alexa Fluor 594 IgG, Alexa Fluor 488 IgG, or Alexa Fluor 647 IgG (Invitrogen) for 1 h. Fluoroshield mounting medium with DAPI (Abcam) was used to stain nuclei. Images of sections were obtained using a confocal laser-scanning microscope (Zeiss). Protein colocalization (13) was analyzed and quantified using Coloc2 ImageJ (NIH).
Statistical analysis.
Data are expressed as means ± SEM from at least three independent experiments. Statistically significant differences among experimental groups were determined by Student's t-tests or one-way ANOVA. A value of P < 0.05 was considered significant.
RESULTS
ATII cell apoptosis in patients with emphysema.
Emphysema is characterized by high oxidative stress and alveolar wall destruction (49). We examined apoptosis in lung tissue and freshly isolated ATII cells obtained from nonsmokers, smokers, and individuals with this disease. A significantly increased active caspase-3 expression was found in smokers and emphysema patients in both lung tissue and ATII cells compared with nonsmokers (Fig. 1). Our results indicate high ATII cell apoptosis in emphysema.
Fig. 1.
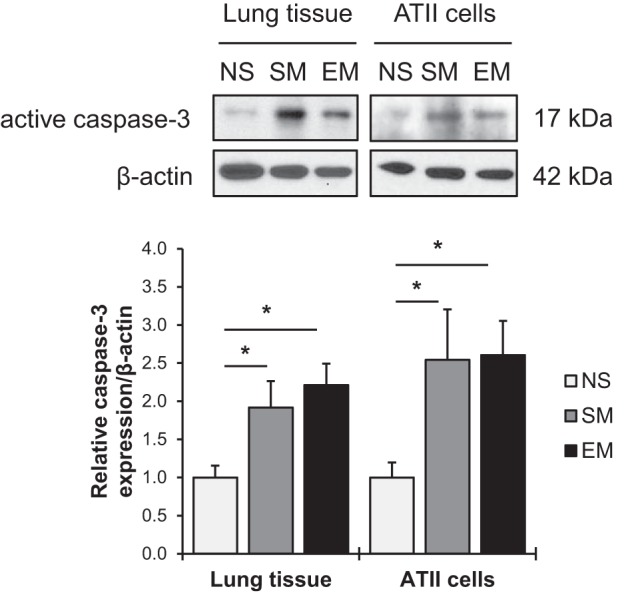
Apoptosis in human lung tissue and alveolar type II (ATII) cells in patients with emphysema. Western blot images of active caspase-3 expression in lung tissue and ATII cells isolated from nonsmokers (NS), smokers (SM), and patients with emphysema (EM). Quantification of active caspase-3 expression was normalized to β-actin and control nonsmokers (n = 4 human lungs per group). Data are expressed as means ± SEM. *P < 0.05.
DJ-1 and S100A8 interaction in human primary ATII cells.
We previously reported that DJ-1 modulates antioxidant defense system (4). The importance of S100A8 in cell homeostasis is supported by observations that the deletion of this gene in mice results in an embryonic lethal phenotype (31). Here, we wanted to determine the functional link between DJ-1 and S100A8 in freshly isolated ATII cells obtained from nonsmokers, smokers, and patients with emphysema. An interaction between DJ-1 and S100A8 was detected in ATII cells using immunoprecipitation followed by Western blotting (Fig. 2A). This interaction was high in smokers and lower in patients with emphysema compared with smokers. Decreased DJ-1 and S100A8 colocalization was also observed in individuals with emphysema in comparison with smokers using immunohistofluorescence (Fig. 2, B and C). Our results suggest a link between emphysema pathophysiology and a decreased DJ-1 and S100A8 expression.
Fig. 2.

DJ-1 and S100A8 interaction in human primary alveolar type II (ATII) cells. A: DJ-1 or control IgG were immunoprecipitated (IP) in freshly isolated ATII cells from nonsmokers (NS), smokers (SM), and patients with emphysema (EM) followed by Western blotting for S100A8 expression and quantification. S100A8 expression was normalized to immunoprecipitated DJ-1 and nonsmokers (n = 3–8 lungs per group). B: human lung sections were costained with prosurfactant protein C (proSP-C; violet), DJ-1 (red), S100A8 (green), and DAPI (blue) and analyzed using a confocal microscopy. C: Pearson’s correlation coefficient was used to determine DJ-1 and S100A8 colocalization in ATII cells in 3 lungs per group. *P < 0.05. Data are expressed as means ± SEM.
Decreased S100A8 sulfination in patients with emphysema.
It has been reported that oxidation of C42 within S100A8 affects protein function in redox signaling (39, 40). Cysteine residues can be oxidized into sulfenic acid (SOH) and their further oxidation leads to sulfinic acid (SO2H). We immunoprecipitated S100A8 in lung tissue and ATII cells followed by Western blotting analysis using an antibody that recognizes cysteine residues with oxidized thiol groups (SOH, SO2H and SO3H). However, we did not observe significant differences between non-smokers, smokers, and emphysema patients in lung tissue (Fig. 3, IA) or ATII cells (Fig. 3, IIA). Therefore, we analyzed S100A8 sulfenation and sulfination using specific chemosensitive probes (28, 32). Our results indicate no significant differences in S100A8 levels with sulfenic acid in lung tissue (Fig. 3, IB) and freshly isolated ATII cells (Fig. 3, IIB) obtained from nonsmokers, smokers, or emphysema patients. However, reduced levels of S100A8 sulfination were observed in lung tissue in individuals with this disease (Fig. 3, IC). Decreased levels of this S100A8 form were detected in ATII cells obtained from emphysema patients (Fig. 3, IIC).
Fig. 3.

S100A8 sulfenylation and sulfinylation in human lung tissue and alveolar type II (ATII) cells. I and II: S100A8 was immunoprecipitated in lung tissue (I) and freshly isolated ATII cells (II) from nonsmokers (NS), smokers (SM), and patients with emphysema (EM) followed by Western blotting. Antibody against oxidized cysteine residues (A) and chemoselective probes to detected sulfenic acids (SOH) (B), and sulfinic acids (SO2H) (C) were used. Densitometric quantifications are also shown. Expression of oxidized cysteine residues, sulfenic, or sulfinic acids within S100A8 was normalized to immunoprecipitated (IP) S100A8 and nonsmokers (n = 4 lungs per group). III: human A549 cell line was exposed to 20% cigarette smoke extract (CSE) for 0, 2, 4, 24, and 48 h. A: active caspase-3 levels were analyzed by Western blotting and normalized to β-actin and control. B: cell lysates were subjected to S100A8 immunoprecipitation followed by detection of S100A8 sulfinylation by Western blotting using chemoselective probes. Quantification of S100A8 sulfinylation was normalized to precipitated S100A8 protein and control. C: correlation analysis of active caspase-3 expression and S100A8 sulfinylation in response to treatment with CSE at 0, 2, 4, 24, and 48 h. N = 3 replicates. Data are expressed as means ± SEM. *P < 0.05, **P < 0.001.
We further provide evidence of an association between S100A8 sulfination and apoptosis by an analysis of active caspase-3 levels in A549 cells treated with cigarette smoke extract. Cell apoptosis was increased in a time-dependent manner (Fig. 3, IIIA). We also observed low S100A8 sulfination after 48 h (Fig. 3, IIIB). In addition, we found that S100A8 sulfination levels negatively correlated with cigarette smoke extract-induced active caspase-3 expression (Fig. 3, IIIC). Our results suggest that decreased S100A8 sulfination may affect cytoprotective protein function and contribute to cell apoptosis and emphysema pathogenesis.
S100A8 modulates cell response to cigarette smoke extract.
To further explore the relationship between S100A8 and DJ-1, we analyzed the effect of S100A8 loss-of-function in A549 cells using siRNA strategy. Apoptosis was found in cells with S100A8 knockdown using acridine orange and ethidium bromide double staining (Fig. 4, IA). S100A8 deficiency increased the percentage of apoptotic cells after treatment with cigarette smoke extract compared with controls. In addition, S100A8 knockdown sensitized cells to apoptosis as shown by increased active caspase-3 levels (Fig. 4, IB). We found that this treatment induced expression of cytoprotective DJ-1 protein. DJ-1 protein levels were decreased in cells with S100A8 knockdown followed by this treatment.
Fig. 4.

S100A8 regulates cell response to cigarette smoke extract. I: A549 cells were transfected with nontargeting (NT) siRNA or S100A8 siRNA followed by exposure to 20% cigarette smoke extract (CSE) for 24 h. A: acridine orange and ethidium bromide double staining was applied to distinguish between early apoptotic (chromatin fragmentation or condensation), late apoptotic (yellow), and necrotic (red) cells using a fluorescent microscope. B: active caspase-3, DJ-1, and S100A8 expression was analyzed by Western blotting. Densitometric quantifications are also shown. II: A549 cells were transfected with empty vector or pcDNA3.1 S100A8 plasmid followed by exposure to 20% CSE for 24 h. A: Representative images of acridine orange and ethidium bromide double staining. B: Active caspase-3, DJ-1 and S100A8 expression was analyzed by Western blotting. Densitometric quantifications are also shown. *P < 0.05, **P < 0.001, #P < 0.05 compared with controls and CSE; n = 3 replicates. Data are expressed as means ± SEM.
Fig. 4.—Continued.

On the other hand, S100A8 overexpression followed by exposure to cigarette smoke extract reduced cell apoptosis in comparison with cigarette smoke extract alone as detected by acridine orange and ethidium bromide double staining (Fig. 4, IIA). Lowered active caspase-3 expression and DJ-1 levels were also observed (Fig. 4, IIB). Our results indicate the cytoprotective function of S100A8.
S100A8 compensates the absence of DJ-1.
It has been shown that DJ-1 knockdown sensitized cells to death induced by hydrogen peroxide, whereas its overexpression and activation protected cells against oxidative stress-induced injury (37). We wanted to further determine the relationship between DJ-1 and S100A8 in A549 cells. Cell transfection with wild-type DJ-1-His tag plasmid significantly increased DJ-1 expression and decreased S100A8 levels compared with empty vector (Fig. 5A). However, we did not observe significant differences in DJ-1 and S100A8 levels in cells transfected with DJ-1 C106 mutant construct (C106A). Annexin V and PI double staining was applied to further evaluate DJ-1 function in cells treated with cigarette smoke extract. DJ-1 overexpression reduced cell apoptosis induced by this exposure. Increased apoptosis was found in A549 cells transfected with DJ-1 C106A followed by treatment with cigarette smoke extract compared with cells transfected with wild type DJ-1 (Fig. 5B). This indicates the critical role of C106 within DJ-1 in the cytoprotective function of this protein. In addition, we overexpressed DJ-1 wild-type or C106A mutant constructs in A549 cells followed by exposure to cigarette smoke extract to determine DJ-1 and S100A8 interaction. We detected that cigarette smoke significantly decreased the interaction between DJ-1 C106A and S100A8 (Fig. 5C).
Fig. 5.

S100A8 overexpression reduced apoptosis induced by cigarette smoke extract in DJ-1-deficient cells. A: A549 cells were transfected with empty vector, pcDNA3.1-DJ-1 wild-type (WT), or C106A mutant construct for 48 h. His-tag, DJ-1, and S100A8 expression was determined by Western blotting. Quantification of protein levels was normalized to β-actin and control. B: cells were transfected with empty vector, DJ-1 WT, or DJ-1 C106A mutant constructs followed by treatment with 20% cigarette smoke extract (CSE) for 24 h. Apoptosis was evaluated by Annexin V and propidium iodide (PI) staining. Representative images of flow cytometric analysis and quantification of apoptotic cells are shown. C: cells were transfected with empty vector, DJ-1 WT, or DJ-1 C106A mutant construct followed by treatment with 20% CSE for 24 h. His-tag or control IgG was immunoprecipitated (IP) followed by Western blotting to analyze S100A8 expression. Protein expression was normalized to His and DJ-1 WT. CTL, control. D: A549 cells were transfected with nontargeting (NT) or DJ-1 siRNA for 24 h. DJ-1 and S100A8 expression were determined by Western blotting. Quantification of DJ-1 and S100A8 levels is shown. E: A549 cells with DJ-1 knockdown were transfected with a pcDNA3.1-S100A8 plasmid for 24 h followed by treatment with 20% CSE for 24 h. Representative images of flow cytometric analysis using Annexin V and PI double staining and quantification of apoptotic A549 cells are shown. Data are expressed as means ± SEM; n = 3 replicates. *P < 0.05, **P < 0.001, #P < 0.05 compared with control.
Fig. 5.—Continued.

To complement this approach, we knocked down DJ-1 in A549 cells using siRNA strategy. Our results indicate that DJ-1 deficiency significantly increased S100A8 expression (Fig. 5D). In addition, DJ-1 knockdown sensitized cells to apoptosis induced by cigarette smoke extract as shown by Annexin V and PI double staining (Fig. 5E). S100A8 in A549 cells was overexpressed using pcDNA3.1-S100A8 plasmid. We found that S100A8 overexpression compensated the lack of DJ-1 and decreased cell apoptosis induced by cigarette smoke extract. Together, our findings indicate the cytoprotective function of S100A8 and the functional relationship between DJ-1 and S100A8 under oxidative stress conditions.
Cigarette smoke increases S100A8 expression in DJ-1 KO mice.
We used wild-type and DJ-1-KO mice to further determine the association between S100A8 and DJ-1. Mice were exposed to cigarette smoke followed by an analysis of active caspase-3 expression in ATII cells by immunofluorescence (Fig. 6A). ATII cell apoptosis was found in both wild-type and DJ-1 KO mice exposed to cigarette smoke (Fig. 6B). Moreover, DJ-1-deficiency increased the percentage of apoptotic ATII cells compared with wild-type mice. Interestingly, cigarette smoke significantly increased S100A8 expression in lung tissue obtained from DJ-1 KO mice in comparison with wild-type mice as detected by Western blotting (Fig. 6C). This exposure also upregulated S100A8 mRNA levels in lung tissue in wild-type and DJ-1 KO mice (Fig. 6D). We further analyzed S100A8 expression in murine ATII cells in wild-type and DJ-1 KO mice by immunohistofluorescence. Higher S100A8 levels were detected in DJ-1 KO mice compared with wild-type mice (Fig. 6E). Furthermore, we found that cigarette smoke increased colocalization of DJ-1 and S100A8 in wild-type mice (Fig. 6F). Our results suggest that DJ-1 deficiency sensitized mice to lung injury induced by cigarette smoke.
Fig. 6.

Apoptosis in DJ-1 knockout (KO) mice exposed to cigarette smoke (CS). Wild-type (WT) and DJ-1 KO mice were exposed to 150 mg/m3 total suspended particles for 2 h/day every day for 3 wk. A: active caspase-3 expression was analyzed in WT and DJ-1 KO mice by immunohistofluorescence. Alveolar type II (ATII) cells were identified using an SP-C antibody. B: percentage of active caspase-3-positive ATII cells is shown. C: Western blot images of S100A8 and DJ-1 expression in lung tissue obtained from WT and DJ-1 KO mice (CTL, control). Densitometric quantification is also shown. S100A8 expression was normalized to β-actin and control WT mice. D: S100A8 mRNA levels in murine lung tissue analyzed by RT-PCR (n = 4 per group). E: S100A8 expression in ATII cells, identified using a prosurfactant protein C (proSP-C) antibody, in WT and DJ-1 KO mice exposed to cigarette smoke by immunohistofluorescence. Relative fluorescence intensity of S100A8 in ATII cells is also shown. F: Pearson’s correlation coefficient was used for DJ-1 (red) and S100A8 (green) colocalization in ATII cells in WT mice (n = 3 per group). *P < 0.05, **P < 0.001. Data are expressed as means ± SEM.
Fig. 6.—Continued.

DISCUSSION
Oxidative stress can cause alveolar epithelial cell and lung injury (15, 24, 44). Oxidant molecules are present in cigarette smoke, which contains free radicals and reactive oxygen species (ROS) and contributes to emphysema pathogenesis (8). In addition, chronic inflammation that persists even after smoking cessation is also a source of oxidative stress (47). This suggests that antioxidant defense systems in the lungs of subjects with this disease can be overwhelmed and contribute to prolonged oxidative cellular damage (30). Emphysema is associated with decreased alveolar surface area (30) and low ATII cell proliferation and ATII cell differentiation to alveolar type I cells (38), suggesting the impaired mechanisms of lung regeneration. In this study, we isolated ATII cells from patients with cigarette smoke-induced emphysema to determine the cytoprotective function of DJ-1 and S100A8 proteins.
We observed high active caspase-3 expression in freshly isolated ATII cells from individuals with emphysema. Decreased S100A8 and DJ-1 expression and interaction were found, as well as the impairment of their cytoprotective activities, which can contribute to cell death and this disease pathogenesis. S100A8 is a scavenger of ROS, which indicates its important role in the antioxidant defense system and anti-inflammatory processes (14). It has been shown that intranasal administration of S100A8 suppressed allergic inflammation in mice by reducing key cytokines affecting eosinophil migration and mucus production (51).
Oxidation of proteins involves the conversion of cysteine residues to sulfenic, sulfinic, or sulfonic acids. These posttranslational modifications can modulate protein functions in cells (11). Cysteine residues modified to sulfenic acids (SOH) are inherently reactive moieties and often intermediates to sulfinic acids (SO2H) or irreversible sulfonic acids (SO3H) (28, 33). However, the functions of oxidized proteins are not well known, owing to major challenges of their detection (1). S100A8 deletion is embryonic lethal, which indicates its critical role in cell homeostasis and development (6). It has been suggested that S100A8 oxidation can regulate anti-inflammatory processes (14). Here, we used recently developed chemoselective specific probes to detect oxidation of cysteine residue(s) within S100A8 to sulfenic or sulfinic acids (28, 32). No changes were detected in S100A8 levels with sulfenic acids in lung tissue or freshly isolated ATII cells from nonsmokers, smokers, and emphysema patients. Interestingly, we found decreased expression of S100A8 with cysteine residue(s) modified to sulfinic acid in individuals with emphysema compared with control nonsmokers. At the molecular level, S100A8 is a two-electron oxidant scavenger, and oxidation of a single, highly reactive C42 can affect its function (16). Oxidation of C42 within S100A8 has been detected by mass spectrometry analysis in sputum obtained from patients with asthma and HOCl-oxidized recombinant S100A8 (14). In our study, a correlation was found between decreased sulfination of cysteine residue(s) within S100A8 protein and increased cigarette smoke extract-induced cell apoptosis. This indicates cytoprotective function of S100A8. Our observations suggest the involvement of cysteine residue(s) within S100A8 protein in response to redox status. Further studies are required to determine the biological function of oxidized S100A8.
DJ-1 is an atypical peroxiredoxin-like peroxidase (2). Oxidation of cysteine at position 106 within this protein, which is a redox-sensitive residue, to sulfinic acid is required for DJ-1 antioxidant activity (9, 34). We previously showed that DJ-1 modulates the defense system in ATII cells under oxidative stress conditions (4). In addition, DJ-1 overexpression using adenovirus strategy restored the antioxidant pathway in primary ATII cells obtained from heavy smokers and decreased apoptosis induced by exposure to cigarette smoke extract. It was also shown that DJ-1 regulates the expression of genes encoding cytoprotective proteins (37, 48). Here, we found increased apoptosis after cell transfection with DJ-1 C106A mutant construct followed by treatment with cigarette smoke extract compared with wild type DJ-1 plasmid. Our results are in agreement with a previous study showing decreased antioxidant and antiapoptotic activity of DJ-1 with C106A in cells treated with hydrogen peroxide (34).
It has been reported that DJ-1 interaction with other molecules affects glutathione metabolism, heat shock and uncoupling proteins, proteasome pathway, and signal transduction (10, 35). Here, we provide a new insight into the functional relationship between DJ-1 and S100A8 proteins and their cytoprotective functions against ATII cell death and emphysema development. We detected an interaction between DJ-1 and S100A8 proteins by immunoprecipitation and their compensatory effects in vitro. DJ-1 overexpression decreased S100A8 levels in A549 cells. We also found that DJ-1 knockdown led to increased S100A8 expression in cells. Cigarette smoke extract induced apoptosis in DJ-1-deficient A549 cells. S100A8 overexpression decreased cell apoptosis induced by this treatment in the absence of DJ-1. Recent findings also indicate that S100A8 suppressed the expression of proinflammatory molecules induced by lipopolysaccharide in the murine lung (16).
Wild-type and DJ-1 KO mice were used to further determine the role of S100A8. First, we found that DJ-1 KO mice are more susceptible to ATII cell apoptosis induced by cigarette smoke compared with wild-type mice. Moreover, this exposure induced higher S100A8 expression in DJ-1 KO mice than wild-type mice, which suggests the compensatory function of S100A8 in the absence of DJ-1. An increased colocalization of DJ-1 and S100A8 was found in ATII cells in smokers and wild type mice exposed to cigarette smoke. Expression and interaction of both proteins were decreased in patients with emphysema compared with smokers. Of note, mice were exposed to cigarette smoke for 2 h per day for 3 wk. Human lung tissue was obtained from smoker organ donors who smoked 10–20 cigarettes per day for at least 3 yr, which can explain variability in response. Further studies are required to determine whether the association between DJ-1 and S100A8 is dependent on the cellular and biological context.
In conclusion, our findings indicate the functional relationship between DJ-1 and S100A8 proteins ex vivo, in vitro, and in vivo. We also observed decreased expression of both DJ-1 and S100A8 proteins in ATII cells in emphysema compared with control smokers, which suggests their decreased cytoprotective activity. Targeting DJ-1 and S100A8 by small molecules may restore their function and protect against this disease development.
GRANTS
This work was supported by National Institutes of Health National Heart, Lung, and Blood Institute Grant R01 HL118171 (B. Kosmider), Grant-in-Aid-Award (Temple University) to B. Kosmider, and the Catalyst Award from the American Lung Association (K. Bahmed).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.-R.L., K.B., and B.K. conceived and designed research; C.-R.L., K.B., D.T., M.A.W., and B.K. performed experiments; C.-R.L., K.B., D.T., and B.K. analyzed data; C.-R.L., K.B., M.A.W., and B.K. interpreted results of experiments; C.-R.L. and D.T. prepared figures; C.-R.L. drafted manuscript; C.-R.L., K.B., and B.K. edited and revised manuscript; C.-R.L., K.B., D.T., N.M., G.J.C., S.B., M.A.W., M.M., and B.K. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank the Lung Center Tissue Bank at Temple University for providing the human lung specimens.
REFERENCES
- 1.Akter S, Fu L, Jung Y, Conte ML, Lawson JR, Lowther WT, Sun R, Liu K, Yang J, Carroll KS. Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat Chem Biol 14: 995–1004, 2018. doi: 10.1038/s41589-018-0116-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci USA 104: 14807–14812, 2007. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahmed K, Boukhenouna S, Karim L, Andrews T, Lin J, Powers R, Wilson MA, Lin CR, Messier E, Reisdorph N, Powell RL, Tang HY, Mason RJ, Criner GJ, Kosmider B. The effect of cysteine oxidation on DJ-1 cytoprotective function in human alveolar type II cells. Cell Death Dis 10: 638, 2019. doi: 10.1038/s41419-019-1833-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bahmed K, Messier EM, Zhou W, Tuder RM, Freed CR, Chu HW, Kelsen SG, Bowler RP, Mason RJ, Kosmider B. DJ-1 modulates nuclear erythroid 2-related factor-2-mediated protection in human primary alveolar type II cells in smokers. Am J Respir Cell Mol Biol 55: 439–449, 2016. doi: 10.1165/rcmb.2015-0304OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai J, Guo C, Sun W, Li M, Meng X, Yu Y, Jin Y, Tong D, Geng J, Huang Q, Qi J, Fu S. DJ-1 may contribute to metastasis of non-small cell lung cancer. Mol Biol Rep 39: 2697–2703, 2012. doi: 10.1007/s11033-011-1024-7. [DOI] [PubMed] [Google Scholar]
- 6.Baker JR, Jeffery R, May RD, Mathies M, Spencer-Dene B, Poulsom R, Hogg N. Distinct roles for S100a8 in early embryo development and in the maternal deciduum. Dev Dyn 240: 2194–2203, 2011. doi: 10.1002/dvdy.22709. [DOI] [PubMed] [Google Scholar]
- 7.Batelli S, Albani D, Rametta R, Polito L, Prato F, Pesaresi M, Negro A, Forloni G. DJ-1 modulates alpha-synuclein aggregation state in a cellular model of oxidative stress: relevance for Parkinson’s disease and involvement of HSP70. PLoS One 3: e1884, 2008. doi: 10.1371/journal.pone.0001884. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Boukhenouna S, Wilson MA, Bahmed K, Kosmider B. Reactive oxygen species in chronic obstructive pulmonary disease. Oxid Med Cell Longev 2018: 5730395, 2018. doi: 10.1155/2018/5730395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canet-Avilés RM, Wilson MA, Miller DW, Ahmad R, McLendon C, Bandyopadhyay S, Baptista MJ, Ringe D, Petsko GA, Cookson MR. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci USA 101: 9103–9108, 2004. doi: 10.1073/pnas.0402959101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan JY, Chan SH. Activation of endogenous antioxidants as a common therapeutic strategy against cancer, neurodegeneration and cardiovascular diseases: a lesson learnt from DJ-1. Pharmacol Ther 156: 69–74, 2015. doi: 10.1016/j.pharmthera.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Chang YC, Huang CN, Lin CH, Chang HC, Wu CC. Mapping protein cysteine sulfonic acid modifications with specific enrichment and mass spectrometry: an integrated approach to explore the cysteine oxidation. Proteomics 10: 2961–2971, 2010. doi: 10.1002/pmic.200900850. [DOI] [PubMed] [Google Scholar]
- 12.Clements CM, McNally RS, Conti BJ, Mak TW, Ting JP. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc Natl Acad Sci USA 103: 15091–15096, 2006. doi: 10.1073/pnas.0607260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300: C723–C742, 2011. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gomes LH, Raftery MJ, Yan WX, Goyette JD, Thomas PS, Geczy CL. S100A8 and S100A9-oxidant scavengers in inflammation. Free Radic Biol Med 58: 170–186, 2013. doi: 10.1016/j.freeradbiomed.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 15.Hecker L. Mechanisms and consequences of oxidative stress in lung disease: therapeutic implications for an aging populace. Am J Physiol Lung Cell Mol Physiol 314: L642–L653, 2018. doi: 10.1152/ajplung.00275.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hiroshima Y, Hsu K, Tedla N, Chung YM, Chow S, Herbert C, Geczy CL. S100A8 induces IL-10 and protects against acute lung injury. J Immunol 192: 2800–2811, 2014. doi: 10.4049/jimmunol.1302556. [DOI] [PubMed] [Google Scholar]
- 17.Hiroshima Y, Hsu K, Tedla N, Wong SW, Chow S, Kawaguchi N, Geczy CL. S100A8/A9 and S100A9 reduce acute lung injury. Immunol Cell Biol 95: 461–472, 2017. doi: 10.1038/icb.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med 47: 1354–1361, 2009. doi: 10.1016/j.freeradbiomed.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 19.Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, DeLuca C, Liepa J, Zhou L, Snow B, Binari RC, Manoukian AS, Bray MR, Liu FF, Tsao MS, Mak TW. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell 7: 263–273, 2005. doi: 10.1016/j.ccr.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 20.Kim V, Criner GJ. Chronic bronchitis and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 187: 228–237, 2013. doi: 10.1164/rccm.201210-1843CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kosmider B, Loader JE, Murphy RC, Mason RJ. Apoptosis induced by ozone and oxysterols in human alveolar epithelial cells. Free Radic Biol Med 48: 1513–1524, 2010. doi: 10.1016/j.freeradbiomed.2010.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kosmider B, Messier EM, Chu HW, Mason RJ. Human alveolar epithelial cell injury induced by cigarette smoke. PLoS One 6: e26059, 2011. doi: 10.1371/journal.pone.0026059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kosmider B, Wells RD. Fragile X repeats are potent inducers of complex, multiple site rearrangements in flanking sequences in Escherichia coli. DNA Repair (Amst) 6: 1850–1863, 2007. doi: 10.1016/j.dnarep.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Kosmider B, Mason RJ, Bahmed K. Isolation and characterization of human alveolar type II cells. Methods Mol Biol 1809: 83–90, 2018. doi: 10.1007/978-1-4939-8570-8_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SH, Hadipour-Lakmehsari S, Miyake T, Gramolini AO. Three-dimensional imaging reveals endo(sarco)plasmic reticulum-containing invaginations within the nucleoplasm of muscle. Am J Physiol Cell Physiol 314: C257–C267, 2018. doi: 10.1152/ajpcell.00141.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin CR, Bahmed K, Criner GJ, Marchetti N, Tuder RM, Kelsen S, Bolla S, Mandapati C, Kosmider B. S100A8 protects human primary alveolar type II cells against injury and emphysema. Am J Respir Cell Mol Biol 60: 299–307, 2019. doi: 10.1165/rcmb.2018-0144OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo Conte M, Lin J, Wilson MA, Carroll KS. A chemical approach for the detection of protein Sulfinylation. ACS Chem Biol 10: 1825–1830, 2015. doi: 10.1021/acschembio.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mason RJ. Biology of alveolar type II cells. Respirology 11, Suppl: S12–S15, 2006. doi: 10.1111/j.1440-1843.2006.00800.x. [DOI] [PubMed] [Google Scholar]
- 30.Morissette MC, Parent J, Milot J. Alveolar epithelial and endothelial cell apoptosis in emphysema: what we know and what we need to know. Int J Chron Obstruct Pulmon Dis 4: 19–31, 2009. [PMC free article] [PubMed] [Google Scholar]
- 31.Passey RJ, Williams E, Lichanska AM, Wells C, Hu S, Geczy CL, Little MH, Hume DA. A null mutation in the inflammation-associated S100 protein S100A8 causes early resorption of the mouse embryo. J Immunol 163: 2209–2216, 1999. [PubMed] [Google Scholar]
- 32.Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat Chem Biol 8: 57–64, 2011. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raftery MJ, Yang Z, Valenzuela SM, Geczy CL. Novel intra- and inter-molecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J Biol Chem 276: 33393–33401, 2001. doi: 10.1074/jbc.M101566200. [DOI] [PubMed] [Google Scholar]
- 34.Ren H, Fu K, Wang D, Mu C, Wang G. Oxidized DJ-1 interacts with the mitochondrial protein BCL-XL. J Biol Chem 286: 35308–35317, 2011. doi: 10.1074/jbc.M110.207134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saito Y, Akazawa-Ogawa Y, Matsumura A, Saigoh K, Itoh S, Sutou K, Kobayashi M, Mita Y, Shichiri M, Hisahara S, Hara Y, Fujimura H, Takamatsu H, Hagihara Y, Yoshida Y, Hamakubo T, Kusunoki S, Shimohama S, Noguchi N. Oxidation and interaction of DJ-1 with 20S proteasome in the erythrocytes of early stage Parkinson’s disease patients. Sci Rep 6: 30793, 2016. doi: 10.1038/srep30793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm 2013: 828354, 2013. doi: 10.1155/2013/828354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sekito A, Koide-Yoshida S, Niki T, Taira T, Iguchi-Ariga SM, Ariga H. DJ-1 interacts with HIPK1 and affects H2O2-induced cell death. Free Radic Res 40: 155–165, 2006. doi: 10.1080/10715760500456847. [DOI] [PubMed] [Google Scholar]
- 38.Skronska-Wasek W, Mutze K, Baarsma HA, Bracke KR, Alsafadi HN, Lehmann M, Costa R, Stornaiuolo M, Novellino E, Brusselle GG, Wagner DE, Yildirim AO, Konigshoff M. Reduced frizzled receptor 4 expression prevents WNT/β-catenin–driven alveolar lung repair in chronic obstructive pulmonary disease. Am J Respir Crit Care Med, 2017. doi: 10.1164/rccm.201605-0904OC. [DOI] [PubMed] [Google Scholar]
- 39.Sroussi HY, Berline J, Dazin P, Green P, Palefsky JM. S100A8 triggers oxidation-sensitive repulsion of neutrophils. J Dent Res 85: 829–833, 2006. doi: 10.1177/154405910608500910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sroussi HY, Köhler GA, Agabian N, Villines D, Palefsky JM. Substitution of methionine 63 or 83 in S100A9 and cysteine 42 in S100A8 abrogate the antifungal activities of S100A8/A9: potential role for oxidative regulation. FEMS Immunol Med Microbiol 55: 55–61, 2009. doi: 10.1111/j.1574-695X.2008.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taira T, Saito Y, Niki T, Iguchi-Ariga SM, Takahashi K, Ariga H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep 5: 213–218, 2004. doi: 10.1038/sj.embor.7400074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan LH, Bahmed K, Lin CR, Marchetti N, Bolla S, Criner GJ, Kelsen S, Madesh M, Kosmider B. The cytoprotective role of DJ-1 and p45 NFE2 against human primary alveolar type II cell injury and emphysema. Sci Rep 8: 3555, 2018. doi: 10.1038/s41598-018-21790-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsuboi Y, Munemoto H, Ishikawa S, Matsumoto K, Iguchi-Ariga SM, Ariga H. DJ-1, a causative gene product of a familial form of Parkinson’s disease, is secreted through microdomains. FEBS Lett 582: 2643–2649, 2008. doi: 10.1016/j.febslet.2008.06.043. [DOI] [PubMed] [Google Scholar]
- 44.Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest 122: 2749–2755, 2012. doi: 10.1172/JCI60324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tuder RM, Yoshida T, Arap W, Pasqualini R, Petrache I. State of the art. Cellular and molecular mechanisms of alveolar destruction in emphysema: an evolutionary perspective. Proc Am Thorac Soc 3: 503–510, 2006. doi: 10.1513/pats.200603-054MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waak J, Weber SS, Görner K, Schall C, Ichijo H, Stehle T, Kahle PJ. Oxidizable residues mediating protein stability and cytoprotective interaction of DJ-1 with apoptosis signal-regulating kinase 1. J Biol Chem 284: 14245–14257, 2009. doi: 10.1074/jbc.M806902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willemse BW, Postma DS, Timens W, ten Hacken NH. The impact of smoking cessation on respiratory symptoms, lung function, airway hyperresponsiveness and inflammation. Eur Respir J 23: 464–476, 2004. doi: 10.1183/09031936.04.00012704. [DOI] [PubMed] [Google Scholar]
- 48.Xu J, Zhong N, Wang H, Elias JE, Kim CY, Woldman I, Pifl C, Gygi SP, Geula C, Yankner BA. The Parkinson’s disease-associated DJ-1 protein is a transcriptional co-activator that protects against neuronal apoptosis. Hum Mol Genet 14: 1231–1241, 2005. doi: 10.1093/hmg/ddi134. [DOI] [PubMed] [Google Scholar]
- 49.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev 87: 1047–1082, 2007. doi: 10.1152/physrev.00048.2006. [DOI] [PubMed] [Google Scholar]
- 50.Zemski Berry KA, Murphy RC, Kosmider B, Mason RJ. Lipidomic characterization and localization of phospholipids in the human lung. J Lipid Res 58: 926–933, 2017. doi: 10.1194/jlr.M074955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao J, Endoh I, Hsu K, Tedla N, Endoh Y, Geczy CL. S100A8 modulates mast cell function and suppresses eosinophil migration in acute asthma. Antioxid Redox Signal 14: 1589–1600, 2011. doi: 10.1089/ars.2010.3583. [DOI] [PubMed] [Google Scholar]