

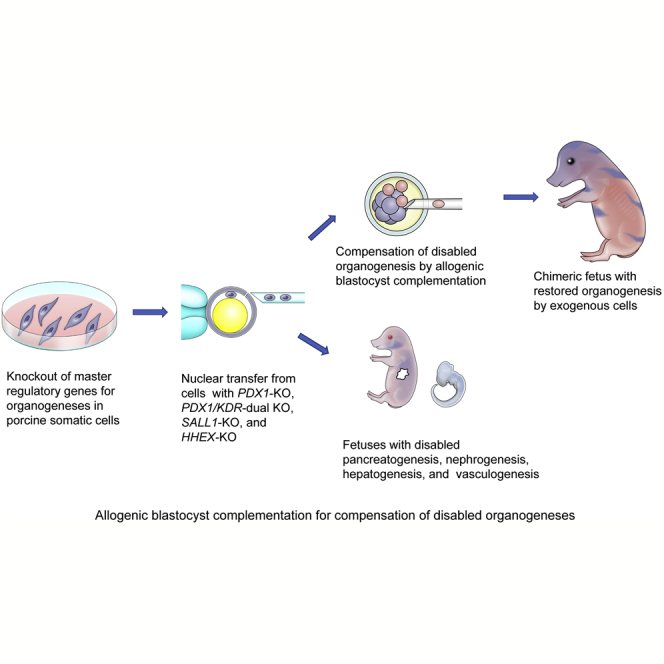
Summary
We have previously established a concept of developing exogenic pancreas in a genetically modified pig fetus with an apancreatic trait, thereby proposing the possibility of in vivo generation of functional human organs in xenogenic large animals. In this study, we aimed to demonstrate a further proof-of-concept of the compensation for disabled organogeneses in pig, including pancreatogenesis, nephrogenesis, hepatogenesis, and vasculogenesis. These dysorganogenetic phenotypes could be efficiently induced via genome editing of the cloned pigs. Induced dysorganogenetic traits could also be compensated by allogenic blastocyst complementation, thereby proving the extended concept of organ regeneration from exogenous pluripotent cells in empty niches during various organogeneses. These results suggest that the feasibility of blastocyst complementation using genome-edited cloned embryos permits experimentation toward the in vivo organ generation in pigs from xenogenic pluripotent cells.
Keywords: blastocyst complementation, organ regeneration, organogenesis, pluripotent stem cells, cloned pig
Graphical Abstract
Highlights
-
•
Dysorganogenetic phenotypes induced via genome editing of cloned pigs
-
•
Apancreatic, anephrogenic, ahepatogenic, and vasculogenesis-disabled traits induced
-
•
Compensation of disabled organogenesis by allogenic blastocyst complementation
-
•
Use of genome-edited cloned pig embryos permits in vivo organ generation studies
In this article, Nagashima and colleagues generated various cloned pig fetuses with dysorganogenetic phenotypes by editing master regulatory genes for pancreatogenesis, nephrogenesis, hepatogenesis, and vasculogenesis. They also demonstrate that these disabled organogeneses can be compensated by allogenic blastocyst complementation, thereby enabling use of the genetically modified pig fetuses for in vivo organ generation studies.
Introduction
Generation of functional organs from pluripotent stem cells (PSCs) is one of the ultimate goals of regenerative medicine. In addition to the in vitro approaches, such as organoid culture and organ-fabrication (Jung et al., 2016, Liu et al., 2013, Lu et al., 2013, Taguchi et al., 2014, Takasato et al., 2015, Takasato and Little, 2016, Takebe et al., 2013, Takebe et al., 2015, Yamanaka and Yokoo, 2015), an alternative in vivo strategy has been proposed. This strategy originally proposed by Kobayashi et al. (2010) uses an organogenesis-disabled animal and a blastocyst complementation technique. They generated an interspecies chimera by introducing rat induced pluripotent stem cells (iPSCs) into mouse embryos with a Pdx1−/− mutation that had caused a pancreatogenesis-disabled phenotype and succeeded in creating a mouse with the pancreas of a rat. These results were interpreted as follows––an intra-embryonic environment lacking endogenous pancreatic cell lineage allowed the exogenous rat iPSCs to colonize in an empty developmental niche to form a xenogeneic pancreas in the mouse body (Kobayashi et al., 2010, Rashid et al., 2014).
This approach is endorsed by the notion that in vitro generation of human organs with complex function and structure is extremely difficult (Rashid et al., 2014), and organ development from PSCs in the natural physiological environment of a xenogeneic fetus would be better supported. Recent studies have been demonstrated compelling evidence for blastocyst complementation in rodents by generating organs, such as kidney, brain, vessels, and blood (Goto et al., 2019, Hamanaka et al., 2018, Kobayashi et al., 2010, Matsunari et al., 2013, Nagashima and Matsunari, 2016, Rashid et al., 2014, Usui et al., 2012, Wu et al., 2016, Yamaguchi et al., 2017).
To fulfill the ultimate aim of generating human organs in an animal body, the use of large animals is essential. We, therefore, established a blastocyst complementation system in pigs (Matsunari et al., 2013). At present, we have demonstrated the production of genetically modified pigs with a pancreatogenesis-disabled phenotype and proved that the missing organ could be restored by exogenous cells through allogenic blastocyst complementation (Matsunari et al., 2013). The concept of blastocyst complementation that chimerizes a cloned dysorganogenetic embryo with functionally normal pluripotent cells needs to be verified for applicability to multifarious organs in pigs to determine the potential value of the technology in the medical setting.
In our previous study (Matsunari et al., 2013), the apancreatic phenotype was induced in pigs by the overexpression of a transgene (mouse Pdx1-Hes1: hairy and enhancer of split-1 gene driven by pancreatic and duodenal homeobox 1 gene promoter). In recent years, gene knockout (KO) by genome editing has significantly advanced its feasibility and practical applicability in mammals (Gupta and Musunuru, 2014, Tan et al., 2012). Thus, knocking out the master regulator gene of a specific organ has become a straightforward method for generating the dysorganogenetic phenotype (Goto et al., 2019, Hamanaka et al., 2018, Kobayashi et al., 2010, Usui et al., 2012, Vilarino et al., 2017). Direct genome editing for zygotes, however, has been reported to result in mosaic mutations (Vilarino et al., 2017), which hinder obtaining conclusive evidence for blastocyst complementation. We, therefore, used, in this study, a somatic cloning technique using nuclear donor cells carrying a defined loss-of-function mutation to produce host embryos with an organogenesis-disabled trait for blastocyst complementation.
We aimed at investigating the efficiency of genetic modification by genome editing in pigs to give rise to an organogenesis-disabled phenotype in various organs, including pancreas, kidney, liver, and blood vessels. We also verified the feasibility of blastocyst complementation for compensating the disabled organogeneses.
Results
Apancreatic Phenotype Generated by PDX1-KO and Restoration of Normal Pancreatogenesis by Blastocyst Complementation
In our previous study (Matsunari et al., 2013), we demonstrated that an apancreatic phenotype in pigs could be achieved by overexpressing the mouse Pdx1-Hes1 transgene. In this study, we knocked out porcine endogenous PDX1 gene, based on previous reports demonstrating apancreatic phenotype caused by Pdx1−/− mutation in rodents (Jonsson et al., 1994, Kobayashi et al., 2010, Offield et al., 1996) and pigs (Kang et al., 2017).
Porcine fetal fibroblast cells (male) carrying TALEN-induced biallelic mutations in exon 1 of PDX1 were used for somatic cell nuclear transfer (SCNT) (Figure S1A). Two types of cloned embryos were generated from two lines of nuclear donor cells with different mutation types (Figure S1B), and these cloned embryos were transferred together to a recipient gilt (Figure S1C). Analysis of four cloned fetuses recovered at mid-gestation (day 55) revealed that both the PDX1-KO mutations gave rise to apancreatic phenotypes (Figures 1A and S1C).
Figure 1.

Apancreatic Phenotype of the PDX1−/− Fetus and Restoration of Pancreatogenesis by Blastocyst Complementation
(A) Apancreatic phenotype of a PDX1−/− cloned fetus at mid-gestation (day 55; WT, day 66).
(B) Chimeric full-term fetuses (three left animals in upper panels) and restored pancreata (lower panels). Fetuses exhibiting huKO fluorescence and male phenotype were determined as chimeras. Chimeric fetuses (∗) indicating higher level of systemic chimerism showed well-restored pancreata (leftmost in lower panels), while development of pancreatic tissue was poor in a fetus with lower chimerism (center in lower panels). The rightmost animal in upper panels (#) presents a PDX1-KO fetus showing typical features of the apancreatic phenotype (lower panels).
(C) Histological features of the pancreatic tissue generated by blastocyst complementation. Upper panels: H&E-stained sections. Lower panels: sections immunostained for insulin (white), glucagon (green), and huKO (red). Restored pancreatic tissue of the fetus (center) with higher chimerism entirely expressed huKO fluorescence. Scale bars, 100 μm.
Primary cultured fibroblasts were established from the PDX1-KO apancreatic fetuses (male) and used for generating cloned embryos as the host embryos for blastocyst complementation. The host PDX1-KO embryos at the morula stage were injected, as reported previously (Matsunari et al., 2013), with donor blastomeres derived from female cloned embryos expressing the fluorescent proteins, Kusabira-Orange (huKO) (Matsunari et al., 2008) or Plum (Watanabe et al., 2015). Transfer of 325 complemented embryos at the blastocyst stage to the uterus of four recipients resulted in three pregnancies, which allowed us to analyze ten full-term fetuses comprising four chimeric and six non-chimeric animals (Table S1). Chimerism was proven by phenotypic features (phenotypically male individuals expressing huKO or Plum) (Matsunari et al., 2018) or genotyping (Figure 1B). Two of the chimeric fetuses showed normally developed pancreata. The restored pancreata entirely expressed huKO fluorescence (Figures 1B and 1C), indicating that they were derived from exogenous cells. The restored pancreatic tissue demonstrated normal histological features, including endocrine and exocrine cells.
However, two of the chimeric fetuses possessed less-developed pancreata (Figures 1B and 1C). The proportion of the exogenous cells in these chimeric fetuses was lower (Figure 1B), suggesting that the restoration of the PDX1-KO apancreatic trait required chimerism with exogenous PDX1+/+ cells in high density in the pancreatogenic progenitor tissue.
Vasculogenesis-Disabled Phenotype Generated by Kinase Insert Domain Receptor-KO and Restoration of Normal Vessels by Blastocyst Complementation
Previous studies (Kobayashi et al., 2010, Usui et al., 2012, Yamaguchi et al., 2017) have revealed that blood vessels had been developed from both donors and host animal-derived cells in the restored organ generated by blastocyst complementation in apancreatic and anephrogenic animals. For clinical transplantation of an organ generated in a host animal by means of interspecies blastocyst complementation, it is necessary to replace the vasculature with exogenous human cells so that the number of xenogenic component cells derived from the host animal can be minimized.
Vasculogenesis can be disrupted by the deficiency of the Flk-1 gene in rodents (Shalaby et al., 1995). Thus, establishing a vasculogenesis-disabled trait in the host animal and restoring the trait by exogenous cells may be a strategy to overcome composite vasculogenesis of the host- and donor-derived cells (Hamanaka et al., 2018). In this study, we, therefore, examined whether deficiency of the FLK1 ortholog or kinase insert domain receptor (KDR) in pigs can cause the vasculogenesis-disabled phenotype as seen in rodents (Sakurai et al., 2005, Shalaby et al., 1995) and whether the trait can be restored by blastocyst complementation.
Additional KDR deficiency was introduced into the PDX1-KO fetal fibroblast cells (#45) using TALEN targeting the exon 1 of the gene (Figures S2A and S2B). The PDX1/KDR dual KO cells (male) were used for SCNT to produce cloned fetuses (Figure S2C). Fetuses at the somite stage (days 15–21) showed distinctly retarded development lacking vasculogenesis and blood flow at all embryonic stages observed (Figures 2A and 3A).
Figure 2.

Vasculogenesis-Disabled Phenotype of KDR−/– Fetus and Its Compensation by Blastocyst Complementation
(A) A PDX1/KDR dual KO (DKO) fetus with vasculogenesis-disabled phenotype (left) and a chimeric fetus with normalized trait (right) obtained after blastocyst complementation.
(B) Left: a PDX1/KDR-KO fetus showing retarded development without blood flow. Center: the appearance of a chimeric fetus showing systemic chimerism with dense distribution of the exogenous huKO-expressing cells. Right: morphology and fluorescence intensity of a normal fetus derived from the huKO transgenic cell. Scale bars, 1 mm.
See also Figure S2.
Figure 3.

Morphological and Histological Features of the PDX1/KDR Dual KO Cloned Fetuses and Its Compensation by Blastocyst Complementation
(A) Immunohistochemical analysis of PDX1/KDR dual KO fetuses and chimeric fetuses with compensated vasculogenesis by blastocyst complementation. Scale bars, 50 μm. Left panels show H&E-stained sagittal section of day 21 fetuses. Top, a PDX1/KDR dual KO (DKO); middle, a chimera obtained by blastocyst complementation; bottom, a non-chimeric fetus derived from the huKO-expressing donor blastomeres. Signals of KDR, PECAM1, and CD34 can be seen (DAB stained) on the vascular endothelial cells of the chimera and donor cell (huKO) derived fetuses. On the other hand, neither vascular structure nor endothelial markers were observed in the tissue of the PDX1/KDR dual KO fetus.
(B) Upper panels: a full-term fetus proven to be chimeric by its phenotypic sex (male) accompanying huKO expression and its restored pancreas entirely expressed huKO fluorescence, indicating that it was generated from the exogenous cells as a result of complementation. Insets show bright-field pictures. Lower panels: the restored pancreatic tissue included well-developed islets stained with α and β cell markers. Scale bars, 100 μm.
(C and D) (C) PECAM1-positive (green) endothelial tissue of a splenic blood vessel in the chimeric fetus exhibiting the donor-cell-derived huKO signal (red). (D) Spleen tissue of the chimeric fetus exhibiting double-positive signals of hematopoietic cell marker (CD45, green) and the donor-cell-derived huKO (red). (C and D) A pair of mirror-image sections were stained due to difficulty of double immunohistochemical staining (lower panels, flipped picture). Scale bars, 10 μm (C) and 50 μm (D).
Blastocyst complementation was performed using the PDX1/KDR-KO cloned morulae as the host embryos and in-vivo-derived morulae expressing huKO as the blastomere donors. Transfer of 38 complemented blastocysts to a recipient gilt resulted in 27 fetuses (day 21), including 9 chimeric and 18 non-chimeric fetuses (Figure 2B; Table S1). Among the nine chimeric fetuses, five showed normal development, including vasculogenesis and blood flow similar to the normal cloned fetuses (Figures 2 and 3A). Four other chimeric fetuses exhibited severe retardation of development, which was typically observed in the PDX1/KDR-KO fetuses. Vasculogenesis was scarce in these chimeras, indicating that contribution of normal exogenous cells was limited in their embryo/fetal development.
Then, we examined the full-term development of the chimeric fetuses generated by complementation of the PDX1/KDR-KO embryos. The PDX1/KDR-KO host embryos were complemented by blastomeres of in-vivo-derived or IVF (in vitro fertilization)-derived morulae expressing huKO. Transfer of 92 complemented blastocysts to two recipients (one miscarried) gave rise to four full-term fetuses (day 106), including a chimera (Figure 3B). This chimeric fetus showed well-developed pancreas, including islets identified by endocrine markers (Figure 3B). Immunohistochemical analysis revealed that endothelial tissue and hematopoietic cells had derived from the donor cells (Figures 3C and 3D). These results demonstrated that the dual organogenesis-disabled phenotype could be simultaneously rescued by blastocyst complementation.
Anephrogenic Phenotype Generated by Spalt-like Transcription Factor 1-KO and Restoration of Normal Nephrogenesis by Blastocyst Complementation
Homozygous KO of spalt-like transcription factor 1 (Sall1) in rodents was reported in a previous study to induce a nephrogenesis-disabled phenotype (Goto et al., 2019, Usui et al., 2012). In this study, we investigated whether SALL1-KO in pigs also results in the anephrogenic phenotype and whether blastocyst complementation is effective in restoring the deficient trait.
Two types of porcine fetal fibroblasts harboring biallelic KO mutations in exon 3 of the SALL1 gene were established using genome editing by ZFN (Figures S3A and S3B). Using these cells for SCNT, we obtained five cloned fetuses at day 43 to 66 (Figure S3C). Figure 4A shows a typical SALL1-KO fetus exhibiting impaired nephrogenesis (day 43) accompanying normally developed ureters and urinary bladder. The size of the vestigial kidney tissue varied among the fetuses with the same mutation type (Figure S4A). Histological development of the vestigial SALL−/– kidney was severely impaired with structural variations (Figure 4B). Renal tissues of the SALL−/– fetuses mainly comprised of interstitial tissues lacking organized structures. Immunohistochemical analysis detected no SALL1 expression in these tissues (Figure 4B). On the other hand, ureters and bladders of the SALL−/– fetuses exhibited no morphological and histological abnormalities. The most developed case we studied was the anephrogenic phenotype in a fetus at day 66. This fetus, however, appeared to be in the process of being aborted, suggesting lethality of the SALL1−/− mutation after the mid-gestation stage.
Figure 4.

Anephrogenic Phenotype of SALL1−/− Fetus and Its Compensation by Blastocyst Complementation
(A) Anephrogenic phenotype of a SALL1−/− cloned fetus at mid-gestation (day 43). WT, kidney of age-matched control.
(B) Histological features of the vestigial kidney tissues. Upper panels indicate phenotypic variation in SALL1−/− fetuses. Immunostaining of the SALL1−/− vestigial kidney (lower right) detected no SALL1 expression. Lower left: SALL1-positive WT tissue. Scale bars, 200 μm.
(C) Normally developed kidneys of a chimeric fetus (day 43, left) obtained by blastocyst complementation. Note that the bladder was torn off from the ureter at the time of excising. huKO-tg, normally formed kidneys of a control fetus expressing huKO fluorescence. SALL1-KO, deficient renal development of a SALL1−/− fetus. Scale bars, 5 mm.
(D) Histological features of the kidney tissues (upper panels) from a chimeric fetus produced by blastocyst complementation. Fluorescence expression shown in the upper right panel indicates that renal development in the chimeric fetus is entirely compensated by exogenous cells by blastocyst complementation. Lower panels: renal tissues of a control WT fetus. Scale bars, 100 μm.
See also Figure S4.
We established primary cultured fibroblasts from the cloned mutant fetuses with anephrogenic phenotype and used them as nuclear donors to produce host embryos for blastocyst complementation. The host embryos at the morula stage were complemented with blastomeres of cloned embryos expressing huKO. Transfer of 291 complemented blastocysts to four recipients (three impregnated) resulted in two chimeric and five non-chimeric fetuses (Table S1). Both chimeric fetuses retained impaired kidney development similar to the cloned SALL1-KO fetuses (Figure S4B). Distribution of huKO-expressing cells in the hypoplastic kidney tissue was limited (Figures S4B and S4C), indicating that contribution of exogenous cells in the development of these chimeric fetuses was not enough, hence resulting in the incomplete restoration of nephrogenesis of the SALL1-KO embryos. A limited number of the huKO-positive cells were seen in the ureter as well (Figure S4B).
In a reattempt at blastocyst complementation of the SALL1-KO cloned embryos, we used donor blastomeres prepared from in-vivo-derived or IVF-derived embryos, instead of the cloned embryos (Table S1). Transfer of 97 complemented blastocysts to two recipients (all impregnated) gave rise to 12 fetuses (Table S1), including a chimeric fetus (day 43) that exhibited normally developed kidneys (Figure 4C). The restored kidney tissue was histologically normal and entirely positive for huKO expression, confirming the role of exogenous cells in nephrogenesis (Figure 4D).
Ahepatogenic Phenotype Generated by Hematopoietically Expressed Homeobox-KO and Restoration of Normal Hepatogenesis by Blastocyst Complementation
Hex deficiency in rodents is known to cause a hepatogenesis-disabled phenotype (Barbera et al., 2000). The aim of the following experiment was to investigate whether hematopoietically expressed homeobox (HHEX)-KO in pig also results in an ahepatogenic phenotype and whether blastocyst complementation is effective in restoring the deficient trait. Porcine fetal fibroblasts (PFFs) carrying biallelic KO mutations in exon 1 of the HHEX gene were established by TALEN (Figures S5A and S5B). Using these cells as nuclear donors for SCNT, cloned fetuses (day 22) were obtained, which exhibited severe developmental retardation, including liver dysplasia (Figures 5A, 5B, and S5C). Varying morphological abnormalities were significant in the anterior portion of the mutant fetuses as previously described for the Hex-KO mouse (Barbera et al., 2000).
Figure 5.

Ahepatogenic Phenotype of HHEX–/– Fetus and Its Compensation by Blastocyst Complementation
(A) An HHEX–/– fetus with a hepatogenesis-disabled phenotype (left) and a fetus with normalized trait obtained after blastocyst complementation (middle). Scale bars, 5 mm.
(B) Transverse section of a HHEX–/– fetus (left) with a nearly empty liver capsule-like structure (arrowhead), indicating failure of liver development. A chimeric fetus obtained by blastocyst complementation (center) exhibited normally developed liver as seen in a WT fetus (right). Inset pictures of fetuses indicate directions of the transverse sections (dotted line). Sagittal sections of the HHEX–/– (left) and WT (right) fetuses. Scale bars, 500 μm.
(C) A chimeric (HHEX–/– ↔ huKO) full-term fetus with normal hepatogenesis obtained by blastocyst complementation, showing fluorescence derived from the donor cells.
(D) Morphologically normal liver developed in the chimeric fetus.
(E) Histological features of the liver developed from the exogenous cells. Scale bars, 100 μm.
HHEX-KO cloned embryos with ahepatogenic trait were used as the host embryos for blastocyst complementation. Host embryos at the morula stage were complemented using blastomeres from the in-vivo-derived morulae. As a result of transferring 37 complemented blastocysts to a recipient gilt, 15 fetuses (day 23), including 4 chimeric and 11 non-chimeric ones were produced (Table S1). Among the four chimeric fetuses, one exhibited organogenesis, including a normal liver (Figures 5A and 5B). Anatomical features of the restored liver were similar to an age-matched wild-type (WT) fetus. The other three chimeric fetuses showed severe developmental retardation as seen in the HHEX-KO fetuses, indicating limited contribution of the exogenous cells to embryogenesis thereby failure in restoring dysplastic hepatogenesis.
We then confirmed the full-term development of the fetuses with complemented hepatogenesis. We produced three chimeric fetuses after the transfer of 95 complemented blastocysts (Figures 5C; Table S1). The chimeric fetuses obtained were all alive at the time of the cesarean section and development of the liver was normal in all the animals (Figures 5D and 5E).
Discussion
To overcome organ shortage in organ transplantation therapy, we have advocated a concept of generating human organs from PSCs by organogenesis in xenogenic animals. This concept consists of two fundamental technologies, i.e., induction of an empty developmental niche in the organogenesis of pig fetus and its compensation by exogenous iPSCs through blastocyst complementation. In this study, we proved that disruption of a gene playing a pivotal role in organogenesis could give rise to an organogenesis-disabled trait in pigs not only for the pancreas, but also for the kidney, liver, and blood vessels. We also demonstrated the applicability of the blastocyst complementation technique in an allogenic setting for compensating the dysorganogenetic phenotypes of the pigs.
We revealed, in this study, that knocking out porcine PDX1 gave rise to an apancreatic phenotype as has been shown in rodents (Jonsson et al., 1994, Kobayashi et al., 2010, Offield et al., 1996). In our previous study (Matsunari et al., 2013), vestigial pancreatic tissue remained in the phenotype induced by Pdx1-Hes1 overexpression. The results of our study, as well as that of Kang et al. (2017), showed that the PDX1-KO in pig gave rise to a complete apancreatic phenotype while permitting the mutant fetus to develop to full term. To our knowledge, this is the first report demonstrating successful blastocyst complementation in pigs with PDX1-KO-induced complete pancreatogenesis-disabled phenotype (Kang et al., 2017).
Blastocyst complementation of the PDX1-KO trait, however, could occasionally result in incomplete restoration of the missing pancreata even in chimeric piglets exhibiting overt coat color chimerism. Pancreatic progenitor cells expressing PDX1 appear in the foregut of the somite stage fetuses (Habener et al., 2005). Dense contribution of exogenous cells in the pancreatic precursor region of the chimeric fetus was probably crucial in determining the fate of pancreatogenesis.
To accomplish successful clinical transplantation of a pig-derived human organ, the presence of porcine tissues, such as blood vessels need to be avoided to prevent xenorejections. We demonstrated for the first time in pigs that generation of host-derived blood vessels could also be replaced by exogenous cells in the KDR-KO host embryos for blastocyst complementation. In this study, we additionally knocked out the KDR gene of the PDX1-KO fetal fibroblast cells. This appears to be a feasible option to add KDR-KO to a preexisting cell line carrying other dysorganogenetic mutations.
Flk-1 null mice have been reported to die between embryonic day E8.5 and E9.5 without any organized blood vessels or yolk sac blood islands (Sakurai et al., 2005, Shalaby et al., 1995). KDR-KO in pigs was also found to be lethal to the embryo at the somite stage around E15 to E17, which corresponds to E8.5/E9.5 in mice (Noden and de Lahunta, 1985). Thus KDR-KO in pigs appeared to become lethal at nearly the same embryonic stage as in the Flk-1−/− mice.
Fetuses with restored vasculogenesis generated by complementation of the PDX1/KDR-KO embryos were chimeric with a systemically higher contribution of exogenous cells. KDR expression is pivotal for the development of multiple tissues that are distributed throughout the whole body, including vascular endothelia, smooth muscles, and hematopoietic cells (Hamanaka et al., 2018, Sakurai et al., 2005, Shalaby et al., 1995). Therefore, dense distribution of exogenous cells throughout the body tissue of the chimeric fetuses may be hardly avoidable when the empty niche of KDR deficiency is to be compensated.
Systemic chimerism is also an emerging challenge in compensation of a gene deficiency, such as HHEX-KO, which is involved in the dysorganogenesis of multiple organs. HHEX plays a pivotal role not only in hepatogenesis, but also in the generation of the thyroid and forebrain (Barbera et al., 2000, Bort et al., 2006, Keng et al., 2000). In the conception of interspecies blastocyst complementation between pigs and humans, the incidence of disordered chimerism involving exogenous human cells needs to be avoided. Developing measures to restrict chimerism (Kobayashi et al., 2015) in the target organ/tissue will be increasingly important.
Deletion of Sall1 in mice has been reported to cause depletion of nephron progenitors in developing kidney (Nishinakamura et al., 2001), thereby resulting in renal agenesis or hypoplasia. Our results demonstrated that SALL1−/− in pigs also gave rise to the anephrogenic phenotype. We also demonstrated for the first time in pigs that the anephrogenic phenotype of SALL1-KO could be compensated by allogenic blastocyst complementation, as shown in the previous rodent study (Usui et al., 2012).
Our data suggested that the degree of chimerism appeared to be critical for successful compensation of the anephrogenic phenotype. In the chimeric fetuses in which compensation of the SALL1−/− anephrogenic phenotype was incomplete, distribution of the donor blastomere-derived cells appeared less in the nephrogenic tissue. Fetal kidneys develop through mutual inductive interaction between the metanephric mesenchyme and the ureteric bud (Nishinakamura et al., 2001). Minor populations of normal cells in the nephrogenic progenitor tissues might have failed to ensure nephrogenesis in the SALL1−/− ↔ WT chimeras.
During blastocyst complementation for the anephrogenic phenotype in Sall1−/− mice, some of the nephric tissues, such as collecting duct and microvascular endothelium, were not entirely replaced by exogenic cells (Usui et al., 2012). These results would imply coexistence of the host-derived cells in the kidney tissue that is generated by interspecies blastocyst complementation. Although residual cells/tissues in the SALL1−/− porcine kidney are yet to be examined, additional genetic modifications to the SALL1−/− trait would enable high-level elimination of the pig-derived cells (Yamanaka et al., 2017).
Autopsy of four recipient females that conceived the SALL1−/− fetuses yielded no anephrogenic fetus that had grown beyond the mid-gestation stage (Figure S3). We conducted a separate study in which the SALL1+/− males and females produced by TALEN-mediated genome editing of zygotes were mated with each other. We obtained compelling data indicating lethality of the SALL1−/− fetuses around the mid-gestation stage (Watanabe et al., 2019). Therefore, it is unlikely that the lethality of the SALL1−/− fetuses obtained in this study is ascribed to the abnormality of the cloned fetuses. Although involvement of off-target mutations in the SALL1−/− fetuses cannot be ruled out, possible influence of SALL1-KO in multiple organs/tissues, including brain, limb buds, and heart (Kohlhase et al., 1996, Usui et al., 2012), is more likely the cause of fetal lethality in pigs. Usui et al. (2012) ascribed neonatal lethality of the Sall1-KO mice to a defect in the nervous system causing abnormal suckling function. The reasons for the lethality of the SALL1−/− pig fetuses are yet to be investigated.
In our previous study, we obtained a proof-of-concept (POC) for blastocyst complementation that compensates for the apancreatic phenotype of the porcine Pdx1-Hes1 transgenic embryos. In this study, we demonstrated that the POC could be extended to a wider range of dysorganogenetic phenotypes induced by an alternative genetic modification approach, i.e., genome editing. Although factors hindering interspecies chimerism between humans and pigs is yet to be clarified, blastocyst complementation has been proposed as a means of compensating the empty developmental niche of organogenesis by exogenous-xenogenic pluripotent cells. Strategies that allow the organogenic region of the target organ in a pig fetus to be highly chimeric with the xenogenic cells will be a crucial part of future research. Attempts using chimera-competent porcine embryonic stem cells (Gao et al., 2019) would secure a bridgehead into human-pig blastocyst complementation. At the same time, developing a strategy for cell fate control is also crucial to ensure compensation of the empty developmental niche, while avoiding unwanted systemic chimerism.
Experimental Procedures
Animal Care and Chemicals
All animal experiments in this study were approved by the Institutional Animal Care and Use Committee of Meiji University (IACUC07-0006, 11-0016, 12-0008, 16-0006, 17-0005). All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Design and Preparation of Genome Editing Tools
Custom TALEN plasmids targeted to porcine PDX1, KDR, and HHEX genes, and ZFN plasmids targeted to porcine SALL1 gene, were obtained from ToolGen (Seoul, South Korea) and Sigma-Aldrich, respectively. The design and validation of these TALENs and ZFNs were performed by the manufacturers. For the production of mRNAs encoding TALENs and ZFNs, each of the linearized plasmids were used as templates for in vitro transcription. The capped and poly(A)-tailed TALEN and ZFN mRNAs were synthesized using a mMESSAGE mMACHINE T7 Ultra Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions. The TALEN- and ZFN-encoding mRNAs were purified using a MEGAclear Kit (Thermo Fisher Scientific) and suspended in RNase-free water. These genome-editing molecules were stored at −80°C until use.
Establishment of Gene-KO Cells and Culture Conditions
A primary culture of male PFF cells that was used to establish gene-KO cells was cultured in Minimum Essential Media α (Thermo Fisher Scientific) supplemented with 15% fetal bovine serum (FBS) and 1% antibiotic-antimycotic solution (Thermo Fisher Scientific) in type-I collagen-coated dishes or plates (AGC Techno Glass, Shizuoka, Japan), under a humidified atmosphere containing 5% CO2 at 37°C. For electroporation, PFF cells were cultured to 70%–90% confluence, washed twice with Dulbecco’s PBS (D-PBS(−)) and detached with 0.05% trypsin-EDTA. These cells (5 × 105) were suspended in 50 μL buffer R (included in the Neon Transfection System, Thermo Fisher Scientific) containing 1 μg TALEN or ZFN mRNAs. The cells were then electroporated with a single DC pulse (1,100 V, 30 ms, pulse number 1; program no. 6 for PDX1-, KDR-, and SALL1-KO, and 1,600 V, 20 ms, pulse number 1; program no. 4 for HHEX-KO), followed by culture at 32°C for 3 days, without antibiotics for the first 24 h and with antibiotics thereafter. Next, the cells were cultured at 37°C until they reached 90% confluence, after which limiting dilution was performed to obtain single-cell-derived clones in five 96-well plates. Twelve to 14 days later, colonies at high confluence (>70%) were selected and divided for subculture and mutation analyses.
Analysis of Mutations in Nuclear Donor Cells and Fetuses
For mutation analysis, the target region of each genome-editing tool was amplified via direct PCR from cell clones using MightyAmp DNA polymerase version 2 (Takara Bio, Shiga, Japan) and then nested PCR was performed using appropriate primers (Table S2). The sequence of the amplicons containing the target region was determined using sequencing primers (Table S2) and the BigDye Terminator Cycle Sequencing Kit (Thermo Fisher Scientific), on an ABI PRISM 3130xl Genetic Analyzer (Thermo Fisher Scientific). For some cell clones, the nested PCR products were cloned and sequenced using a Zero Blunt TOPO PCR Cloning Kit for Sequencing (Thermo Fisher Scientific).
Production of Gene-KO Pig Fetuses by Somatic Cell Nuclear Transfer
SCNT of gene-KO cells was performed as described previously (Kurome et al., 2008, Matsunari et al., 2013), with slight modifications. In brief, nuclear donor cells were used for SCNT following cell-cycle synchronization through serum starvation for 2 days. A single donor cell was electrically fused with each enucleated cytoplast prepared from in-vitro-matured oocytes. The reconstructed embryos were electrically activated and cultured in porcine zygote medium-5 ([PZM-5], Research Institute for Functional Peptides, Yamagata, Japan) (Yoshioka et al., 2008) for 3 h in the presence of 5 μg/mL cytochalasin B and 500 nM scriptaid (Zhao et al., 2009), and embryos were then cultured with 500 nM scriptaid for another 12–15 h. After these treatments, the cloned embryos were cultured in PZM-5 under a humidified atmosphere of 5% CO2, 5% O2, and 90% N2 at 38.5°C. After reaching the morula stage (day 4), the embryos were cultured in PZM-5 supplemented with 10% FBS. Cloned embryos at days 1–2 or days 5–6 were surgically transferred to the oviducts or uterine horns, respectively, of estrus-synchronized recipients. SCNT embryos at the morula stage were used to generate chimeric embryos.
Production of Chimeric Pig Fetuses by Blastocyst Complementation
Lines of mutant fibroblast cells carrying the PDX1-KO, PDX1/KDR-KO, SALL1-KO, and HHEX-KO traits were employed for nuclear transfer to produce cloned host embryos. Cloned donor embryos were produced through nuclear transfer using female fibroblasts isolated from a pig carrying the huKO (Matsunari et al., 2008) or Plum (Watanabe et al., 2015) transgenes. In some experiments, the donor blastomeres carrying huKO transgene were prepared from in-vivo-derived or IVF-derived morulae that had been cryopreserved previously (Maehara et al., 2012). WT embryos were also used for complementing the HHEX-KO embryos.
Donor embryos at the morula stage (day 4) were de-compacted with 0.1 mM EDTA-2Na (in Ca2+/Mg2+-free PBS supplemented with 0.01% polyvinyl alcohol) for 20 min, followed by the removal of the zonae pellucidae via digestion with a 0.25% pronase solution (in PBS(−)). Blastomeres were isolated from the embryos by gentle pipetting using a finely drawn glass capillary. Host embryos at the morula stage were similarly de-compacted. Six to eight donor blastomeres were injected into the center of each host embryo through micromanipulation. The injected embryos were cultured in vitro for 24 or 48 h to obtain chimeric blastocysts. Developing blastocysts were surgically transferred to the uteri of estrus-synchronized recipients on day 5 or 6.
Detection of Chimerism and Genotyping of Fetuses Using PCR
Genomic DNA was extracted from biopsy specimens from the fetuses employing a DNA extraction kit (DNeasy Blood & Tissue Kit, QIAGEN, Hilden, Germany). All chimeras were identified by the detection of host and donor embryo-derived sequences by PCR analysis using appropriate primers (Table S2) and DNA sequence analysis.
In brief, to identify PDX1-KO ↔ huKO (or Plum) chimeras, nested PCR was conducted to detect sequences derived from the host embryos (PDX1-KO) and donor embryos (huKO or Plum), respectively.
To identify PDX1/KDR-KO ↔ huKO chimeras, KDR-KO mutant sequences (host embryo derived), were amplified by PCR, cloned, and sequenced as described above, in addition to the detection of the PDX1-KO mutant and huKO transgene sequences (Table S2). For some chimeric fetuses, allele-specific PCR using Hi-Di DNA polymerase (myPOLS Biotec, Konstanz, Germany) was performed using allele-specific primers, which allowed to amplify the KDR-KO allele (host embryo derived) efficiently.
To identify SALL1-KO ↔ huKO chimeras, nested PCR was performed to detect host embryo-derived (SALL1-KO) cells. To detect huKO transgene sequence (donor embryo-derived) and amelogenin (AMEL X and Y for sex determination) sequences, nested PCR was performed as described previously (Matsunari et al., 2018).
To identify HHEX-KO ↔ WT (or huKO) chimeras, DNA sequence analyses to detect both WT and HHEX sequences (donor embryo derived) and HHEX-KO mutant sequence (host embryo derived) were conducted. The target regions in the HHEX gene were amplified by PCR, cloned, and sequenced, as described above (Table S2). Nested PCR was conducted to detect huKO (donor embryo derived) and amelogenin sequences.
Histological Analyses
Histological examination of mutated and complemented organogenesis was performed on tissue samples from the PDX1-KO, PDX1/KDR-KO, SALL1-KO, HHEX-KO, and chimeric fetuses. Control tissues were obtained from age-matched male normal (WT) fetuses. Tissue samples were fixed for 24 h in a neutrally buffered paraformaldehyde solution (4%) and embedded in paraffin blocks. The sections were stained with H&E or analyzed immunohistochemically using secondary antibodies conjugated with horseradish peroxidase (DAB stain) or fluorescent labels.
The primary antibodies used in this study were anti-insulin (no. LS-C24686, LifeSpan BioSciences, WA), anti-glucagon (no. G2654), and anti-VEGFR2 (no. 2479, Cell Signaling Technology, MA), anti-PECAM-1 (no. sc-1506, Santa Cruz Biotechnology, TX), anti-CD34 (no. 250591, Abbiotec, CA), anti-CD45 (no. 60287-1-Ig, Proteintech, IL), anti-SALL1 (no. PP-K9814-00, Perseus Proteomics, Tokyo, Japan), anti-albumin (no. A100-110A, Bethyl Laboratories, TX), and anti-huKO (no. PM051M, Medical & Biological Laboratories, Nagoya, Japan). The isotype-specific secondary antibodies were anti-guinea pig IgG coupled to Alexa Fluor 488 (no. ab150185, Abcam, Cambridge, UK) and anti-rabbit IgG coupled to Alexa Fluor 594 (no. A21207, Thermo Fisher Scientific). After antibody treatment, the sections were mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA) containing DAPI for nuclear counterstaining and observed by fluorescence microscopy (BZ-X710; Keyence, Osaka, Japan).
Author Contributions
H. Nagashima designed the overall study. H. Nagashima, H. Matsunari, and M.W. wrote the manuscript with contributions from H. Nakauchi, R.N., H. Masaki, S.H., T.Y., and M.N. H. Matsunari, and H. Nagashima performed the experiments for somatic cell cloning with technical help from K.H., A.U., and K.N. M.W. and K.U. generated gene knockout cells and performed genetic and biochemical analysis of samples.
Acknowledgments
We thank H. Kadoi for help with the maintenance of pigs and S. Fujimura, T. Ohmori, and S. Takayanagi for experimental/technical assistance. This work was supported by grants from the Japan Science and Technology Agency (ERATO, Nakauchi Stem Cell and Organ Regeneration Project), the Japan Agency for Medical Research and Development (LEAP, Generation of Functional Organs using developmental niche, JP19gm0010002), and Meiji University International Institute for Bio-Resource Research. H.Nagashima is a co-founder and shareholder of ChimaERA Corporation and PorMedTec Co., Ltd. H.Nakauchi. is a co-founder and shareholder of iCELL Inc., ChimaERA Corporation, and ReproCELL Inc.
Published: December 26, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.11.008.
Supplemental Information
References
- Barbera J.P.M., Clements M., Thomas P., Rodriguez T., Meloy D., Kioussis D., Beddington R.S.P. The homeobox gene Hex is required in definitive endodermal tissues for normal forebrain, liver and thyroid formation. Development. 2000;127:2433–2445. doi: 10.1242/dev.127.11.2433. [DOI] [PubMed] [Google Scholar]
- Bort R., Signore M., Tremblay K., Barbera J.P.M., Zaret K.S. Hex homeobox gene controls the transition of the endoderm to a pseudostratified, cell emergent epithelium for liver bud development. Dev. Biol. 2006;290:44–56. doi: 10.1016/j.ydbio.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Gao X., Nowak-Imialek M., Chen X., Hermann D., Ruan D., Chen D.C.H., Eckersley-Maslin M.A., Ahmad S., Lee Y.L., Kobayashi T. Establishment of porcine and human expanded potential stem cells. Nat. Cell Biol. 2019;21:687–699. doi: 10.1038/s41556-019-0333-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto T., Hara H., Sanbo M., Masaki H., Sato H., Yamaguchi T., Hochi S., Kobayashi T., Nakauchi H., Hirabayashi M. Generation of pluripotent stem cell-derived mouse kidneys in Sall1-targeted anephric rats. Nat. Commun. 2019;10:451. doi: 10.1038/s41467-019-08394-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.M., Musunuru K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Invest. 2014;124:4154–4161. doi: 10.1172/JCI72992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habener J.F., Kemp D.M., Thomas M.K. Minireview: transcriptional regulation in pancreatic development. Endocrinology. 2005;146:1025–1034. doi: 10.1210/en.2004-1576. [DOI] [PubMed] [Google Scholar]
- Hamanaka S., Umino A., Sato H., Hayama T., Yanagida A., Mizuno N., Kobayashi T., Kasai M., Suchy F.P., Yamazaki S. Generation of vascular endothelial cells and hematopoietic cells by blastocyst complementation. Stem Cell Reports. 2018;11:988–997. doi: 10.1016/j.stemcr.2018.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson J., Carlsson L., Edlund T., Edlund H. Insulin-promoter-factor-1 is required for pancreas development in mice. Nature. 1994;371:606–609. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- Jung J.P., Bhuiyan D.B., Ogle B.M. Solid organ fabrication: comparison of decellularization to 3D bioprinting. Biomater. Res. 2016;20:27. doi: 10.1186/s40824-016-0074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J.D., Kim H., Jin L., Guo Q., Cui C.D., Li W.X., Kim S., Kim J.S., Yin X.J. Apancreatic pigs cloned using Pdx1-disrupted fibroblasts created via TALEN-mediated mutagenesis. Oncotarget. 2017;8:115480–115489. doi: 10.18632/oncotarget.23301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng V.W., Yagi H., Ikawa M., Nagano T., Myint Z., Yamada K., Tanaka T., Sato A., Muramatsu I., Okabe M. Homeobox gene Hex is essential for onset of mouse embryonic liver development and differentiation of the monocyte lineage. Biochem. Biophys. Res. Commun. 2000;279:739. doi: 10.1006/bbrc.2000.3548. [DOI] [PubMed] [Google Scholar]
- Kobayashi T., Kato-Itoh M., Nakauchi H. Targeted organ generation using Mixl1-inducible mouse pluripotent stem cells in blastocyst complementation. Stem Cells Dev. 2015;24:182–189. doi: 10.1089/scd.2014.0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T., Yamaguchi T., Hamanaka S., Kato-Itoh M., Yamazaki Y., Ibata M., Sato H., Lee Y.S., Usui J., Knisely A.S. Generation of rat pancreas in mouse by interspecific blastocyst injection of pluripotent stem cells. Cell. 2010;142:787–799. doi: 10.1016/j.cell.2010.07.039. [DOI] [PubMed] [Google Scholar]
- Kohlhase J., Schuh R., Dowe G., Kühnlein R.P., Jäckle H., Schroeder B., Schulz-Schaeffer W., Kretzschmar H.A., Köhler A., Müller U. Isolation, characterization, and organ-specific expression of two novel human zinc finger genes related to the Drosophila gene spalt. Genomics. 1996;38:291–298. doi: 10.1006/geno.1996.0631. [DOI] [PubMed] [Google Scholar]
- Kurome M., Ishikawa T., Tomii R., Ueno S., Shimada A., Yazawa H., Nagashima H. Production of transgenic and non-transgenic clones in miniature pigs by somatic cell nuclear transfer. J. Reprod. Dev. 2008;54:156–163. doi: 10.1262/jrd.19165. [DOI] [PubMed] [Google Scholar]
- Liu Y., Yang R., He Z., Gao W.Q. Generation of functional organs from stem cells. Cell Renen. (Lond.) 2013;2:1. doi: 10.1186/2045-9769-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T.Y., Lin B., Kim J., Sullivan M., Tobita K., Salama G., Yang L. Repopulation of decellularized mouse heart with human induced pluripotent stem cell-derived cardiovascular progenitor cells. Nat. Commun. 2013;4:2307. doi: 10.1038/ncomms3307. [DOI] [PubMed] [Google Scholar]
- Maehara M., Matsunari H., Honda K., Nakano K., Takeuchi Y., Kanai T., Matsuda T., Matsumura Y., Hagiwara Y., Sasayama N. Hollow fiber vitrification provides a novel method for cryopreserving in vitro maturation/fertilization-derived porcine embryos. Biol. Reprod. 2012;87:133. doi: 10.1095/biolreprod.112.100339. [DOI] [PubMed] [Google Scholar]
- Matsunari H., Nagashima H., Watanabe M., Umeyama K., Nakano K., Nagaya M., Kobayashi T., Yamaguchi T., Sumazaki R., Herzenberg L.A. Blastocyst complementation generates exogenic pancreas in vivo in apancreatic cloned pigs. Proc. Natl. Acad. Sci. U S A. 2013;110:4557–4562. doi: 10.1073/pnas.1222902110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunari H., Onodera M., Tada N., Mochizuki H., Karasawa S., Haruyama E., Nakayama N., Saito H., Ueno S., Kurome M. Transgenic-cloned pigs systemically expressing red fluorescent protein, Kusabira-Orange. Cloning Stem Cells. 2008;10:313–323. doi: 10.1089/clo.2008.0024. [DOI] [PubMed] [Google Scholar]
- Matsunari H., Watanabe M., Nakano K., Enosawa S., Umeyama K., Uchikura A., Yashima S., Fukuda T., Klymiuk N., Kurome M. Modeling lethal X-linked genetic disorders in pigs with ensured fertility. Proc. Natl. Acad. Sci. U S A. 2018;115:708–713. doi: 10.1073/pnas.1715940115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima H., Matsunari H. Growing human organs in pigs––a dream or reality? Theriogenology. 2016;86:422–426. doi: 10.1016/j.theriogenology.2016.04.056. [DOI] [PubMed] [Google Scholar]
- Nishinakamura R., Matsumoto Y., Nakao K., Nakamura K., Sato A., Copeland N.G., Gilbert D.J., Jenkins N.A., Scully S., Lacey D.L. Murine homolog of SALL1 is essential for ureteric bud invasion in kidney development. Development. 2001;128:3105–3115. doi: 10.1242/dev.128.16.3105. [DOI] [PubMed] [Google Scholar]
- Noden D.M., de Lahunta A. Williams & Wilkins; 1985. The Embryology of Domestic Animals: Developmental Mechanisms and Malformations. [Google Scholar]
- Offield M.F., Jetton T.L., Labosky P.A., Ray M., Stein R.W., Magnuson M.A., Hogan B.L., Wright C.V. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122:983–995. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- Rashid T., Kobayashi T., Nakauchi H. Revisiting the flight of Icarus: making human organs from PSCs with large animal chimeras. Cell Stem Cell. 2014;15:406–409. doi: 10.1016/j.stem.2014.09.013. [DOI] [PubMed] [Google Scholar]
- Sakurai Y., Ohgimoto K., Kataoka Y., Yoshida N., Shibuya M. Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc. Natl. Acad. Sci. U S A. 2005;102:1076–1081. doi: 10.1073/pnas.0404984102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby F., Rossant J., Yamaguchi T.P., Gertsenstein M., Wu X.F., Breitman M.L., Schuh A.C. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Taguchi A., Kaku Y., Ohmori T., Sharmin S., Ogawa M., Sasaki H., Nishinakamura R. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell. 2014;14:53–67. doi: 10.1016/j.stem.2013.11.010. [DOI] [PubMed] [Google Scholar]
- Takasato M., Er P.X., Chiu H.S., Maier B., Baillie G.J., Ferguson C., Parton R.G., Wolvetang E.J., Roost M.S., Chuva de Sousa Lopes S.M. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature. 2015;526:564–568. doi: 10.1038/nature15695. [DOI] [PubMed] [Google Scholar]
- Takasato M., Little M.H. A strategy for generating kidney organoids: recapitulating the development in human pluripotent stem cells. Dev. Biol. 2016;420:210–220. doi: 10.1016/j.ydbio.2016.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebe T., Enomura M., Yoshizawa E., Kimura M., Koike H., Ueno Y., Matsuzaki T., Yamazaki T., Toyohara T., Osafune K. Vascularized and complex organ buds from diverse tissues via mesenchymal cell-driven condensation. Cell Stem Cell. 2015;16:556–565. doi: 10.1016/j.stem.2015.03.004. [DOI] [PubMed] [Google Scholar]
- Takebe T., Sekine K., Enomura M., Koike H., Kimura M., Ogaeri T., Zhang R.R., Ueno Y., Zheng Y.W., Koike N. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature. 2013;499:481–484. doi: 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- Tan W.S., Carlson D.F., Walton M.W., Fahrenkrug S.C., Hackett P.B. Precision editing of large animal genomes. Adv. Genet. 2012;80:37–97. doi: 10.1016/B978-0-12-404742-6.00002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui J.I., Kobayashi T., Yamaguchi T., Knisely A.S., Nishinakamura R., Nakauchi H. Generation of kidney from pluripotent stem cells via blastocyst complementation. Am. J. Pathol. 2012;180:2417–2426. doi: 10.1016/j.ajpath.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Vilarino M., Rashid S.T., Suchy F.P., McNabb B.R., van der Meulen T., Fine E.J., Ahsan S., Mursaliyev N., Sebastiano V., Diab S.S. CRISPR/Cas9 microinjection in oocytes disables pancreas development in sheep. Sci. Rep. 2017;7:17472. doi: 10.1038/s41598-017-17805-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M., Kobayashi M., Nagaya M., Matsunari H., Nakano K., Maehara M., Hayashida G., Takayanagi S., Sakai R., Umeyama K. Production of transgenic cloned pigs expressing the far-red fluorescent protein monomeric Plum. J. Reprod. Dev. 2015;61:169–177. doi: 10.1262/jrd.2014-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M., Nakano K., Uchikura A., Matsunari H., Yashima S., Umeyama K., Takayanagi S., Sakuma T., Yamamoto T., Morita S. Anephrogenic phenotype induced by SALL1 gene knockout in pigs. Sci. Rep. 2019;9:8016. doi: 10.1038/s41598-019-44387-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J., Greely H.T., Jaenisch R., Nakauchi H., Rossant J., Belmonte J.C. Stem cells and interspecies chimaeras. Nature. 2016;540:51–59. doi: 10.1038/nature20573. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T., Sato H., Kato-Itoh M., Goto T., Hara H., Sanbo M., Mizuno N., Kobayashi T., Yanagida A., Umino A. Interspecies organogenesis generates autologous functional islets. Nature. 2017;542:191–196. doi: 10.1038/nature21070. [DOI] [PubMed] [Google Scholar]
- Yamanaka S., Tajiri S., Fujimoto T., Matsumoto K., Fukunaga S., Kim B.S., Okano H.J., Yokoo T. Generation of interspecies limited chimeric nephrons using a conditional nephron progenitor cell replacement system. Nat. Commun. 2017;8:1719. doi: 10.1038/s41467-017-01922-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S., Yokoo T. Current bioengineering methods for whole kidney regeneration. Stem Cells Int. 2015;2015:724047. doi: 10.1155/2015/724047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka K., Suzuki C., Onishi A. Defined system for in vitro production of porcine embryos using a single basic medium. J. Reprod. Dev. 2008;54:208–213. doi: 10.1262/jrd.20001. [DOI] [PubMed] [Google Scholar]
- Zhao J.G., Ross J.W., Hao Y.H., Spate L.D., Walters E.M., Samuel M.S., Rieke A., Murphy C.N., Prather R.S. Significant improvement in cloning efficiency of an inbred miniature pig by histone deacetylase inhibitor treatment after somatic cell nuclear transfer. Biol. Reprod. 2009;81:525–530. doi: 10.1095/biolreprod.109.077016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.