

Abstract
Maternal cigarette smoking is a major perinatal insult that contributes to an increased risk of cardiovascular and neurodevelopmental diseases in offspring. Our previous studies revealed that perinatal nicotine exposure reprograms a sensitive phenotype in neonatal hypoxic-ischemic encephalopathy (HIE), yet the underlying molecular mechanisms remain largely elusive. The present study tested the hypothesis that perinatal nicotine exposure impacts autophagy signaling in the developing brain, resulting in enhanced susceptibility to neonatal HIE. Nicotine was administered to pregnant rats via subcutaneous osmotic minipumps. Neonatal HIE was conducted in 9-day-old male rat pups. Protein kinase B/glycogen synthase kinase-3β/mammalian target of rapamycin (Akt/GSK-3β/mTOR) signaling and key autophagy markers were determined by Western blotting analysis. Rapamycin and MK2206 were administered via intracerebroventricular injection. Nicotine exposure significantly inhibited autophagy activities in neonatal brain tissues, characterized by an increased ratio of phosphoylated (p-) to total mTOR protein expression but reduced levels of autophagy-related 5, Beclin 1, and LC3βI/II. Treatment with mTOR inhibitor rapamycin effectively blocked nicotine-mediated autophagy deficiency and, more importantly, reversed the nicotine-induced increase in HI brain infarction. In addition, nicotine exposure significantly upregulated p-Akt and p-GSK-3β. Treatment with the Akt selective inhibitor MK2206 reversed the enhanced p-Akt and p-GSK-3β, restored basal autophagic flux, and abolished nicotine-mediated HI brain injury. These findings suggest that perinatal nicotine-mediated alteration of Akt/GSK-3β/mTOR signaling plays a key role in downregulation of autophagic flux, which contributes to the development of hypoxia/ischemia-sensitive phenotype in the neonatal brain.
Keywords: Akt, autophagy, mTOR, neonatal HIE, perinatal nicotine
INTRODUCTION
Maternal smoking is closely correlated with increased risks of cardiovascular and neurological diseases (3, 4, 23, 33, 44, 47). Neonatal hypoxic-ischemic encephalopathy (HIE) is a catastrophic neurological disease, with incidence of one to six per 1,000 term newborns (10, 41). Despite continued advances in perinatal medicine, neonatal HIE is still one of the leading causes of acute brain damage, associated with a much higher mortality and morbidity, along with severe long-lasting neuropsychiatric deficits, including cerebral palsy, seizure, and cognitive retardation in infants and children (10, 41). Currently, universally accepted interventions for HIE are still lacking, which may be largely attributable to the poor understanding of its pathogenesis (1, 20). Our previous studies revealed that perinatal nicotine exposure induces development of a hypoxia/ischemia (HI)-sensitive phenotype of neonatal male brain in a sex-dependent manner. However, the underlying mechanisms are still not fully elucidated (26, 27, 42). Thus, it is warranted to further investigate its underlying molecular mechanisms and explore potential molecular therapeutic targets for the prevention and effective treatment of neonatal HIE.
Autophagy is an evolutionarily ancient mechanism responsible for the lysosomal degradation of old, supernumerary or ectopic cytoplasmic entities and ensures the maintenance of homeostasis in response to stress (12–14, 21). Aberrant autophagy is well documented to be implicated in diverse human pathological processes, which are highly context specific (12–14, 21). Mild to moderate adaptive autophagic responses may confer neuroprotective effects in several forms of acute brain damage, including methamphetamine intoxication, spinal cord injury, and subarachnoid hemorrhage (12, 22). However, defective autophagic responses contribute to various human pathologies, including cardiovascular and renal diseases as well as acute and chronic neurological disorders (8, 9, 12, 30). Nowadays, more and more evidence also begins to reveal the disturbance of autophagy flux in the pathogenesis of ischemic brain injury (2, 12, 15, 29, 37).
In the present study, we first examined the effect of perinatal nicotine exposure on autophagy-associated signaling pathways in the developing brain. Then, we employed a HIE neonatal model to investigate the cause and effect of autophagy signaling in perinatal nicotine exposure-mediated development of a HI-sensitive phenotype of neonatal brain. In this study, we present new evidence of the adverse programming effects of maternal nicotine exposure on fetal and neonatal brain development and suggest that autophagy defect may be one of the important molecular mechanisms linking maternal cigarette smoking/nicotine exposure-induced heightened HIE vulnerability in newborns, which may serve as a novel promising therapeutic target for the intervention of neonatal HIE.
MATERIALS AND METHODS
Experimental animals.
Pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Portage, MI) and were randomly divided into two groups: 1) saline control and 2) nicotine administration through osmotic minipumps implanted subcutaneously from day 4 of gestation to day 10 after birth (26, 27, 42). All pregnant rats were allowed to give birth. Given that our previous studies had demonstrated that perinatal nicotine exposure enhances brain vulnerability to hypoxia/ischemia (HI) injury in a sex-dependent manner, exerting significant effects in neonatal male pups but no marked effects in female pups (26, 42), all of the experiments in this study were conducted in 9-day-old male neonatal (P9) pups. All procedures and protocols were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Nicotine pump implantation.
As described previously (45), on the fourth day of pregnancy, rats were anesthetized with isoflurane, and an incision was made on the back to insert osmotic minipumps (type 2ML4; Alza, Palo Alto, CA). The incision was closed with four sutures. About one-half of the pregnant rats were implanted with minipumps containing nicotine at a concentration of 102 mg/mL, and the other half were implanted with minipumps containing only saline, which served as the vehicle control. The flow rate of the minipumps was 60 μL/day, which delivered a dose of 2.1 mg of nicotine free-base per day. In rats of an average of 350 g body wt, this corresponds to a dose rate of 6 mg·kg−1·day−1. Based on previous studies in a similar pregnant rat model (11, 39), this dosage produces an average maternal plasma nicotine concentration of ~35 ng/mL, which closely resembles the doses occurring in moderate to heavy human smokers (39). According to the manufacturer’s specifications, the delivery period for the pumps is 28 days; thus, delivery continued after birth until P10. Nicotine treatment did not affect the litter size or the length of gestation, and all of the pregnancies reached full term (23, 45).
HIE induction.
A modified Rice-Vannucci model was conducted on P9 pups as described previously (40). The rationale for HIE induction on P9 is that the rat brain at this stage is more susceptible to hypoxic-ischemic insult (40). Briefly, the offspring at P9 were anesthetized with isoflurane (4–5% for induction, 2% for maintenance). A small midline neck incision was made on the anterior neck. The right common carotid artery was gently isolated from surrounding structures, including the vagus nerve, trachea, and esophagus, double ligated, and cut. After recovery for 1 h on a heating pad, the pups were exposed to 8% O2 for 2 h. Inhalation of 8% oxygen-balance nitrogen is the most common concentration used to produce systemic hypoxia in the rat pups. The rationale for using 8% O2 was that the rat pups are capable of surviving this severity of hypoxia, which allowed us to measure the pathophysiological variables in the rat pups. Pups were returned to their dams for recovery after the hypoxic treatment.
Intracerebroventricular injection.
The mTOR inhibitor rapamycin (1 pmol, R0395, Sigma) and the selective Akt inhibitor, MK2206 (5 μg, S1078; Selleck Chemicals, Houston, TX) were administered intracerebroventricularly on P8, 24 h before the HI treatment. The rationale for intracerebroventricular injection is that it can confer effects mainly in local brain tissues and minimize the confounding systemic effects, which could provide more direct effects of the injected drugs or chemicals on brain function. The intracerebroventricular dosages of these two compounds were based on previous studies in rats and a slight adjustment was made for neonatal rat pups (37, 50). As previously described (26, 27, 42), pups were anesthetized (4–5% for induction and 1.5–2% for maintenance) and fixed on a stereotaxic apparatus (Stoelting, Wood Dale, IL). An incision was made on the skull surface, and bregma was exposed. A small burr hole (0.5 mm diameter; Fine Science Tools) was drilled into the right skull relative to bregma: 1.5 mm lateral, 2.0 mm posterior. All agents were prepared in solvents (ethanol for rapamycin and sterile saline for MK2206) and injected into the right lateral ventricle at a rate of 1 µL/min, with a total volume of 2 µl, via a 10-µL syringe (Stoelting) following the coordinates relative to bregma: 2.0 mm posterior, 1.5 mm lateral, and 3.0 mm below the skull surface (17). The vehicle control was injected following the same procedures. The injection lasted 2 min, and the needle was kept for an additional 5 min and then slowly withdrawn over 5 min to prevent backflow. The burr hole was sealed with bone wax. The wound was closed with sutures, and pups were left to wake up on a heating pad and returned to their mothers.
Infarct size measurement.
2,3,5-Triphenyltetrazolium chloride monohydrate (TTC) staining is a convenient procedure for detection of brain infarction (26, 42). The staining action is based on the presence of a dehydrogenase. Normal brain tissue with adequate activity of this enzyme is stained red. However, the dehydrogenase activity is reduced or inactivated in the infarcted brain tissues, and then the infarcted areas will be unstained, i.e., white. Based on such color changes, infarcted or normal brain tissues can be identified. Briefly, pups were deeply anesthetized and euthanized at 48 h after the HIE treatment. Coronal slices of the brain (2 mm thick) were cut and immersed in a 2% solution of TTC (Sigma-Aldrich) for 5 min at 37°C and then fixed with 10% formaldehyde overnight. Each slice was photographed separately, and the percentage of infarction area for each slice was analyzed by ImageJ software (version 1.40; National Institutes of Health, Bethesda, MD), summed for each brain, and expressed as a percentage of the whole brain (26, 42).
Brain weight assessment.
Rat pups were deeply anesthetized with isoflurane and decapitated. The skin and muscles were removed and torn from the dorsal and posterior part of the skull. Then, the skull at the upper edge of the foramen magnum was opened with scissors. The whole brain was removed with a small-bladed spatula. The entire dissection was performed carefully to avoid stinging or pricking the brain tissues. The extracted brain was then rinsed with ice-cold PBS to remove surface blood. The whole brain weight was determined with a Mettler Toledo AE100 Analytical Balance (Marschal Scientific).
Western blotting analysis.
Brain samples were isolated from P9 rat pups and homogenized with a lysis buffer containing 150 mmol/L NaCl, 50 mmol/L Tris·HCl, 10 mmol/L EDTA, 0.1% Tween 20, 1% Triton, 0.1% mercaptoethanol, 0.1 mmol/L phenylmethylsulfonyl fluoride, 5 g/mL leupeptin, and 5 g/mL aprotinin, pH 7.4. Homogenates were centrifuged at 4°C for 15 min at 14,000 g. The supernatants were then collected, and protein concentrations were determined via a protein assay kit (Bio-Rad, Hercules, CA). Samples with equal amounts of protein (30 μg) were loaded onto 10% or 12% polyacrylamide gel with 0.1% sodium dodecyl sulfate and separated by electrophoresis at 100 V for 90–120 min. Proteins were then transferred onto nitrocellulose membranes. Nonspecific binding sites were blocked for 3–4 h at room temperature in a Tris-buffered saline solution containing 5% dry milk. The membranes were probed with primary antibodies against mTOR (1:500), phosphorylated (p-)mTOR (1:500), ATG5 (1:1,000), Beclin 1 (1:1,000), p62 (1:1,000), LC-3β (1:500), Akt (1:1,000), p-Akt (1:500), glycogen synthase kinase-3β (GSK-3β; 1:1,000), and p-GSK-3β (1:500) (all of these from Cell Signaling), as described previously (26, 27, 42). After washing was completed, membranes were incubated with secondary horseradish peroxidase-conjugated antibodies. Proteins were visualized with enhanced chemiluminescence reagents, and blots were exposed to hyperfilm. The results were analyzed with Kodak ID image analysis software. Band intensities were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Statistical analysis.
All statistical analysis was performed using GraphPad Prism 5. Data are expressed as means ± SE. Experimental number (n) represents neonates from different dams. Statistical significance (P < 0.05) was determined by analysis of variance followed by Newman-Keuls post hoc testing or Student’s t test, where appropriate.
RESULTS
Perinatal nicotine exposure induces asymmetric growth restriction in neonatal rats.
As shown in Fig. 1, consistent with our previous findings (26, 42), perinatal nicotine exposure induced a decrease in both the body weight and the brain weight but a significant increase in the brain-to-body weight ratio in neonatal rat pups. This suggests a pathological process of intrauterine growth restriction (IUGR) and adversely asymmetric growth development in postnatal life in response to perinatal nicotine exposure.
Fig. 1.

Perinatal nicotine exposure induced asymmetric growth restriction in neonatal rats. Perinatal nicotine exposure resulted in a decrease in body weight and brain weight but a significant increase in brain weight-to-body weight ratio in 9-day-old (P9) neonatal rat pups compared with saline controls. Data are means ± SE of animals (n = 9–10) from each group. *P < 0.05 vs. control, as determined by Student’s t test.
Perinatal nicotine exposure represses baseline autophagy signaling in neonatal rat brain.
Autophagy is a vital process that degrades cytoplasmic components in response to various stressful conditions and is linked to diverse cellular processes such as cell survival, cell death, pathogen clearance, etc. (7, 24, 31). Recent studies also documented that autophagy has been implicated in several types of acute brain injury pathogenesis and confers neuroprotective properties in the ischemic challenge (5, 6, 48). Therefore, in the present study, we first determined the potential effects of perinatal nicotine on autophagic signaling pathways in neonatal rat brain. As shown in Fig. 2, perinatal nicotine exposure significantly enhanced the ratio of the mTOR phosphorylation level (p-mTOR) to the total mTOR protein level compared with the saline control group (Fig. 2A). Furthermore, perinatal nicotine exposure attenuated ATG5, Beclin 1, and LC-3βI/II protein expressions (Fig. 2, B, C, and D) compared with the saline control group. These data indicated that nicotine exposure induced a defective autophagic flux at baseline conditions in the developing rat brain.
Fig. 2.

Perinatal nicotine exposure altered mammalian target of rapamycin (mTOR)/autophagy biomarker expression patterns in neonatal rat brain. Brain tissues were isolated from 9-day-old (P9) neonatal rat pups in both nicotine-exposed and saline control groups. Protein abundance in brain tissue was determined by Western blot analysis. GAPDH protein density served as internal normalized control. A: the ratio of phosphorylated (p-)mTOR to mTOR protein density is shown in the saline control (▲) and nicotine-exposed (■) groups. Protein densities of the autophagy biomarkers ATG5 (B), Beclin-1 (C), and LC3βI/II (D) are shown in saline control (▲) and nicotine-exposed (■) groups. Data are means ± SE of animals (n = 6) from each group. *P < 0.05 vs. control, as determined by Student’s t test.
Inhibition of mTOR activation eliminates nicotine-mediated changes of autophagy-related biomarkers and reverses nicotine-mediated exaggerated neonatal brain injury to HIE.
Our previous studies (26, 27, 42) had demonstrated that perinatal nicotine exposure induces an ischemic sensitive phenotype in neonatal rat pups. Given that perinatal nicotine also induced a defective autophagic flux as observed in the present study, we further explored whether the impaired autophagy plays a causal role in the nicotine-induced HI-sensitive phenotype in postnatal offspring. We treated the offspring with the mTOR inhibitor rapamycin. As shown in Fig. 3, treatment with rapamycin significantly inhibited the phosphorylation level of mTOR and eliminated the difference of p-mTOR/mTOR ratio between the nicotine-exposed and saline control groups (Fig. 3A). In addition, after treatment with rapamycin, all of the autophagy-related biomarkers, including ATG5 (Fig. 3B), Beclin 1 (Fig. 3C), and LC3β I/II (Fig. 3D) protein levels, did not have significant differences between the nicotine-exposed group and the saline control group. Most importantly, as shown in Fig. 4, in the absence of rapamycin perinatal nicotine exposure enhanced HI-induced brain infarction in neonatal offspring compared with the saline controls. However, in the presence of rapamycin there was no significant difference in HI-induced brain infarct size between the perinatal nicotine-exposed group and saline controls.
Fig. 3.

Effect of rapamycin on nicotine-mediated autophagy in neonatal rat brain. Rapamycin or vehicle was administered intracerebraoventricularly to perinatal nicotine-exposed and saline control offspring on postnatal day 8. After 24 h of treatment, brains were isolated from those offspring, and protein abundance in brain tissue was determined by Western blot analysis. GAPDH protein density served as internal normalized control. A: the ratio of phosphorylated mammalian target of rapamycin (p-mTOR) to mTOR protein density is shown in saline control (▲) and nicotine-exposed (■) groups. Protein densities of the autophagy biomarkers ATG5 (B), Beclin-1 (C), and LC3βI/II (D) are shown in saline control (▲) and nicotine-exposed (■) groups. Data are means ± SE of animals (n = 6) from each group.
Fig. 4.
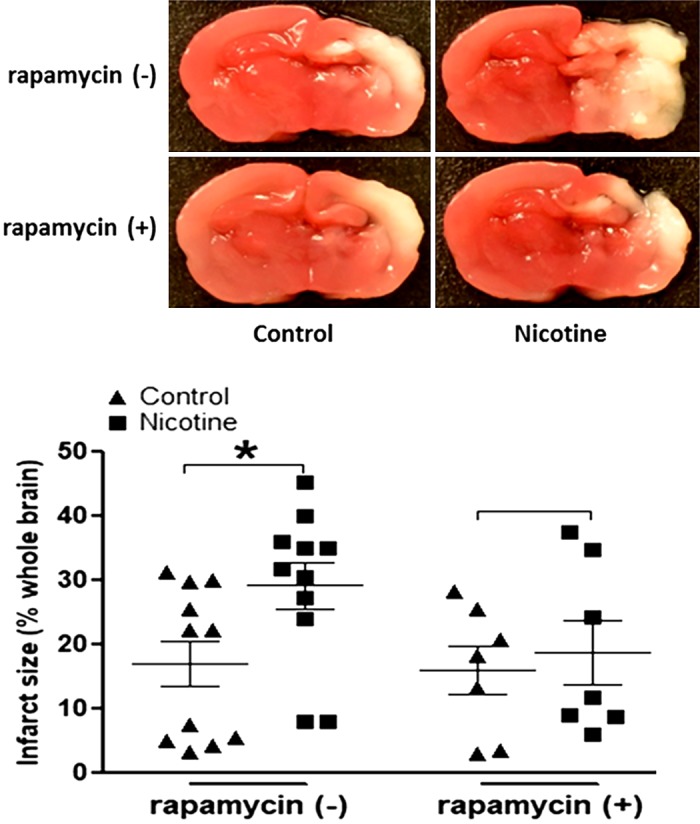
Effect of rapamycin on hypoxia/ischemia (HI)-induced brain infarct size in both nicotine-exposed and saline control offspring. Rapamycin or vehicle was administered intracerebraoventricularly to perinatal nicotine-exposed and saline control offspring on postnatal day 8. After 24 h of treatment, the pups were exposed to HI procedures. Then, HI-induced brain infarct size with/without treatment of rapamycin was measured by triphenyltetrazolium chloride monohydrate (TTC) staining 48 h after the HI procedures. Top: images represent HI-induced brain infarct size determined by 2% TTC staining. Summed infarct sizes, expressed as percent total brain weight, are shown at bottom. Data are means ± SE of animals (n = 9–11) from each group. Data were analyzed by 2-way ANOVA. *P < 0.05 vs. saline control.
Perinatal nicotine exposure enhances Akt/GSK-3β signaling in neonatal rat brain.
Emerging evidence has indicated that Akt/GSK-3β signaling cascades serve as one of common modulators of autophagy, mainly by influencing activation of mTOR (12, 16, 18, 28, 35). Therefore, in the present study, we examined the effect of perinatal nicotine exposure on Akt/GSK-3β signaling in the neonatal rat brain. As shown in Fig. 5, perinatal nicotine exposure had no effect on total Akt protein expression (Fig. 5A) but significantly enhanced the phosphorylation level of Akt (p-Akt) (Fig. 5B) in the neonatal brain tissues compared with the saline controls. Similarly, perinatal nicotine exposure had no effect on total GSK-3β protein expression (Fig. 5C) but significantly enhanced its phosphorylation level (Fig. 5D) in the neonatal brain tissues compared with the saline controls.
Fig. 5.

Effect of perinatal nicotine exposure on protein kinase B/glycogen synthase kinase-3β (Akt/GSK-3β) protein abundances in neonatal rat brain. Brain tissues were isolated from 9-day-old (P9) neonatal rat pups in both nicotine-exposed and saline control groups. Protein abundance in brain tissue was determined by Western blot analysis. GAPDH protein density served as internal normalized control. Total Akt (A), phosphorylated (p-)Akt (B), total GSK-3β (C), and p-GSK-3β (D) protein densities are shown in saline control (▲) and nicotine-exposed (■) groups. Data are means ± SE of animals (n = 6) from each group. *P < 0.05 vs. control, as determined by Student’s t test.
Inhibition of Akt/GSK-3β restores basal autophagic flux and abolishes nicotine-mediated HI-induced brain infarct size in neonatal offspring.
MK2206, a selective Akt inhibitor, was utilized in the current study to further determine the causal role of activated Akt/GSK-3β signaling cascades in nicotine exposure-induced defects of autophagic flux and neonatal brain ischemic injury. As shown in Fig. 6, MK2206 treatment eliminated the differences of total Akt (Fig. 6A) and p-Akt (Fig. 6B) between the nicotine-exposed group and the saline controls. Similarly, MK2206 treatment also eliminated the differences of total GSK-3β (Fig. 6C) and p-GSK-3β (Fig. 6D) between the nicotine-exposed group and the saline controls. Furthermore, as shown in Fig. 7, inhibition of Akt with MK2206 also restored the nicotine-mediated changes of autophagy-associated markers and eliminated the differences of these autophagy-related protein expressions between the control and nicotine-exposed offspring. In addition, as shown in Fig. 8, in the absence of MK2206 treatment, perinatal nicotine exposure enhanced HI-induced brain infarction in neonatal offspring compared with the saline controls. However, MK2206 treatment significantly attenuated HI-induced brain infarct size in perinatal nicotine-exposed offspring and eliminated the difference of HI-induced brain infarct size between the perinatal nicotine-exposed group and the saline controls.
Fig. 6.

Effect of MK2206 on nicotine-mediated changes of protein kinase B/glycogen synthase kinase-3β (Akt/GSK-3β) in neonatal rat brain. MK2206 or vehicle was administered intracerebraoventricularly to perinatal nicotine-exposed and saline control offspring on postnatal day 8. After 24 h of treatment, brains were isolated from those offspring, and protein abundance in brain tissue was determined by Western blot analysis. GAPDH protein density served as internal normalized control. Total Akt (A), phosphorylated (p-)Akt (B), total GSK-3β (C), and p-GSK-3β (D) protein densities are shown in saline control (▲) and nicotine-exposed (■) groups. Data are means ± SE of animals (n = 6) from each group.
Fig. 7.

Effect of MK2206 on nicotine-mediated changes of autophagy biomarkers in neonatal rat brain. MK2206 or vehicle was administered intracerebraoventricularly to perinatal nicotine-exposed and saline control offspring on postnatal day 8. After 24 h of treatment, brains were isolated from those offspring, and protein abundance in brain tissue was determined by Western blot analysis. GAPDH protein density served as internal normalized control. The ratio of phosphorylated mammalian target of rapamycin (p-mTOR) to mTOR protein density is shown in the saline control (▲) and nicotine-exposed (■) groups (A). The protein densities of the autophagy biomarkers ATG5 (B), Beclin 1 (C), and LC3βI/II (D) are shown in saline control and nicotine-exposed groups. Data are means ± SE of animals (n = 6) from each group.
Fig. 8.
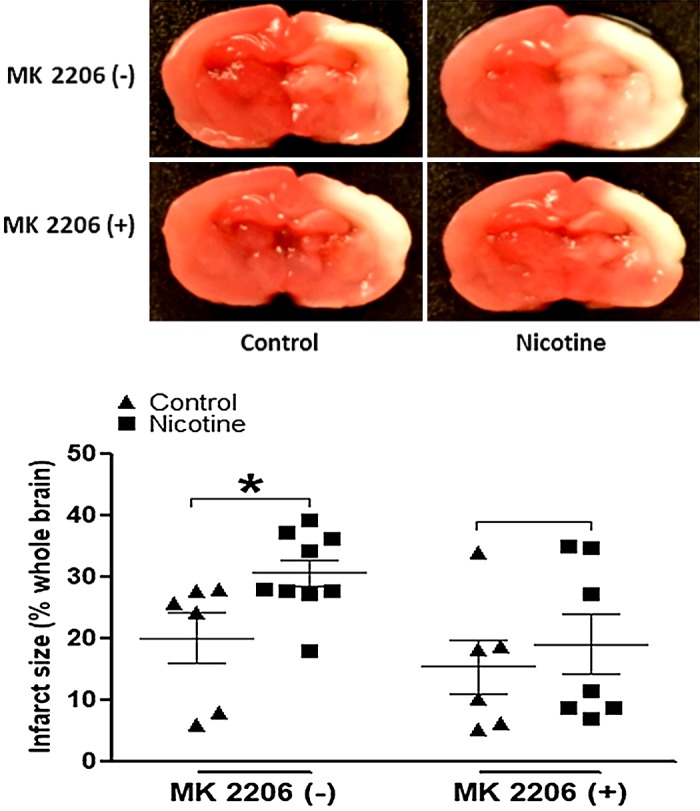
Effect of MK2206 on hypoxia/ischemia (HI)-induced brain infarct size in both nicotine-exposed and saline control offspring. MK2206 or vehicle was administered intracerebraoventricularly to perinatal nicotine-exposed and saline control offspring on postnatal day 8. After 24 h of treatment, pups were exposed to HI procedures. Then, HI-induced brain infarct size with/without treatment of MK2206 was measured by triphenyltetrazolium chloride monohydrate (TTC) staining 48 h after HI procedures. Top: images represent HI-induced brain infarct size determined by 2% TTC staining. Summed infarct sizes, expressed as precent total brain weight, are shown at bottom. Data are means ± SE of animals (n = 8–11) from each group. Data were analyzed by 2-way ANOVA. *P < 0.05 vs. saline control.
DISCUSSION
The present study provides novel evidence that perinatal nicotine exposure-mediated alteration of autophagy signaling plays a key role in the development of HI-sensitive brain phenotype in neonates. There are several new observations in the present study. 1) Perinatal nicotine exposure decreased body weight and brain weight but increased the brain weight/body weight ratio in neonates compared with the saline controls. 2) Perinatal nicotine exposure enhanced mTOR phosphorylation levels in neonatal brain tissues compared with the saline controls. 3) Perinatal nicotine exposure downregulated autophagy-related proteins including ATG5, Beclin 1, and LC3β I/II expression. 4) Inhibition of mTOR activation by rapamycin abolished the nicotine-mediated changes of autophagy-related proteins and reversed nicotine-mediated HI-induced brain infarction. 5) Perinatal nicotine exposure enhanced the phosphorylation levels of Akt and GSK-3β genes, and inhibition of Akt activation by MK2206 abolished the nicotine-mediated changes of autophagy-related proteins and reversed nicotine-mediated HI-induced brain infarction in offspring.
It is well documented that maternal tobacco smoking is associated with IUGR (36). In the current study, our data indicated that nicotine exposure during pregnancy significantly decreased neonatal body weight and brain weight, which is consistent with our previous studies (26, 42). These results suggest that, as one of the major components in cigarette smoking, nicotine contributes to maternal tobacco smoking-induced IUGR. Previous studies had reported that fetal growth restriction is one of the important risk factors that contribute significantly to the development of neonatal encephalopathy (49). In the present study, we demonstrated that perinatal nicotine exposure enhanced HI-induced brain injury associated with neonatal growth restriction, which further suggests that growth restriction may be the link between perinatal nicotine exposure and development of the brain HI-sensitive phenotype in postnatal life.
The molecular mechanisms underlying the perinatal nicotine-mediated increase in HI-induced neonatal brain injury are still not fully understood. In the present study, we observed that perinatal nicotine exposure attenuated autophagy biomarkers (including LC3βI/II, Beclin-1, and ATG5) protein abundance in neonatal brain tissues. This finding suggests that perinatal nicotine exposure reprograms the autophagy signaling pathway, resulting in autophagy deficiency in the developing brain. Accumulating evidence has shown a vital role of autophagy in the pathogenesis of ischemic brain insults (2, 12, 15, 29, 37). The major function of autophagy is to remove the bulk of cellular proteins or damaged organelles, responsible for maintaining an intricate balance between cell survival and cell death. However, whether autophagy plays a protective or a destructive role in the pathogenesis of ischemic brain injury is debatable and controversial. In the mouse HI model and rat neonatal HI model, activation of autophagy contributed to cell death and brain ischemic injury (34, 43), whereas other studies demonstrated that autophagy contributes to a protective effect for neurons during ischemic challenge (5, 6, 48). Our present finding that perinatal nicotine exposure enhanced HI-induced brain injury associated with a decrease in autophagy biomarkers (LC3βI/II, Beclin-1, and ATG5) in neonatal brain tissues suggests a protective role of autophagy signaling in the pathogenesis of brain ischemic injury. The deficiency of autophagy will diminish the protective effect, leading to an increase the perinatal nicotine-mediated brain ischemic injury in neonatal offspring.
Several cellular signaling pathways have been reported to regulate autophagy in brain ischemic injury. Of these, mTOR is one of the major signaling pathways under stress to regulate the process of autophagy. It is well known that activation of mTOR results in a negative regulation of autophagy (12, 35, 48). In the present study, we observed that perinatal nicotine exposure enhanced the phosphorylation level of mTOR in the neonatal brain tissues, which suggests an exaggerated activation of mTOR signaling in response to perinatal nicotine exposure. In addition, our present study demonstrated that treatment with the mTOR inhibitor rapamycin enhanced autophagy biomarkers (Beclin-1, ATG5, LC3βI/II) protein expressions and eliminated the differences of these biomarkers between the perinatal nicotine-exposed and saline control groups. This suggests that mTOR is a key upstream negative regulator of the autophagy signaling pathway and contributes significantly to the perinatal nicotine-mediated autophagy deficiency in the developing brain. Furthermore, our present finding that treatment with rapamycin eliminated the differences of HI-induced brain infarct size between the perinatal nicotine-exposed and saline control groups suggests a causal role of the mTOR/autophagy signaling pathway in nicotine-induced fetal programming of the brain ischemia-sensitive phenotype in postnatal life. Consistently, a recent preclinical study demonstrated that administration of a negative feedback regulator of mTOR not only reduced infarct size and brain atrophy but also significantly improved neurological function after a neonatal HIE challenge (38). These studies suggest that mTOR/autophagy signaling may act as a key switching point in the pathogenesis of perinatal brain ischemic injury, and pharmacological manipulation of mTOR signaling may be considered an attractive therapeutic target in perinatal nicotine-mediated neonatal HIE.
Another novel finding of the present study is that activation of Akt/GSK-3β signaling cascades may play an important role in perinatal nicotine exposure-induced autophagy deficiency in the neonatal brain. Emerging evidence has indicated that Akt and GSK-3β are involved in the modulation of autophagy in response to various stimuli (16, 18, 28, 35). In the present study, we observed that there were remarkably increased levels of p-Akt and p-GSK-3β in nicotine-treated animals compared with the saline controls, which suggests a heightened activation pattern of Akt/GSK-3β signaling in the developing brain in response to the perinatal nicotine exposure. Furthermore, the finding that administration of the selective Akt inhibitor MK2206 effectively abolished the upregulation of p-Akt/p-GSK-3β, as well as p-mTOR, and blocked nicotine-induced autophagy deficiency suggests that activation of Akt/GSK-3β signaling cascades serves as an important upstream operator in perinatal nicotine exposure-induced defective autophagic responses in the developing brain. In addition, our present finding that inhibition of Akt via MK2206 significantly improved nicotine-mediated brain injury in neonatal HIE further suggests a causal role of Akt/mTOR/autophagy signaling in the perinatal nicotine-mediated enhanced brain ischemic injury in neonates.
Perspectives and Significance
The present study for the first time not only demonstrated that perinatal nicotine exposure altered the Akt/mTOR/autophagy signaling pathway but also confirmed that the altered Akt/mTOR/autophagy signaling played a causal role in nicotine-mediated development of brain ischemia-sensitive phenotype in neonatal offspring. Our findings provide a molecular basis and therapeutic target of autophagy signaling for the effective intervention of perinatal brain ischemic injury. In the present study, we observed that perinatal nicotine exposure induced a defective autophagic flux via activation of Akt/GSK-3β/mTOR signaling pathway. However, the precise molecular mechanisms underlying perinatal stress-induced pathogenesis of HIE cannot be clearly interpreted based only on the data available in the current study. Taking into account the complexity of the pathogenesis in ischemic brain injury, the autophagy deficiency observed in our present study may not be the only pathway contributing to the nicotine exposure-induced enhanced HIE injury. Given the facts that autophagic flux and apoptotic pathway are tightly linked together, it is highly worthwhile to further investigate whether perinatal nicotine exposure programs apoptosis signaling and to test whether autophagy interacting with apoptosis signaling contributes to the development of ischemic-sensitive phenotype to HIE in offspring. Our previous studies had demonstrated that adverse prenatal stresses, such as maternal hypoxia and smoking, can induce aberrant gene expression patterns via epigenetic molecular mechanisms in offspring, leading to increased risk of cardiovascular and neurological disorders (19, 25–27, 32, 42, 46). Therefore, we speculate that changes of epigenetic pathways such as DNA methylation, histone modification, and microRNA regulation may play a key role in perinatal nicotine-induced aberrant autophagy biomarker expression patterns in neonatal offspring, which open a wide door for our future studies. In addition, given the high mortality and morbidity of neonatal HIE, as well as the extensive abuse of nicotine products during pregnancy, it is highly warranted to further explore the potential epigenetic mechanisms underlying the perinatal stresses-induced programming of brain ischemic injury in postnatal life.
GRANTS
This work was supported by National Institutes of Health Grants HL-135623 (D. Xiao), DA-041492 (D. Xiao), and HD-088039 (D. Xiao). This project was partially supported by funds provided by The Regents of the University of California, Research Grants Program Office, Tobacco Related Disease Research Program (TRDRP) Grant T29IR0437. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.L., A.M.S., L.Z., and D.X. conceived and designed research; Y.L., Y.F., M.Y., J.J., B.L., and L.X. performed experiments; Y.L., Y.F., A.W., M.Y., J.J., B.L., L.X., and D.X. analyzed data; Y.L., A.M.S., A.W., M.Y., J.J., B.L., L.X., L.Z., and D.X. interpreted results of experiments; Y.L., Y.F., and D.X. prepared figures; Y.L., A.M.S., A.W., and D.X. drafted manuscript; Y.L., A.M.S., A.W., M.Y., J.J., B.L., L.X., L.Z., and D.X. edited and revised manuscript; Y.L. and D.X. approved final version of manuscript.
REFERENCES
- 1.Azzopardi DV, Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, Kapellou O, Levene M, Marlow N, Porter E, Thoresen M, Whitelaw A, Brocklehurst P; TOBY Study Group . Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med 361: 1349–1358, 2009. doi: 10.1056/NEJMoa0900854. [DOI] [PubMed] [Google Scholar]
- 2.Baek SH, Noh AR, Kim KA, Akram M, Shin YJ, Kim ES, Yu SW, Majid A, Bae ON. Modulation of mitochondrial function and autophagy mediates carnosine neuroprotection against ischemic brain damage. Stroke 45: 2438–2443, 2014. doi: 10.1161/STROKEAHA.114.005183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beratis NG, Panagoulias D, Varvarigou A. Increased blood pressure in neonates and infants whose mothers smoked during pregnancy. J Pediatr 128: 806–812, 1996. doi: 10.1016/S0022-3476(96)70333-5. [DOI] [PubMed] [Google Scholar]
- 4.Blake KV, Gurrin LC, Evans SF, Beilin LJ, Landau LI, Stanley FJ, Newnham JP. Maternal cigarette smoking during pregnancy, low birth weight and subsequent blood pressure in early childhood. Early Hum Dev 57: 137–147, 2000. doi: 10.1016/S0378-3782(99)00064-X. [DOI] [PubMed] [Google Scholar]
- 5.Carloni S, Albertini MC, Galluzzi L, Buonocore G, Proietti F, Balduini W. Increased autophagy reduces endoplasmic reticulum stress after neonatal hypoxia-ischemia: role of protein synthesis and autophagic pathways. Exp Neurol 255: 103–112, 2014. doi: 10.1016/j.expneurol.2014.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol Dis 32: 329–339, 2008. doi: 10.1016/j.nbd.2008.07.022. [DOI] [PubMed] [Google Scholar]
- 7.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy 6: 322–329, 2010. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol 13: 722–737, 2013. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doria A, Gatto M, Punzi L. Autophagy in human health and disease. N Engl J Med 368: 1845–1846, 2013. doi: 10.1056/NEJMc1303158. [DOI] [PubMed] [Google Scholar]
- 10.Ferriero DM. Neonatal brain injury. N Engl J Med 351: 1985–1995, 2004. doi: 10.1056/NEJMra041996. [DOI] [PubMed] [Google Scholar]
- 11.Fewell JE, Smith FG, Ng VK. Prenatal exposure to nicotine impairs protective responses of rat pups to hypoxia in an age-dependent manner. Respir Physiol 127: 61–73, 2001. doi: 10.1016/S0034-5687(01)00232-8. [DOI] [PubMed] [Google Scholar]
- 12.Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci 17: 467–484, 2016. doi: 10.1038/nrn.2016.51. [DOI] [PubMed] [Google Scholar]
- 13.Galluzzi L, Bravo-San Pedro JM, Kroemer G. Organelle-specific initiation of cell death. Nat Cell Biol 16: 728–736, 2014. doi: 10.1038/ncb3005. [DOI] [PubMed] [Google Scholar]
- 14.Galluzzi L, Pietrocola F, Levine B, Kroemer G. Metabolic control of autophagy. Cell 159: 1263–1276, 2014. doi: 10.1016/j.cell.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ginet V, Pittet MP, Rummel C, Osterheld MC, Meuli R, Clarke PG, Puyal J, Truttmann AC. Dying neurons in thalamus of asphyxiated term newborns and rats are autophagic. Ann Neurol 76: 695–711, 2014. doi: 10.1002/ana.24257. [DOI] [PubMed] [Google Scholar]
- 16.Guo D, Xie J, Zhao J, Huang T, Guo X, Song J. Resveratrol protects early brain injury after subarachnoid hemorrhage by activating autophagy and inhibiting apoptosis mediated by the Akt/mTOR pathway. Neuroreport 29: 368–379, 2018. doi: 10.1097/WNR.0000000000000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci 20: 5775–5781, 2000. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang L, Lin M, Zhong X, Yang H, Deng M. Galangin decreases p-tau, Aβ42 and β-secretase levels, and suppresses autophagy in okadaic acid-induced PC12 cells via an Akt/GSK3β/mTOR signaling-dependent mechanism. Mol Med Rep 19: 1767–1774, 2019. doi: 10.3892/mmr.2019.9824. [DOI] [PubMed] [Google Scholar]
- 19.Ke J, Dong N, Wang L, Li Y, Dasgupta C, Zhang L, Xiao D. Role of DNA methylation in perinatal nicotine-induced development of heart ischemia-sensitive phenotype in rat offspring. Oncotarget 8: 76865–76880, 2017. doi: 10.18632/oncotarget.20172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelen D, Robertson NJ. Experimental treatments for hypoxic ischaemic encephalopathy. Early Hum Dev 86: 369–377, 2010. doi: 10.1016/j.earlhumdev.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 21.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884, 2006. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 22.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 40: 280–293, 2010. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence J, Xiao D, Xue Q, Rejali M, Yang S, Zhang L. Prenatal nicotine exposure increases heart susceptibility to ischemia/reperfusion injury in adult offspring. J Pharmacol Exp Ther 324: 331–341, 2008. doi: 10.1124/jpet.107.132175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 469: 323–335, 2011. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu B, Hu X, Li Y, Ke J, Dasgupta C, Huang X, Walayat A, Zhang L, Xiao D. Epigenetic downregulation of BKCa channel by miR-181a contributes to the fetal and neonatal nicotine-mediated exaggerated coronary vascular tone in adult life. Int J Cardiol 281: 82–89, 2019. doi: 10.1016/j.ijcard.2019.01.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Xiao D, Dasgupta C, Xiong F, Tong W, Yang S, Zhang L. Perinatal nicotine exposure increases vulnerability of hypoxic-ischemic brain injury in neonatal rats: role of angiotensin II receptors. Stroke 43: 2483–2490, 2012. doi: 10.1161/STROKEAHA.112.664698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Y, Xiao D, Yang S, Zhang L. Promoter methylation represses AT2R gene and increases brain hypoxic-ischemic injury in neonatal rats. Neurobiol Dis 60: 32–38, 2013. doi: 10.1016/j.nbd.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Long Q, Li X, He H, He D. Autophagy activation protects shock wave induced renal tubular epithelial cell apoptosis may through modulation of Akt/GSK-3β pathway. Int J Biol Sci 12: 1461–1471, 2016. doi: 10.7150/ijbs.16864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehta SL, Lin Y, Chen W, Yu F, Cao L, He Q, Chan PH, Li PA. Manganese superoxide dismutase deficiency exacerbates ischemic brain damage under hyperglycemic conditions by altering autophagy. Transl Stroke Res 2: 42–50, 2011. doi: 10.1007/s12975-010-0027-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 16: 345–357, 2015. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 31.Münz C. Enhancing immunity through autophagy. Annu Rev Immunol 27: 423–449, 2009. doi: 10.1146/annurev.immunol.021908.132537. [DOI] [PubMed] [Google Scholar]
- 32.Patterson AJ, Chen M, Xue Q, Xiao D, Zhang L. Chronic prenatal hypoxia induces epigenetic programming of PKCepsilon gene repression in rat hearts. Circ Res 107: 365–373, 2010. doi: 10.1161/CIRCRESAHA.110.221259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pauly JR, Slotkin TA. Maternal tobacco smoking, nicotine replacement and neurobehavioural development. Acta Paediatr 97: 1331–1337, 2008. doi: 10.1111/j.1651-2227.2008.00852.x. [DOI] [PubMed] [Google Scholar]
- 34.Qin AP, Liu CF, Qin YY, Hong LZ, Xu M, Yang L, Liu J, Qin ZH, Zhang HL. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy 6: 738–753, 2010. doi: 10.4161/auto.6.6.12573. [DOI] [PubMed] [Google Scholar]
- 35.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90: 1383–1435, 2010. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 36.Reeves S, Bernstein I. Effects of maternal tobacco-smoke exposure on fetal growth and neonatal size. Expert Rev Obstet Gynecol 3: 719–730, 2008. doi: 10.1586/17474108.3.6.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheng R, Zhang LS, Han R, Liu XQ, Gao B, Qin ZH. Autophagy activation is associated with neuroprotection in a rat model of focal cerebral ischemic preconditioning. Autophagy 6: 482–494, 2010. doi: 10.4161/auto.6.4.11737. [DOI] [PubMed] [Google Scholar]
- 38.Shi X, Xu L, Doycheva DM, Tang J, Yan M, Zhang JH. Sestrin2, as a negative feedback regulator of mTOR, provides neuroprotection by activation AMPK phosphorylation in neonatal hypoxic-ischemic encephalopathy in rat pups. J Cereb Blood Flow Metab 37: 1447–1460, 2017. doi: 10.1177/0271678X16656201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther 285: 931–945, 1998. [PubMed] [Google Scholar]
- 40.Vannucci RC, Connor JR, Mauger DT, Palmer C, Smith MB, Towfighi J, Vannucci SJ. Rat model of perinatal hypoxic-ischemic brain damage. J Neurosci Res 55: 158–163, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 41.Verklan MT. The chilling details: hypoxic-ischemic encephalopathy. J Perinat Neonatal Nurs 23: 59–68, 2009. doi: 10.1097/01.JPN.0000346221.48202.7e. [DOI] [PubMed] [Google Scholar]
- 42.Wang L, Ke J, Li Y, Ma Q, Dasgupta C, Huang X, Zhang L, Xiao D. Inhibition of miRNA-210 reverses nicotine-induced brain hypoxic-ischemic injury in neonatal rats. Int J Biol Sci 13: 76–84, 2017. doi: 10.7150/ijbs.17278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen YD, Sheng R, Zhang LS, Han R, Zhang X, Zhang XD, Han F, Fukunaga K, Qin ZH. Neuronal injury in rat model of permanent focal cerebral ischemia is associated with activation of autophagic and lysosomal pathways. Autophagy 4: 762–769, 2008. doi: 10.4161/auto.6412. [DOI] [PubMed] [Google Scholar]
- 44.Wickström R. Effects of nicotine during pregnancy: human and experimental evidence. Curr Neuropharmacol 5: 213–222, 2007. doi: 10.2174/157015907781695955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiao D, Huang X, Lawrence J, Yang S, Zhang L. Fetal and neonatal nicotine exposure differentially regulates vascular contractility in adult male and female offspring. J Pharmacol Exp Ther 320: 654–661, 2007. doi: 10.1124/jpet.106.113332. [DOI] [PubMed] [Google Scholar]
- 46.Xiao D, Huang X, Yang S, Zhang L. Antenatal nicotine induces heightened oxidative stress and vascular dysfunction in rat offspring. Br J Pharmacol 164: 1400–1409, 2011. doi: 10.1111/j.1476-5381.2011.01437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao D, Xu Z, Huang X, Longo LD, Yang S, Zhang L. Prenatal gender-related nicotine exposure increases blood pressure response to angiotensin II in adult offspring. Hypertension 51: 1239–1247, 2008. doi: 10.1161/HYPERTENSIONAHA.107.106203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xin D, Chu X, Bai X, Ma W, Yuan H, Qiu J, Liu C, Li T, Zhou X, Chen W, Liu D, Wang Z. l-Cysteine suppresses hypoxia-ischemia injury in neonatal mice by reducing glial activation, promoting autophagic flux and mediating synaptic modification via H2S formation. Brain Behav Immun 73: 222–234, 2018. doi: 10.1016/j.bbi.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 49.Yager JY, Ashwal S. Animal models of perinatal hypoxic-ischemic brain damage. Pediatr Neurol 40: 156–167, 2009. doi: 10.1016/j.pediatrneurol.2008.10.025. [DOI] [PubMed] [Google Scholar]
- 50.Yan F, Cao S, Li J, Dixon B, Yu X, Chen J, Gu C, Lin W, Chen G. Pharmacological inhibition of PERK attenuates early brain injury after subarachnoid hemorrhage in rats through the activation of Akt. Mol Neurobiol 54: 1808–1817, 2017. doi: 10.1007/s12035-016-9790-9. [DOI] [PubMed] [Google Scholar]