

Abstract
Cancer progression is marked by the infiltration of immunosuppressive cells, such as tumor-associated macrophages (TAMs), regulatory T lymphocytes (Tregs), and myeloid-derived suppressor cells (MDSCs). These cells play a key role in abrogating the cytotoxic T lymphocyte-mediated (CTL) immune response, allowing tumor growth to proceed unabated. Furthermore, targeting these immunosuppressive cells through the use of peptides and peptide-based nanomedicine has shown promising results. Here we review the origins and functions of immunosuppressive cells in cancer progression, peptide-based systems used in their targeting, and explore future avenues of research regarding cancer immunotherapy. The success of these studies demonstrates the importance of the tumor immune microenvironment in the propagation of cancer and the potential of peptide-based nanomaterials as immunomodulatory agents.
Keywords: Immunosuppression, Tumor microenvironment, Peptide, Nanoparticle
Graphical abstract
Highlights
-
•
Describe the role of immunosuppressive cells in supporting cancer progression.
-
•
Discuss immunosuppressive cell-targeting through peptide-based approaches.
-
•
Highlight in vivo studies that demonstrate efficacy of peptide-based nanomedicine.
1. Introduction
Cancer is a leading cause of death worldwide, with an estimated 9.6 million deaths in 2018 [1]. The lethality of the disease is in part attributed to the infiltration of immune-derived immunosuppressive cells, such as tumor-associated macrophages (TAMs), regulatory T lymphocytes (Tregs), and myeloid-derived suppressor cells (MDSCs) into the tumor microenvironment (TME). The recruitment of these cells into the TME has been linked to poor patient outcomes in a variety of cancers, such as skin, prostate, lung, and ovarian cancer [[2], [3], [4], [5], [6], [7], [8], [9], [10]]. These cells support cancer progression by suppressing the body's tumoricidal immune response, namely CD8+ cytotoxic T lymphocytes (CTLs), and such immunosuppression is mediated largely through the expression of immune checkpoint molecules, as well as the secretion of anti-inflammatory cytokines [11]. For example, programmed death ligand 1 (PD-L1) and CD80/CD86 are expressed on the surfaces of TAMs and interact with the immune checkpoints programmed death protein 1 (PD-1) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), respectively, present on the surfaces of activated CTLs and impairs their potency and proliferation [12].
1.1. Brief overview of CTLs
CTLs are a major component of the adaptive immune system, along with B lymphocytes. In contrast to the natural killer (NK) cells, macrophages, and dendritic cells (DCs) that comprise the innate immune system, CTLs must first be primed and expanded against a specific antigen before exhibiting cytotoxicity. Tumor-associated antigens (TAAs) are short peptide sequences (~8–11 amino acids) scavenged from cancer cells by antigen-presenting cells (APCs) that then present these peptides on a complex of surface proteins, called the major histocompatibility complex (MHC) [13]. Antigen-loaded MHCs are used to activate naïve CTLs through the T cell receptor (TCR), priming them to react specifically to the presented antigen. The activation of CTLs occurs in a co-stimulatory manner, contingent on the simultaneous activation of the TCR by the MHC, as well as the activation of another surface protein, CD28, by CD80/CD86 also expressed by APCs. Following activation, CTLs undergo clonal expansion in the presence of IL-2 and exhibit cytotoxicity towards antigen-expressing cells. The cytotoxic effector functions of CTLs are mediated by the release of cytotoxins such as granzymes, perforin, and granulysin that induce pore formation and membrane lysis in target cells [14]. Additionally, CTLs can directly induce apoptosis through cell-cell contact via the expression of the FAS (FS-7-associated surface antigen) death ligand. Following activation, CTLs exhibit downregulated expression of CD28 and upregulated expression of immune checkpoints such as PD-1 and CTLA-4. Interaction of these checkpoints with their associated ligands (PD-L1 and CD80/CD86, respectively) expressed on macrophages, DCs, Tregs, and cancer cells can halt the immune response by impairing effector function and proliferation of CTLs. As CTLs comprise the bulk of the adaptive immune response, re-stimulation of CTL-mediated tumor immunity has been the major focus of the cancer immunotherapy field.
1.2. Current immunotherapeutic strategies
The field of immunotherapy has been developed in an effort to stimulate the CTLs that are suppressed in cancer. Although the mechanisms behind cancer-driven immunosuppression have only been discovered recently, the link between immune stimulation and therapy has been empirically observed as far back as the nineteenth century. The first study documenting this concept occurred in 1868 by Wilhelm Busch, in which he observed tumor regression following a bacterial infection mediated by Streptococcus pyogenes [15]. Twenty-five years later, William Coley published a report corroborating the efficacy of bacterial infections in treating cancer in 10 different patient cases [16]. A bacterial vaccine including this strain was named “Coley's toxin” in homage of his work and was used for several decades as an anti-cancer remedy [17]. The concept of immunotherapy was set aside in favor of small-molecule cell cycle inhibitors and radiotherapies until the 1990s when advances in immunology identified the crucial role of immune cells in controlling cancer growth [[18], [19], [20]].
The use of monoclonal antibodies as immune checkpoint inhibitors comprises most immunotherapies, with the first successful pre-clinical application reported in 1996 by Allison et al. [21]. Results from a clinical trial utilizing a CTLA-4-targeted monoclonal antibody (ipilimumab) was published in 2010, showing improved survival in patients with metastatic melanoma compared to the standard of care gp100 peptide vaccine (10.1 months vs. 6.4 months) [22]. These results led to ipilimumab gaining FDA-approval the following year under the trade name Yervoy for use in metastatic melanoma [23]. Not long after, the FDA also approved two PD-1 immune checkpoint inhibitors, pembrolizumab/lambrolizumab (Keytruda) and nivolumab (Opdivo), for melanoma, non-small cell lung cancer, and renal cell carcinoma [24,25]. Additionally, an anti-PD-L1 monoclonal antibody, atezolizumab (Tecentriq), was approved in 2016 for use in bladder cancer, and then again in 2019 for small cell lung cancer and triple-negative breast cancer [26]. Notably, the 2018 Nobel Prize in Physiology or Medicine was jointly awarded to Allison and Honjo, researchers who first demonstrated the efficacy of CTLA-4 and PD-1 immunotherapies [27,28].
While immune checkpoint inhibitors function by preventing the premature shut-down of the immune response, other immunotherapies focus on assisting the priming of CTLs to mount a greater immune response. Peptide vaccines have been explored in both pre-clinical models and clinical trials [[29], [30], [31]]. The purpose of peptide vaccines is to synthesize a peptide sequence identical to the TAAs presented on cancer cells and deliver it to CTLs to increase their activation and priming against cancer cells expressing these antigens. This concept can be extrapolated to engineer CTLs in vitro that express chimeric antigen receptors (CARs) that have antigen-binding and T cell-activating moieties (CAR T cells) [32]. CAR T cells are generated by adoptive cell transfer, in which autologous T lymphocytes are taken from the patient, engineered to express CARs, primed against a patient-specific antigen, expanded in vitro, and re-introduced into the patient [33,34].
Although both peptide vaccines and adoptive cell therapies have shown clinical efficacy, they are not without limitations. Both treatment options require the expression of specific TAAs by the cancer cells, but cancer cells can rapidly evolve to downregulate or even eliminate their expression of TAAs [35]. Additionally, peptide vaccines are limited by their weak immunogenicity and instability in vivo, as they are prone to degradation by proteases [36]. Moreover, CAR T cell therapy is hindered by drawbacks inherent to the procedure of adoptive cell therapy, including a limited amount of autologous T cells derived from patients that is necessary for the procedure [37]. Although these immunotherapies have shown clinical efficacy, their drawbacks have pushed researchers to investigate other alternatives.
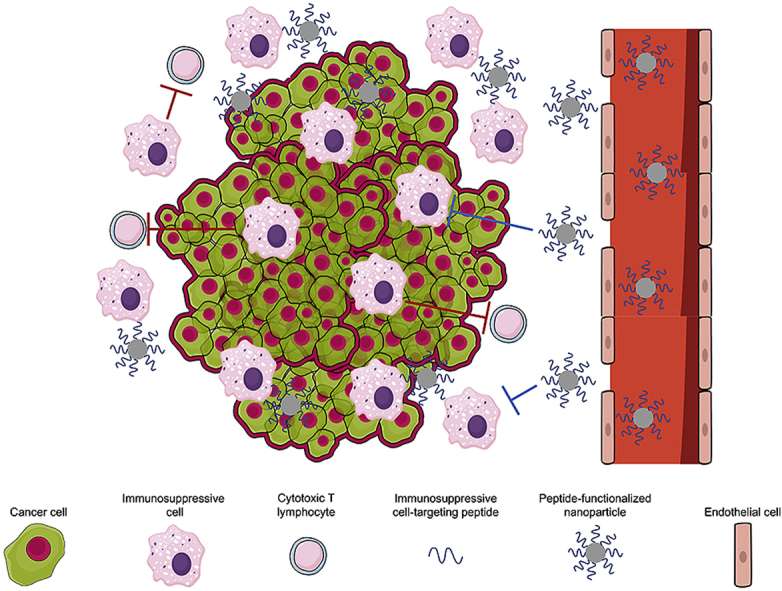
An alternative and promising immunotherapeutic approach is to target and deliver therapeutic agents such as peptides, monoclonal antibodies, and nucleic acid aptamers to immunosuppressive TAMs, Tregs, and MDSCs [[38], [39], [40], [41]]. In particular, peptides are strong candidates for immunotherapy and have been used in a variety of studies targeting immunosuppressive cells, as they possess a number of attractive qualities, such as biocompatibility, cost-efficiency, and versatility as both targeting moieties and therapeutic agents [42,43]. However, peptides are limited by their poor stability in vivo, as they are vulnerable to degradation by proteases present in the serum and tissues. Nanoparticle systems are often used to circumvent this issue, allowing the safe delivery of peptides to target cells. Furthermore, nanoparticles functionalized with peptides exhibiting specificity for immunosuppressive cells have been used to manipulate these small cell populations, even though they sit within a highly heterogeneous microenvironment.
This mini-review details the origins, biomarkers, and functions of immunosuppressive cells pertinent to cancer propagation and then highlights the use of peptides and peptide-functionalized nanoparticles in targeting these cells for immunotherapeutic response. We direct the reader to other reviews that extensively describe general immunotherapy and nanomedicine strategies for immunotherapy [18,34,44,45].
2. Immunosuppressive cells in cancer
Infiltrating immune cells such as M2-like TAMs, Tregs, and MDSCs adopt suppressive roles in cancer, inhibiting CTL-mediated tumor immunity [[46], [47], [48]] (Fig. 1). The endogenous functions of M2 macrophages and Tregs are to halt the immune response once an infection has been dealt with, as well as to prevent autoimmunity. However, in the context of cancer, these cells are affiliated with disease progression by inhibiting the CTL-mediated immune response to the disease. MDSCs are a unique sub-population of cells observed only in pathological scenarios, including cancer. The origins, markers, and roles of TAMs, Tregs, and MDSCs in cancer progression will be further discussed below.
Fig. 1.

Cancer progression is mediated by the infiltration of immunosuppressive cell types into the tumor that aid the developing tumor in avoiding eradication. Adapted and reprinted with permission from the National Cancer Institute.
2.1. Tumor-associated macrophages (TAMs)
Macrophages are immune cells that phagocytose abnormal cells and foreign invaders, as well as present antigens and secrete immunomodulatory cytokines [49]. Monocyte-derived macrophages are recruited from the blood stream as undifferentiated monocytes in accordance to inflammatory cues, such as CCL2 (or monocyte chemoattractant protein-1, MCP-1), CCL3 (or macrophage inflammatory protein-1a, MIP-1a), CXCL12 (or stromal-derived factor-1, SDF-1), and CX3CL1 (or fractalkine) [[50], [51], [52], [53], [54], [55]]. Depending on the immunological landscape into which these monocytes extravasate, they may differentiate into the pro-inflammatory M1 phenotype or anti-inflammatory M2 phenotype macrophage. M1 macrophages are polarized by lipopolysaccharides and Th1 cytokines, such as interferon gamma (IFN-y) and granulocyte-macrophage colony-stimulating factor (GM-CSF) [56]. They are associated with the killing of pathogens and release of pro-inflammatory cytokines (TNF-α, CCL3, IL-6, IL-12), making them tumoricidal [[57], [58], [59]]. M2 macrophages, on the other hand, are activated by Th2 cytokines like IL-4 and IL-13 [60,61] and associated with extracellular matrix remodeling, as well as anti-inflammatory and immunosuppressive cytokine secretion (IL-10, TGF-β) that serves to support tumor growth [62]. As M1 and M2 macrophages serve different functions in the immune regime, discrimination between the two can be done by evaluating metabolic output. For example, the disparity in macrophage function is elegantly demonstrated in the metabolism of the amino acid arginine. M1 macrophages metabolize arginine through nitric oxide synthase to produce nitric oxide, which diffuses through the membranes of target cells and mediates cytotoxicity [63,64]. On the other hand, M2 macrophages have been observed to utilize arginase to convert arginine into ornithine, an amino acid involved in the urea cycle that promotes cell proliferation and tissue repair by stimulating the generation of matrix proteins, such as collagen [65,66]. TAMs typically fall under the M2 classification and play a major role in cancer progression, as they are linked to poor patient outcomes [67]. Studies have shown that infiltration of TAMs into the tumor and surrounding microenvironment is mediated by chemical signaling from cancer cells, which recruit undifferentiated monocytes from the peripheral blood largely through the MCP-1 signaling axis [68]. Cancer-secreted IL-4 and IL-13 then promote the polarization of these infiltrating monocytes towards the M2 phenotype [69].
TAMs have been observed to modulate the TME by actively inhibiting CTL responses (Fig. 2). This is done primarily through the expression of inhibitory ligands on the cell surface, as well as the release of immunosuppressive cytokines that impair CTL expansion and function. TAMs also play a role in the recruitment of immunosuppressive Tregs, whose function and role in contributing to the immunosuppressive microenvironment will be discussed further in later sections. TAMs express many ligands that interact with immune checkpoints displayed on the surfaces of CTLs, resulting in CTL suppression. For example, TAMs express PD-L1, which binds to PD-1 receptors that populate the surfaces of activated CTLs, down-regulating their function and proliferation [70,71]. TAMs also express the CD80 and CD86 ligands, which can be immunostimulatory or immunosuppressive, depending on which receptor they interact with [72]. CD80/CD86 can interact with CD28 for co-stimulation of naive CTLs [73]. However, these ligands have a much stronger affinity for the CTLA-4 on activated CTLs [74,75]. CTLA-4 is expressed at very low levels in naïve CTLs, but its expression becomes up-regulated upon activation/priming of these cells [76,77]. Thus, CTLA-4 can act as a braking mechanism, allowing CD80-and CD86-expressing TAMs that have been attracted to the site by CTL effector activity, to shut down the immune response before it can eliminate the cancer [78]. This dynamic is often exploited to prematurely suppress CTL function before the disease is cleared, as in the case of cancer with the heavy infiltration of CD80+/CD86+ TAMs.
Fig. 2.

TAMs inhibit effector function of T cells through the expression of inhibitor ligands such as PD-L1, B7 (CD80/CD86), and prostaglandins, secretion of T cell-suppressing IL-10 and TGF-β, and metabolic starvation through arginine depletion. Adapted and reprinted with permission from ref. 69.
The promotion of an immunosuppressive microenvironment by TAMs is one of the key reasons they are associated with poor patient prognosis and outcome. These macrophages also perform a number of other tumor-promoting functions, such as angiogenesis induction and extracellular matrix remodeling, but these are outside the scope of this review, and are covered in detail in other studies [[79], [80], [81], [82]].
2.2. Regulatory T cells (Tregs)
Tregs comprise an important sub-type of CD4+ T cells that serve to maintain immunogenic self-tolerance and suppress adaptive immune responses after the clearance of foreign bodies [83]. Disruption of the balance between CTLs and Tregs has been observed in many pathologies, including cancer. In the case of autoimmune diseases such as type I diabetes, multiple sclerosis, and rheumatoid arthritis, the failure of Tregs to suppress CTLs leads to autoimmunity [84]. On the opposite end of the spectrum, hyper-activity of Tregs renders the CTL population incapable of neutralizing pathogens or tumors. It is this upregulation in Treg activity that allows cancers to avoid eradication by the CTL response.
Tregs originate as naïve CD4+ T cells that continue to mature in the thymus until TCR activation and forkhead box P3 (FoxP3) expression promotes a suppressive phenotype [85]. Tregs can be identified by their expression of CD4, CD25, CTLA-4, lymphocyte activation gene 3 (LAG-3), neuropilin-1 (Nrp1), and FoxP3 [[86], [87], [88]]. Tregs are able to suppress activated CTL function directly through contact-dependent inhibition, as well as through the release of regulatory cytokines. Tregs are attracted to sites of inflammation and activated via IL-2, an inflammatory cytokine secreted by active CTLs [89]. Since IL-2 is also necessary for the activation and expansion of CTLs, the high expression of the IL-2 receptor, IL-2R, on Tregs allows them to rapidly deplete the surrounding microenvironment of IL-2, inhibiting CTL activation. Tregs may also inhibit effector function more directly through contact-dependent mechanisms. Nakamura et al. has shown that the expression of transforming growth factor beta-1 (TGF-β1) on the surfaces of Tregs contributes to immunosuppression in a contact-dependent manner, although the biomolecular mechanisms governing this process are yet unknown [90]. Tregs also express LAG3, another immunosuppressive cell surface ligand with an affinity for MHC class II and CD4. Although LAG3 is an inhibitory receptor primarily associated with CTLs, recent studies have shown it to be vital to the immunosuppressive function of Tregs, although the cause for this remains to be elucidated [91]. This phenomenon in which LAG3 is associated with inhibition in one T lymphocyte subset (CTLs) but associated with the activation of another (Tregs), has been observed in other cell-surface markers, such as PD-1 and CTLA-4. Immunosuppressive cytokines secreted by Tregs also play a role in down-regulating the immune response. These regulatory cells have been observed to release high levels of soluble TGF-β1 and IL-10, both of which have negative effects on CTL activity and proliferation. TGF-β1 acts by inhibiting the activation of the TCR complex necessary for CTL expansion [92]. IL-10 operates by inhibiting the phosphorylation of CD28, the surface protein complementary to the TCR in the co-stimulatory pathway of CTL activation [93].
The induction of cancer is oftentimes accompanied by the establishment of an immunosuppressive environment spearheaded by the recruitment and activation of naïve T lymphocytes into Tregs. Similar to TAM recruitment, Tregs follow a chemotactic gradient established by cancer cells, based largely on the CCL22 chemokine [94]. Growth factors like TGF-β and IL-10 stimulate the rapid proliferation of Tregs in the TME [95]. TGF-β plays an additional role by promoting the conversion of non-suppressive CD25− T lymphocytes into the suppressive CD4+, CD25+, FoxP3+ Treg phenotype. The trafficking of Tregs into a TME that stimulates their expansion results in a synergistic cycle in which cancer cells and Tregs promote the other's growth and proliferation, establishing a heavily immunosuppressive environment.
2.3. Myeloid-derived suppressor cells (MDSCs)
MDSCs are immunosuppressive myeloid cells found in TMEs that have been linked to poor patient prognoses [96]. In a healthy individual, MDSCs do not exist. These cells, unique to pathological conditions such as cancer, are derived from myeloid progenitor cells whose differentiation into mature myeloid lineages has been inhibited (Fig. 3) [97]. Normally, immature myeloid cells travel from the bone marrow to peripheral organs, where they quickly mature into macrophages, dendritic cells, or granulocytes. However, in cancer, the differentiation of these immature cells is inhibited by various signaling factors present in the tumor, such as GM-CSF, macrophage colony-stimulating factor (M-CSF), IL-6, IL-10, and TNF-α [98,99]. Furthermore, these factors also induce the activation of immature myeloid cells into an immunosuppressive phenotype. These immature, immunosuppressive myeloid cells are dubbed MDSCs.
Fig. 3.

Cancer cells inhibit the maturation of immature myeloid cells into dendritic cells, macrophages, and granulocytes, and instead promote their activation into MDSCs. Adapted and reprinted with permission from ref. 97.
MDSCs do not have an MDSC-specific biomarker. Rather, they express biomarkers indicative of the myeloid lineage, such as CD11b and CD33, but lack markers of fully differentiated cells [100]. Utilizing these criteria, MDSCs are generally characterized by the CD11b+CD14−CD33+ biomarker profile in humans and CD11b+Gr1+ profile in mice [100]. MDSCs are recruited into the TME via chemokine secretion. In particular, CCL2, CXCL8, and CXCL12 produced by cancer cells have been implicated in the majority of MDSC trafficking [101].
Immunosuppression by MDSCs is mediated by reactive oxygen species (ROS) and cytokine production, as well as arginine depletion. In a study by Corzo et al., increased generation of ROS in MDSCs led to DNA damage and apoptosis in CTLs [102]. ROS also downregulates CTL activity by interfering with their ability to recognize antigens through the TCR. For example, superoxide (O2−) can react with nitric oxide (NO) to form peroxynitrite (ONOO−), a species observed to inhibit CTL activity by inducing apoptosis and inhibiting phosphorylation pathways that govern the proper formation of the TCR [103]. Metabolic starvation of CTLs is another means through which MDSCs can inhibit the immune response. Arginine is an amino acid essential for protein synthesis in and expansion of CTLs [104]. MDSCs express nitric oxide synthase (NOS) and arginase, two major metabolizers of arginine, at high levels, severely lowering the amount of arginine available to CTLs, impairing their ability to proliferate and control cancer growth [105].
In addition, MDSCs are capable recruiters of other immunosuppressive cells. Adoptive cell transfer studies from Huang et al. show that IL-10 and TGF-β by MDSCs are necessary for the induction of Tregs and their associated immunosuppression [106]. Additionally, Sinha et al. co-cultured MDSCs with M1 macrophages and showed polarization toward a tumor-promoting M2 phenotype in a contact-dependent manner [107]. This observation was marked by a reduction in the release of T cell-stimulatory IL-12 by the macrophages. Moreover, their studies revealed that depletion of MDSCs through the use of the chemotherapy drug gemcitabine restored IL-12 production and tumor immunity.
As described above, immunosuppressive cells are not segregated cell populations individually contributing to cancer progression. Rather, they interact with each other and the surrounding environment in a synergistic and interdependent manner. There is much overlap in the biochemical signals, particularly chemokines, that they are reliant on, which makes these signaling molecules potential targets for immunotherapy.
3. Peptides for targeting immunosuppressive cells
Peptides are strong candidates for immunotherapy, as they are capable of binding to and inducing responses from target cells/receptors on a highly specific level. Furthermore, recent advances in peptide chemistry have made their synthesis faster, cost-effective, and more convenient [108]. Additionally, peptides can be incorporated into nanoparticle systems, enhancing their stability in vivo and allowing for their inclusion in multimodal therapies [[53], [54], [55],[109], [110], [111], [112], [113]]. For example, nanoparticles may be functionalized with peptides to facilitate interaction with the cell membrane and mediate endocytosis [114]. Incorporation into nanoparticles also serves to enhance the therapeutic efficacy of peptides by concentrating them into compact nanoparticles [115]. This is generally done through the engraftment of peptide moieties onto a nanomaterial substrate through chemical conjugation. For example, a commonly used conjugation strategy is to create an amide linker between the peptide and nanoparticle substrate by using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) to react the free amines found in the N-termini of peptides with nanoparticle substrates functionalized with carboxylic acid groups [[116], [117], [118], [119]]. As peptides also have free carboxylic acids displayed on their C-terminus, this same strategy can be leveraged to conjugate peptides to amine-terminated nanoparticles [120]. Another means of directly conjugating peptides to nanoparticle substrates is to terminate the peptide sequence with a sulfur-rich cysteine group and react with a maleimide-functionalized nanoparticle [54,55,109,121]. High-affinity, non-covalent interactions may also be used to link peptides to nanoparticle substrates. For example, biotinylated peptides can be used to associate closely to nanoparticles displaying streptavidin moieties [122,123]. Moreover, electrostatic interactions can be tuned to achieve the desired interaction between peptide and nanoparticle [116,[124], [125], [126]] as reported by Blank-Shim et al. who used a strongly positively-charged arginine homo-peptide (isoelectric point of 11.15) to bind to negatively-charged magnetic nanoparticles.
Confirmation of nanoparticle functionalization can be obtained through characterization of nanoparticle properties via dynamic light scattering (DLS) and transmission electron microscopy (TEM) to examine nanoparticle size, morphology, and polydispersity before and after peptide conjugation [[127], [128], [129]]. Additionally, nanoparticle zeta potential is often characterized to examine differences in surface charge conferred by peptide functionalization [130,131]. Shifts in nuclear magnetic resonance (NMR) and circular dichroism (CD) spectra are also used to confirm peptide conjugation, as well as examine peptide secondary structure [109,127,132].
The rest of this mini-review will highlight peptide-focused studies that have been used for cancer immunotherapeutic strategies focused on addressing the immunosuppressive TAMs, Tregs, and MDSCs, and evaluate the outlook for immunotherapeutic peptides and peptide nanoparticles.
3.1. Targeting TAMs
Given the immunosuppressive roles of TAMs and their negative correlation to cancer prognoses, a variety of novel peptides have been developed to target not only the macrophages themselves, but also their monocyte precursors, as well as the biochemical pathways that facilitate their induction and pathological behavior. Pun et al. developed and validated a novel M2 macrophage-targeting peptide, M2pep (YEQDPWGVKWWY), through phage display [133]. The targeting ability of M2pep was determined through injection of Alexa Fluor 660-tagged M2pep into the intraperitoneal cavity of mice. Harvesting of intraperitoneal cavity cells, as well as those from the spleen, show the ability of M2pep to target F4/80+, CD301+, CD11c+ M2 macrophages in a mixed population of cells including B cells, T cells, and neutrophils (Fig. 4a and b). Moreover, M2pep exhibited increased binding compared to the non-targeting (scrambled peptide) control.
Fig. 4.

Targeting of M2-like TAMs by M2pep and scM2pep in the peritoneal cavity (A) and spleen (B). Adapted and reprinted with permission from ref. 133.
A number of other studies have utilized the same M2pep sequence for TAM targeting and observed positive results. For example, M2pep-functionalized poly (lactic-co-glycolic acid) PLGA nanoparticles loaded with CSF-1R (colony-stimulating factor 1 receptor) inhibitors were used to block proliferative and pro-survival signaling in TAMs in a B16F10 murine melanoma model. After tumors became palpable (170 mm3 size), treatment was administered every two days for ten days, showing an approximately 50% decrease in tumor growth rate compared to free inhibitor [134]. In addition, Qian et al. linked the M2pep to apolipoprotein A1-mimetic α-helical peptide (α-peptide) to form α-M2pep [135]. The α-peptide moiety was included to target scavenger receptor B type 1 (SR-B1), a surface receptor highly expressed in TAMs. The addition of phospholipids induced self-assembly into lipid nanoparticles in which a hydrophobic core was surrounded by the two peptide moieties to facilitate interaction with TAMs. The resultant nanoparticle was then loaded with a cholesterol-modified anti–CSF–1R siRNA (siCD115) to interfere with pro-tumor CSF-1 signaling, forming M2NP-siCD115s. In order to evaluate M2 TAM-targeting, M2NPs (no siRNA) loaded with the near-infrared fluorescent dye DiR-BOA (1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbo-cyanine iodide bisoleate), as well as NPs incorporating a scrambled (non-targeting) peptide sequence, were incubated with M2 macrophages for 1 h, observing a 7.5-fold increase in M2 uptake of M2NPs compared to the scrambled control. Additionally, in vivo studies using the B16 murine melanoma model revealed an 87% decrease in tumor size of mice treated with M2NP-siCD115s every 2 days, compared to a saline-treated control.
Phage display experiments performed by Scodeller et al. identified a short peptide sequence, CSPGAKVRC (dubbed “UNO”), capable of targeting the M2 macrophage-specific CD206 surface marker [136]. The targeting capability of UNO was validated through injection of fluorescently labeled UNO into 4T1 tumor-bearing mice. Mice were sacrificed, and their organs were harvested 2 h post-injection. Confocal microscopy of harvested tissues shows high colocalization of UNO and CD206, with 96% of UNO positive cells also being positive for CD206 staining. Additionally, Lee et al. identified melittin (MEL), a 26-amino acid peptide found in the venom of honey bees, as a CD206-targeting sequence [137,138]. MEL is an amphipathic peptide that has been studied as an anti-cancer drug for its ability to induce apoptosis in cancer cells through mitochondrial pore formation, as well as inhibition of angiogenesis via the downregulation of VEGF expression [139,140]. The authors investigated the anti-tumor efficacy of both MEL alone and MEL fused with the cytotoxic (KLAKLAK)2 peptide (KLA) (MEL-KLA) by administering MEL or MEL-KLA to a Lewis lung carcinoma (LLC) mouse model [138]. Treatment began 5 days after tumor inoculation and consisted of an injection every 3 days until the mice were sacrificed 12 days after inoculation. Results showed a significant decrease in tumor weight upon treatment of MEL-KLA compared to a PBS-treated control, as well as KLA and MEL mono-therapies. To further investigate the M2-targeting ability of MEL, another study from this group showed an increase of M1/M2 macrophage ratio from 0.65 to 1.55 in an LLC model upon MEL treatment [137]. The M1/M2 ratio is an increasingly used metric that has been reported to be positively correlated to prognosis in human cancers [141]. The change in M1/M2 ratio was attributed to cytotoxicity of MEL to M2 macrophages, validated through the reduction of M2 macrophage marker CD206 in flow cytometry and qPCR.
3.2. Targeting Tregs
The neuropilin-1 (Nrp1) receptor was first identified as a potential Treg marker in 2004 by Bruder et al. [88]. Originally identified as a co-receptor for VEGF, Nrp1 has now been identified as essential for Treg function. Its expression on these T cells has been correlated to FoxP3 expression, implicating its role as a mediator of the immunosuppressive phenotype [142]. Moreover, a recent study by Delgoffe et al. has identified its role in maintaining Treg stability, as destabilization of this population through the upregulation of Nrp1 is a common characteristic of cancers [143]. Unsurprisingly, preclinical studies of Nrp1 involving genetic knockout models and antibody blockades have resulted in decreased tumor growth in skin, lung, and prostate cancers [144]. The discovery of an Nrp1 peptide, LyP-1 (CGNKRTRGC), has led to the development of NP systems targeting Tregs specifically through Nrp1 [145,146]. LyP-1 is part of a family of peptides, denoted as C-terminal C-end Rule (CendR) motif peptides, which share tumor penetrative ability. In a study by Ou et al., a nanoparticle incorporating a PLGA core loaded with anti-CTLA-4, the tyrosine kinase inhibitor imatinib (IMT), and LyP-1 as a targeting ligand, significantly reduced tumor growth in a B16 murine melanoma model compared to conventional anti-CTLA-4 immunotherapy. Mice were treated every 2 days for 15 days, starting 10 days post-tumor inoculation with either PBS, free IMT, free anti-CTLA-4, non-targeting IMT-loaded nanoparticles, targeting IMT-loaded nanoparticles, and targeted anti-CTLA-4- and IMT-loaded nanoparticles. The full nanoparticle incorporating LyP-1, IMT, and anti-CTLA-4 proved most effective, decreasing tumor volume by over 50% compared to free anti-CTLA-4, demonstrating the potential of peptide-based nanoparticles to augment the clinical standard [144].
Another Treg-targeting nanoparticle was created by Pastor et al. through conjugation of a FoxP3-inhibiting peptide (P60) to a CD28-targeting aptamer (AptCD28-P60) [147,148]. The conjugated molecule was shown to be capable of countering Treg-mediated immunosuppression of CTLs at a 200-fold lower concentration to unconjugated P60 (100 μM vs. 0.5 μM). Furthermore, treatment of CT-26 carcinoma models with AptCD28-P60 in conjunction with a tumor antigen peptide vaccine (AH1) was shown to eradicate tumors compared to saline control, peptide vaccine alone, peptide vaccine and unconjugated P60, and peptide vaccine and unconjugated P60 with unconjugated AptCD28, showing the necessity in conjugating P60 to AptCD28 in achieving synergistic therapy (Fig. 5).
Fig. 5.

Tumor volume of CT-26 carcinoma following treatment of saline, AH1 peptide vaccine, AH1 + unconjugated P60 FoxP3-inhibiting peptide, AH1 + unconjugated P60 + unconjugated CD28Apt, or AH1 + conjugated CD28Apt-P60 show prevention of tumors upon vaccination of AH1 + CD28-Apt-P60. Injections were given at a dose of 125 pmol every 2 days for 10 days before tumor inoculation. Adapted and reprinted with permission from ref. 148.
3.3. Targeting MDSCs
Given MDSCs’ status as an immature cell population, they have not been found to express an MDSC-specific biomarker that can be used for peptide binding. However, Qin et al. has used phage display to screen for MDSC-binding peptides [149]. The selected peptide (MEWSLEKGYTIK) was fused with the Fc region of murine IgG2b to create a peptide-antibody conjugate (H6 peptibody). Treatment with H6 peptibody led to the depletion of MDSCs from the spleen and in circulation in multiple murine lymphoma models (EL4, EG.7, A20), and exhibited antitumor efficacy in an EL4 model. In addition, Wang et al. investigated the effectiveness of chemokine blockade as a means of indirectly affecting MDSC populations [150]. They used a CCL2 agonist to inhibit CCL2 signaling, a major chemoattractant implicated in MDSC migration [151,152]. This resulted in significantly decreased MDSC infiltration into the TME of a lung cancer model. Notably, it was observed that CCL2 blockade also improved the efficacy of anti-PDL1 treatment, likely via MDSC depletion.
4. Current outlook and conclusions
Advancements in understanding the interactions between immunosuppressive cells and cancer cells in establishing and maintaining an immunosuppressive microenvironment have been made in recent years. Furthermore, there have been strides in the application of this knowledge into peptide-based therapies as described herein. The application of peptides and nanomedicine targeting immunosuppressive cells for cancer immunotherapy, however, is only in its early stages. Consequently, clinical translation of these technologies for human use has been limited. This may be due to the lack of understanding regarding the peptide nanoparticle interactions with the biological milieu and the formation of a protein shell that alters the nanoparticle fate, efficacy, and toxicity [153,154]. Another challenge includes scaling up peptide-functionalized nanoparticles, as well as batch-to-batch variation, limiting commercialization potential. Moreover, before immunotherapeutic targeting of immunosuppressive cells can be translated to the clinic, a greater understanding of the body's immunologic network needs to be achieved. For instance, although the interactions of TAMs, Tregs, and MDSCs with the classical CTL have been well-studied, other immune cells, such as natural killer (NK) cells, B lymphocytes, and dendritic cells, all have unique and complex sets of functions and interactions that merit further study. Peptide-based nanoparticles are tools that offer researchers and clinicians a means of targeting specific molecular pathways and intercellular interactions that contribute to cancer progression, but it is crucial to expand our understanding of immunology to utilize these tools effectively. Furthering our knowledge with regards to the immunological network will allow researchers to not only better develop novel nanoparticle systems, but also better predict their efficacy in vivo.
A major shortcoming in our knowledge of cancer immunotherapy lies in the identification of predictive biomarkers for clinical response to treatment. Although there have been cases in which immunotherapy eradicated chemotherapy-resistant tumors, the reality is that only a minority of patients respond to treatment. The inconsistency in patient response can be attributed to the heterogeneity of cancer but also to the lack of predictive biomarkers [155]. Although some markers, such as immune checkpoints and immunomodulatory cytokines, have been identified, the discovery of other prognostic markers can serve to optimize patient selection, as well as further the understanding of the mechanisms behind immunotherapy [156]. Furthermore, the use of these biomarkers in combination can hold more accurate predictive power.
Nanomedicine makes the use of combinatorial therapies more feasible, as nanoparticles can be engineered to include multiple biologically active moieties. Additionally, packaging of bio-active materials into nanoparticles ensures they are delivered to their desired targets, reducing off-target delivery and toxicity. Many of the studies highlighted here have demonstrated that combination therapies can improve the efficacy and safety of mono-therapeutic clinical standards [137,148]. As scientific research continues to fill in the knowledge gaps discussed above, peptide-based nanoparticles will continue to develop and comprise a growing class of nanomaterials capable of targeting immunosuppressive cell populations.
Declaration of competing interest
None.
Acknowledgements
The authors would like to acknowledge the financial support from the National Heart, Lung, and Blood Institute (NHLBI, R00HL124279), NIH New Innovator Award (DP2DK121328), L.K. Whittier Foundation, Ming Hsieh Institute for Research on Engineering-Medicine for Cancer, the Women in Science and Engineering Gabilan Assistant Professorship, and the University of Southern California startup funds awarded to EJC.
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Cao J., Liu J., Xu R., Zhu X., Zhao X., Qian B.Z. Prognostic role of tumour-associated macrophages and macrophage scavenger receptor 1 in prostate cancer: a systematic review and meta-analysis. Oncotarget. 2017;8(47):83261–83269. doi: 10.18632/oncotarget.18743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gollapudi K., Galet C., Grogan T., Zhang H., Said J.W., Huang J.…Aronson W.J. Association between tumor-associated macrophage infiltration, high grade prostate cancer, and biochemical recurrence after radical prostatectomy. Am J Cancer Res. 2013;3(5):523–529. [PMC free article] [PubMed] [Google Scholar]
- 4.Lissbrant I.F., Stattin P., Wikstrom P., Damber J.E., Egevad L., Bergh A. Tumor associated macrophages in human prostate cancer: relation to clinicopathological variables and survival. Int. J. Oncol. 2000;17(3):445–451. doi: 10.3892/ijo.17.3.445. [DOI] [PubMed] [Google Scholar]
- 5.Nonomura N., Takayama H., Nakayama M., Nakai Y., Kawashima A., Mukai M.…Tsujimura A. Infiltration of tumour-associated macrophages in prostate biopsy specimens is predictive of disease progression after hormonal therapy for prostate cancer. BJU Int. 2011;107(12):1918–1922. doi: 10.1111/j.1464-410X.2010.09804.x. [DOI] [PubMed] [Google Scholar]
- 6.Flammiger A., Weisbach L., Huland H., Tennstedt P., Simon R., Minner S.…Trepel M. High tissue density of FOXP3+ T cells is associated with clinical outcome in prostate cancer. Eur. J. Cancer. 2013;49(6):1273–1279. doi: 10.1016/j.ejca.2012.11.035. [DOI] [PubMed] [Google Scholar]
- 7.Huen N.Y., Pang A.L., Tucker J.A., Lee T.L., Vergati M., Jochems C.…Tsang K.Y. Up-regulation of proliferative and migratory genes in regulatory T cells from patients with metastatic castration-resistant prostate cancer. Int. J. Cancer. 2013;133(2):373–382. doi: 10.1002/ijc.28026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu C., Workman C.J., Vignali D.A. Targeting regulatory T cells in tumors. FEBS J. 2016;283(14):2731–2748. doi: 10.1111/febs.13656. [DOI] [PubMed] [Google Scholar]
- 9.Miller A.M., Lundberg K., Ozenci V., Banham A.H., Hellstrom M., Egevad L., Pisa P. CD4+CD25high T cells are enriched in the tumor and peripheral blood of prostate cancer patients. J. Immunol. 2006;177(10):7398–7405. doi: 10.4049/jimmunol.177.10.7398. [DOI] [PubMed] [Google Scholar]
- 10.Markman J.L., Shiao S.L. Impact of the immune system and immunotherapy in colorectal cancer. J. Gastrointest. Oncol. 2015;6(2):208–223. doi: 10.3978/j.issn.2078-6891.2014.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rabinovich G.A., Gabrilovich D., Sotomayor E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Darvin P., Toor S.M., Sasidharan Nair V., Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med. 2018;50(12):165. doi: 10.1038/s12276-018-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen L., Flies D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13(4):227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golstein P., Griffiths G.M. An early history of T cell-mediated cytotoxicity. Nat. Rev. Immunol. 2018;18(8):527–535. doi: 10.1038/s41577-018-0009-3. [DOI] [PubMed] [Google Scholar]
- 15.Busch W. Aus der sitzung der medicinischen section vom 13.November 1867. Berliner Klinische Wochenschrift. 1868;5:137. [Google Scholar]
- 16.Coley W.B. The treatment of malignant tumors by repeated inoculations of erysipelas: with a report of ten original cases. Am. J. Med. Sci. 1893 May;105:487–511. [PubMed] [Google Scholar]
- 17.Nauts H.C., McLaren J.R. Coley toxins — the first century. In: Bicher H.I., McLaren J.R., Pigliucci G.M., editors. Consensus on Hyperthermia for the 1990s. vol. 267. Springer; Boston, MA: 1990. (Advances in Experimental Medicine and Biology). [DOI] [PubMed] [Google Scholar]
- 18.Oiseth S.J., Aziz M.S. Cancer immunotherapy: a brief review of the history, possibilities, and challenges ahead. J Cancer Metastasis Treat. 2017;3:250–261. [Google Scholar]
- 19.Allison J.P., McIntyre B.W., Bloch D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J. Immunol. 1982;129(5):2293–2300. [PubMed] [Google Scholar]
- 20.van der Bruggen P., Traversari C., Chomez P., Lurquin C., De Plaen E., Van den Eynde B.…Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991;254(5038):1643–1647. doi: 10.1126/science.1840703. [DOI] [PubMed] [Google Scholar]
- 21.Leach D.R., Krummel M.F., Allison J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 22.Hodi F.S., O'Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B.…Urba W.J. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alexander W. The checkpoint immunotherapy revolution: what started as a trickle has become a flood, despite some daunting adverse effects; new drugs, indications, and combinations continue to emerge. PT. 2016;41(3):185–191. [PMC free article] [PubMed] [Google Scholar]
- 24.Hamid O., Robert C., Daud A., Hodi F.S., Hwu W.J., Kefford R.…Ribas A. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013;369(2):134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Topalian S.L., Hodi F.S., Brahmer J.R., Gettinger S.N., Smith D.C., McDermott D.F.…Sznol M. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.FDA Approves New, Targeted Treatment for Bladder Cancer. FDA; 18 May 2016. Retrieved 12 August 2019. [Google Scholar]
- 27.The Nobel Prize in Physiology or Medicine 2018". NobelPrize.org. Retrieved 14 August 2019.
- 28.Ishida Y., Agata Y., Shibahara K., Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Obara W., Sato F., Takeda K., Kato R., Kato Y., Kanehira M.…Fujioka T. Phase I clinical trial of cell division associated 1 (CDCA1) peptide vaccination for castration resistant prostate cancer. Cancer Sci. 2017;108(7):1452–1457. doi: 10.1111/cas.13278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noguchi M., Moriya F., Suekane S., Matsuoka K., Arai G., Matsueda S.…Itoh K. Phase II study of personalized peptide vaccination for castration-resistant prostate cancer patients who failed in docetaxel-based chemotherapy. The Prostate. 2012;72(8):834–845. doi: 10.1002/pros.21485. [DOI] [PubMed] [Google Scholar]
- 31.Rudra J.S., Tian Y.F., Jung J.P., Collier J.H. A self-assembling peptide acting as an immune adjuvant. Proc. Natl. Acad. Sci. U. S. A. 2010;107(2):622–627. doi: 10.1073/pnas.0912124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McHayleh W., Bedi P., Sehgal R., Solh M. Chimeric antigen receptor T-cells: the future is now. J. Clin. Med. 2019;8(2) doi: 10.3390/jcm8020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg S.A., Restifo N.P., Yang J.C., Morgan R.A., Dudley M.E. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat. Rev. Cancer. 2008;8(4):299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Riley R.S., June C.H., Langer R., Mitchell M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019;18(3):175–196. doi: 10.1038/s41573-018-0006-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bodey B., Bodey B., Jr., Siegel S.E., Kaiser H.E. Failure of cancer vaccines: the significant limitations of this approach to immunotherapy. Anticancer Res. 2000;20(4):2665–2676. [PubMed] [Google Scholar]
- 36.Slingluff C.L., Jr. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination? Cancer J. 2011;17(5):343–350. doi: 10.1097/PPO.0b013e318233e5b2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hucks G., Rheingold S.R. The journey to CAR T cell therapy: the pediatric and young adult experience with relapsed or refractory B-ALL. Blood Canc. J. 2019;9(2):10. doi: 10.1038/s41408-018-0164-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pathria P., Louis T.L., Varner J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40(4):310–327. doi: 10.1016/j.it.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 39.Cassetta L., Pollard J.W. Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018;17(12):887–904. doi: 10.1038/nrd.2018.169. [DOI] [PubMed] [Google Scholar]
- 40.Shitara K., Nishikawa H. Regulatory T cells: a potential target in cancer immunotherapy. Ann. N. Y. Acad. Sci. 2018;1417(1):104–115. doi: 10.1111/nyas.13625. [DOI] [PubMed] [Google Scholar]
- 41.Fleming V., Hu X., Weber R., Nagibin V., Groth C., Altevogt P.…Umansky V. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front. Immunol. 2018;9:398. doi: 10.3389/fimmu.2018.00398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gokhale A.S., Satyanarayanajois S. Peptides and peptidomimetics as immunomodulators. Immunotherapy. 2014;6(6):755–774. doi: 10.2217/imt.14.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lau J.L., Dunn M.K. Therapeutic peptides: historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018;26(10):2700–2707. doi: 10.1016/j.bmc.2017.06.052. [DOI] [PubMed] [Google Scholar]
- 44.Velpurisiva P., Gad A., Piel B., Jadia R., Rai P. Nanoparticle design strategies for effective cancer immunotherapy. J. Biom. 2017;2(2):64–77. doi: 10.7150/jbm.18877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park W., Heo Y.J., Han D.K. New opportunities for nanoparticles in cancer immunotherapy. Biomater. Res. 2018;22:24. doi: 10.1186/s40824-018-0133-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Candeias S.M., Gaipl U.S. The immune system in cancer prevention, development and therapy. Anti Cancer Agents Med. Chem. 2016;16(1):101–107. doi: 10.2174/1871520615666150824153523. [DOI] [PubMed] [Google Scholar]
- 47.Mohme M., Neidert M.C., Regli L., Weller M., Martin R. Immunological challenges for peptide-based immunotherapy in glioblastoma. Cancer Treat Rev. 2014;40(2):248–258. doi: 10.1016/j.ctrv.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 48.Shankaran V., Ikeda H., Bruce A.T., White J.M., Swanson P.E., Old L.J., Schreiber R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–1111. doi: 10.1038/35074122. [DOI] [PubMed] [Google Scholar]
- 49.Gordon S., Martinez-Pomares L. Physiological roles of macrophages. Pflüg. Arch. 2017;469(3–4):365–374. doi: 10.1007/s00424-017-1945-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Imhof B.A., Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat. Rev. Immunol. 2004;4(6):432–444. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- 51.Deshmane S.L., Kremlev S., Amini S., Sawaya B.E. Monocyte chemoattractant protein-1 (MCP-1): an overview. J. Interferon Cytokine Res. 2009;29(6):313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sokol C.L., Luster A.D. The chemokine system in innate immunity. Cold Spring Harb Perspect Biol. 2015;7(5) doi: 10.1101/cshperspect.a016303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joo J., Poon C., Yoo S.P., Chung E.J. Shape effects of peptide amphiphile micelles for targeting monocytes. Molecules. 2018;23(11) doi: 10.3390/molecules23112786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poon C., Sarkar M., Chung E.J. Synthesis of monocyte-targeting peptide amphiphile micelles for imaging of atherosclerosis. J. Vis. Exp. 2017;129 doi: 10.3791/56625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Poon C., Chowdhuri S., Kuo C.H., Fang Y., Alenghat F.J., Hyatt D.…Chung E.J. Protein mimetic and anticancer properties of monocyte-targeting peptide amphiphile micelles. ACS Biomater. Sci. Eng. 2017;3(12):3273–3282. doi: 10.1021/acsbiomaterials.7b00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shapouri-Moghaddam A., Mohammadian S., Vazini H., Taghadosi M., Esmaeili S.A., Mardani F.…Sahebkar A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018;233(9):6425–6440. doi: 10.1002/jcp.26429. [DOI] [PubMed] [Google Scholar]
- 57.Ley K. M1 means kill; M2 means heal. J. Immunol. 2017;199(7):2191–2193. doi: 10.4049/jimmunol.1701135. [DOI] [PubMed] [Google Scholar]
- 58.Lu H.L., Huang X.Y., Luo Y.F., Tan W.P., Chen P.F., Guo Y.B. Activation of M1 macrophages plays a critical role in the initiation of acute lung injury. Biosci. Rep. 2018;38(2) doi: 10.1042/BSR20171555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Y.C., He F., Feng F., Liu X.W., Dong G.Y., Qin H.Y.…Han H. Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 2010;70(12):4840–4849. doi: 10.1158/0008-5472.CAN-10-0269. [DOI] [PubMed] [Google Scholar]
- 60.Stein M., Keshav S., Harris N., Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J. Exp. Med. 1992;176(1):287–292. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Doyle A.G., Herbein G., Montaner L.J., Minty A.J., Caput D., Ferrara P., Gordon S. Interleukin-13 alters the activation state of murine macrophages in vitro: comparison with interleukin-4 and interferon-gamma. Eur. J. Immunol. 1994;24(6):1441–1445. doi: 10.1002/eji.1830240630. [DOI] [PubMed] [Google Scholar]
- 62.Arango Duque G., Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front. Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rath M., Muller I., Kropf P., Closs E.I., Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front. Immunol. 2014;5:532. doi: 10.3389/fimmu.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fauskanger M., Haabeth O.A.W., Skjeldal F.M., Bogen B., Tveita A.A. Tumor killing by CD4(+) T cells is mediated via induction of inducible nitric oxide synthase-dependent macrophage cytotoxicity. Front. Immunol. 2018;9:1684. doi: 10.3389/fimmu.2018.01684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mills C.D. M1 and M2 macrophages: oracles of health and disease. Crit. Rev. Immunol. 2012;32(6):463–488. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 66.Yang Z., Ming X.F. Functions of arginase isoforms in macrophage inflammatory responses: impact on cardiovascular diseases and metabolic disorders. Front. Immunol. 2014;5:533. doi: 10.3389/fimmu.2014.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Poh A.R., Ernst M. Targeting macrophages in cancer: from bench to bedside. Front Oncol. 2018;8:49. doi: 10.3389/fonc.2018.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ueno T., Toi M., Saji H., Muta M., Bando H., Kuroi K.…Matsushima K. Significance of macrophage chemoattractant protein-1 in macrophage recruitment, angiogenesis, and survival in human breast cancer. Clin. Cancer Res. 2000;6(8):3282–3289. [PubMed] [Google Scholar]
- 69.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017;14(7):399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Noy R., Pollard J.W. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hartley G.P., Chow L., Ammons D.T., Wheat W.H., Dow S.W. Programmed cell death ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res. 2018;6(10):1260–1273. doi: 10.1158/2326-6066.CIR-17-0537. [DOI] [PubMed] [Google Scholar]
- 72.Cassetta L., Kitamura T. Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front Cell Dev Biol. 2018;6:38. doi: 10.3389/fcell.2018.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sansom D.M. CD28, CTLA-4 and their ligands: who does what and to whom? Immunology. 2000;101(2):169–177. doi: 10.1046/j.1365-2567.2000.00121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wong C.K., Lit L.C., Tam L.S., Li E.K., Lam C.W. Aberrant production of soluble costimulatory molecules CTLA-4, CD28, CD80 and CD86 in patients with systemic lupus erythematosus. Rheumatology. 2005;44(8):989–994. doi: 10.1093/rheumatology/keh663. [DOI] [PubMed] [Google Scholar]
- 75.Linsley P.S., Brady W., Urnes M., Grosmaire L.S., Damle N.K., Ledbetter J.A. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 1991;174(3):561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jago C.B., Yates J., Camara N.O., Lechler R.I., Lombardi G. Differential expression of CTLA-4 among T cell subsets. Clin. Exp. Immunol. 2004;136(3):463–471. doi: 10.1111/j.1365-2249.2004.02478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chan D.V., Gibson H.M., Aufiero B.M., Wilson A.J., Hafner M.S., Mi Q.S., Wong H.K. Differential CTLA-4 expression in human CD4+ versus CD8+ T cells is associated with increased NFAT1 and inhibition of CD4+ proliferation. Genes Immun. 2014;15(1):25–32. doi: 10.1038/gene.2013.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bertani F.R., Mozetic P., Fioramonti M., Iuliani M., Ribelli G., Pantano F.…Rainer A. Classification of M1/M2-polarized human macrophages by label-free hyperspectral reflectance confocal microscopy and multivariate analysis. Sci. Rep. 2017;7(1):8965. doi: 10.1038/s41598-017-08121-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Riabov V., Gudima A., Wang N., Mickley A., Orekhov A., Kzhyshkowska J. Role of tumor associated macrophages in tumor angiogenesis and lymphangiogenesis. Front. Physiol. 2014;5:75. doi: 10.3389/fphys.2014.00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Henze A.T., Mazzone M. The impact of hypoxia on tumor-associated macrophages. J. Clin. Investig. 2016;126(10):3672–3679. doi: 10.1172/JCI84427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Afik R., Zigmond E., Vugman M., Klepfish M., Shimshoni E., Pasmanik-Chor M.…Varol C. Tumor macrophages are pivotal constructors of tumor collagenous matrix. J. Exp. Med. 2016;213(11):2315–2331. doi: 10.1084/jem.20151193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Madsen D.H., Jurgensen H.J., Siersbaek M.S., Kuczek D.E., Grey Cloud L., Liu S.…Bugge T.H. Tumor-associated macrophages derived from circulating inflammatory monocytes degrade collagen through cellular uptake. Cell Rep. 2017;21(13):3662–3671. doi: 10.1016/j.celrep.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sakaguchi S., Yamaguchi T., Nomura T., Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 84.Dominguez-Villar M., Hafler D.A. Regulatory T cells in autoimmune disease. Nat. Immunol. 2018;19(7):665–673. doi: 10.1038/s41590-018-0120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang Z., Zhang W., Guo J., Gu Q., Zhu X., Zhou X. Activation and functional specialization of regulatory T cells lead to the generation of Foxp3 instability. J. Immunol. 2017;198(7):2612–2625. doi: 10.4049/jimmunol.1601409. [DOI] [PubMed] [Google Scholar]
- 86.Corthay A. How do regulatory T cells work? Scand. J. Immunol. 2009;70(4):326–336. doi: 10.1111/j.1365-3083.2009.02308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hori S., Nomura T., Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 88.Bruder D., Probst-Kepper M., Westendorf A.M., Geffers R., Beissert S., Loser K.…Hansen W. Neuropilin-1: a surface marker of regulatory T cells. Eur. J. Immunol. 2004;34(3):623–630. doi: 10.1002/eji.200324799. [DOI] [PubMed] [Google Scholar]
- 89.Chinen T., Kannan A.K., Levine A.G., Fan X., Klein U., Zheng Y.…Rudensky A.Y. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016;17(11):1322–1333. doi: 10.1038/ni.3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nakamura K., Kitani A., Strober W. Cell contact-dependent immunosuppression by CD4(+)CD25(+) regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J. Exp. Med. 2001;194(5):629–644. doi: 10.1084/jem.194.5.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Goldberg M.V., Drake C.G. LAG-3 in cancer immunotherapy. Curr. Top. Microbiol. Immunol. 2011;344:269–278. doi: 10.1007/82_2010_114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Oh S.A., Li M.O. TGF-beta: guardian of T cell function. J. Immunol. 2013;191(8):3973–3979. doi: 10.4049/jimmunol.1301843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Akdis C.A., Blaser K. Mechanisms of interleukin-10-mediated immune suppression. Immunology. 2001;103(2):131–136. doi: 10.1046/j.1365-2567.2001.01235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Curiel T.J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P.…Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004;10(9):942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 95.Chaudhary B., Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel) 2016;4(3) doi: 10.3390/vaccines4030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Safarzadeh E., Hashemzadeh S., Duijf P.H.G., Mansoori B., Khaze V., Mohammadi A.…Baradaran B. Circulating myeloid-derived suppressor cells: an independent prognostic factor in patients with breast cancer. J. Cell. Physiol. 2019;234(4):3515–3525. doi: 10.1002/jcp.26896. [DOI] [PubMed] [Google Scholar]
- 97.Wynn T.A. Myeloid-cell differentiation redefined in cancer. Nat. Immunol. 2013;14(3):197–199. doi: 10.1038/ni.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lechner M.G., Liebertz D.J., Epstein A.L. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J. Immunol. 2010;185(4):2273–2284. doi: 10.4049/jimmunol.1000901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bah I., Kumbhare A., Nguyen L., McCall C.E., El Gazzar M. IL-10 induces an immune repressor pathway in sepsis by promoting S100A9 nuclear localization and MDSC development. Cell. Immunol. 2018;332:32–38. doi: 10.1016/j.cellimm.2018.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gabrilovich D.I., Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Umansky V., Blattner C., Gebhardt C., Utikal J. The role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines (Basel) 2016;4(4) doi: 10.3390/vaccines4040036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Corzo C.A., Cotter M.J., Cheng P., Cheng F., Kusmartsev S., Sotomayor E.…Gabrilovich D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009;182(9):5693–5701. doi: 10.4049/jimmunol.0900092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kusmartsev S.A., Li Y., Chen S.H. Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J. Immunol. 2000;165(2):779–785. doi: 10.4049/jimmunol.165.2.779. [DOI] [PubMed] [Google Scholar]
- 104.Rodriguez P.C., Ochoa A.C., Al-Khami A.A. Arginine metabolism in myeloid cells shapes innate and adaptive immunity. Front. Immunol. 2017;8:93. doi: 10.3389/fimmu.2017.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Geiger R., Rieckmann J.C., Wolf T., Basso C., Feng Y., Fuhrer T.…Lanzavecchia A. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3):829–842. doi: 10.1016/j.cell.2016.09.031. e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huang B., Pan P.Y., Li Q., Sato A.I., Levy D.E., Bromberg J.…Chen S.H. Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 2006;66(2):1123–1131. doi: 10.1158/0008-5472.CAN-05-1299. [DOI] [PubMed] [Google Scholar]
- 107.Sinha P., Clements V.K., Bunt S.K., Albelda S.M., Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J. Immunol. 2007;179(2):977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 108.Behrendt R., White P., Offer J. Advances in Fmoc solid-phase peptide synthesis. J. Pept. Sci. 2016;22(1):4–27. doi: 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang Jonathan. Design and in vivo characterization of kidney-targeting multimodal micelles for renal drug delivery. Nano Research. 2018;11(10):5584–5595. [Google Scholar]
- 110.Le Joncour V., Laakkonen P. Seek & Destroy, use of targeting peptides for cancer detection and drug delivery. Bioorg. Med. Chem. 2018;26(10):2797–2806. doi: 10.1016/j.bmc.2017.08.052. [DOI] [PubMed] [Google Scholar]
- 111.Chung E.J. Targeting and therapeutic peptides in nanomedicine for atherosclerosis. Exp. Biol. Med. 2016;241(9):891–898. doi: 10.1177/1535370216640940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Baig M.H., Ahmad K., Saeed M., Alharbi A.M., Barreto G.E., Ashraf G.M., Choi I. Peptide based therapeutics and their use for the treatment of neurodegenerative and other diseases. Biomed. Pharmacother. 2018;103:574–581. doi: 10.1016/j.biopha.2018.04.025. [DOI] [PubMed] [Google Scholar]
- 113.Si Y., Wen Y., Kelly S.H., Chong A.S., Collier J.H. Intranasal delivery of adjuvant-free peptide nanofibers elicits resident CD8(+) T cell responses. J. Control. Release. 2018;282:120–130. doi: 10.1016/j.jconrel.2018.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Behzadi S., Serpooshan V., Tao W., Hamaly M.A., Alkawareek M.Y., Dreaden E.C.…Mahmoudi M. Cellular uptake of nanoparticles: journey inside the cell. Chem. Soc. Rev. 2017;46(14):4218–4244. doi: 10.1039/c6cs00636a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang R., Leeper C.N., Wang X., White T.A., Ulery B.D. Immunomodulatory vasoactive intestinal peptide amphiphile micelles. Biomater Sci. 2018;6(7):1717–1722. doi: 10.1039/c8bm00466h. [DOI] [PubMed] [Google Scholar]
- 116.Delehanty J.B., Boeneman K., Bradburne C.E., Robertson K., Bongard J.E., Medintz I.L. Peptides for specific intracellular delivery and targeting of nanoparticles: implications for developing nanoparticle-mediated drug delivery. Ther. Deliv. 2010;1(3):411–433. doi: 10.4155/tde.10.27. [DOI] [PubMed] [Google Scholar]
- 117.Zong J., Cobb S.L., Cameron N.R. Peptide-functionalized gold nanoparticles: versatile biomaterials for diagnostic and therapeutic applications. Biomater Sci. 2017;5(5):872–886. doi: 10.1039/c7bm00006e. [DOI] [PubMed] [Google Scholar]
- 118.Giljohann D.A., Seferos D.S., Daniel W.L., Massich M.D., Patel P.C., Mirkin C.A. Gold nanoparticles for biology and medicine. Angew Chem. Int. Ed. Engl. 2010;49(19):3280–3294. doi: 10.1002/anie.200904359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bartczak D., Kanaras A.G. Preparation of peptide-functionalized gold nanoparticles using one pot EDC/sulfo-NHS coupling. Langmuir. 2011;27(16):10119–10123. doi: 10.1021/la2022177. [DOI] [PubMed] [Google Scholar]
- 120.Hauser A.K., Anderson K.W., Hilt J.Z. Peptide conjugated magnetic nanoparticles for magnetically mediated energy delivery to lung cancer cells. Nanomedicine. 2016;11(14):1769–1785. doi: 10.2217/nnm-2016-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martinez-Jothar L., Doulkeridou S., Schiffelers R.M., Sastre Torano J., Oliveira S., van Nostrum C.F., Hennink W.E. Insights into maleimide-thiol conjugation chemistry: conditions for efficient surface functionalization of nanoparticles for receptor targeting. J. Control. Release. 2018;282:101–109. doi: 10.1016/j.jconrel.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 122.Wang Y., Liu X., Zhang J., Aili D., Liedberg B. Time-resolved botulinum neurotoxin A activity monitored using peptide-functionalized Au nanoparticle energy transfer sensors. Chem. Sci. 2014;5:2651–2656. [Google Scholar]
- 123.Wang C., Wang J., Liu D., Wang Z. Gold nanoparticle-based colorimetric sensor for studying the interactions of beta-amyloid peptide with metallic ions. Talanta. 2010;80(5):1626–1631. doi: 10.1016/j.talanta.2009.09.052. [DOI] [PubMed] [Google Scholar]
- 124.Kaasalainen M., Rytkonen J., Makila E., Narvanen A., Salonen J. Electrostatic interaction on loading of therapeutic peptide GLP-1 into porous silicon nanoparticles. Langmuir. 2015;31(5):1722–1729. doi: 10.1021/la5047047. [DOI] [PubMed] [Google Scholar]
- 125.Wagner S.C., Roskamp M., Colfen H., Bottcher C., Schlecht S., Koksch B. Switchable electrostatic interactions between gold nanoparticles and coiled coil peptides direct colloid assembly. Org. Biomol. Chem. 2009;7(1):46–51. doi: 10.1039/b813429d. [DOI] [PubMed] [Google Scholar]
- 126.Blank-Shim S.A., Schwaminger S.P., Borkowska-Panek M., Anand P., Yamin P., Fraga-Garcia P.…Berensmeier S. Binding patterns of homo-peptides on bare magnetic nanoparticles: insights into environmental dependence. Sci. Rep. 2017;7(1):14047. doi: 10.1038/s41598-017-13928-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Taylor S., Qu L., Kitaygorodskiy A., Teske J., Latour R.A., Sun Y.P. Synthesis and characterization of peptide-functionalized polymeric nanoparticles. Biomacromolecules. 2004;5(1):245–248. doi: 10.1021/bm034326m. [DOI] [PubMed] [Google Scholar]
- 128.Mourdikoudis S., Pallares R.M., Thanh N.T.K. Characterization techniques for nanoparticles: comparison and complementarity upon studying nanoparticle properties. Nanoscale. 2018;10(27):12871–12934. doi: 10.1039/c8nr02278j. [DOI] [PubMed] [Google Scholar]
- 129.Hassan N., Cordero M.L., Sierpe R., Almada M., Juarez J., Valdez M., Riveros A., Vargas E., Abou-Hassan A., Ruso J.M., Kogan M.J. Peptide functionalized magneto-plasmonic nanoparticles obtained by microfluidics for inhibition of β-amyloid aggregation. J. Mater. Chem. B. 2018;6(31):5091–5099. doi: 10.1039/c8tb00206a. [DOI] [PubMed] [Google Scholar]
- 130.Kaasalainen M., Makila E., Riikonen J., Kovalainen M., Jarvinen K., Herzig K.H.…Salonen J. Effect of isotonic solutions and peptide adsorption on zeta potential of porous silicon nanoparticle drug delivery formulations. Int. J. Pharm. 2012;431(1–2):230–236. doi: 10.1016/j.ijpharm.2012.04.059. [DOI] [PubMed] [Google Scholar]
- 131.Wan X., Liu C., Lin Y., Fu J., Lu G., Lu Z. pH sensitive peptide functionalized nanoparticles for co-delivery of erlotinib and DAPT to restrict the progress of triple negative breast cancer. Drug Deliv. 2019;26(1):470–480. doi: 10.1080/10717544.2019.1576801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Slocik J.M., Govorov A.O., Naik R.R. Plasmonic circular dichroism of Peptide-functionalized gold nanoparticles. Nano Lett. 2011;11(2):701–705. doi: 10.1021/nl1038242. [DOI] [PubMed] [Google Scholar]
- 133.Cieslewicz M., Tang J., Yu J.L., Cao H., Zavaljevski M., Motoyama K.…Pun S.H. Targeted delivery of proapoptotic peptides to tumor-associated macrophages improves survival. Proc. Natl. Acad. Sci. U. S. A. 2013;110(40):15919–15924. doi: 10.1073/pnas.1312197110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pang L., Pei Y., Uzunalli G., Hyun H., Lyle L.T., Yeo Y. Surface modification of polymeric nanoparticles with M2pep peptide for drug delivery to tumor-associated macrophages. Pharm. Res. 2019;36(4):65. doi: 10.1007/s11095-019-2596-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Qian Y., Qiao S., Dai Y., Xu G., Dai B., Lu L.…Zhang Z. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano. 2017;11(9):9536–9549. doi: 10.1021/acsnano.7b05465. [DOI] [PubMed] [Google Scholar]
- 136.Scodeller P., Simon-Gracia L., Kopanchuk S., Tobi A., Kilk K., Saalik P.…Teesalu T. Precision targeting of tumor macrophages with a CD206 binding peptide. Sci. Rep. 2017;7(1):14655. doi: 10.1038/s41598-017-14709-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lee C., Bae S.S., Joo H., Bae H. Melittin suppresses tumor progression by regulating tumor-associated macrophages in a Lewis lung carcinoma mouse model. Oncotarget. 2017;8(33):54951–54965. doi: 10.18632/oncotarget.18627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lee C., Jeong H., Bae Y., Shin K., Kang S., Kim H.…Bae H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J Immunother Cancer. 2019;7(1):147. doi: 10.1186/s40425-019-0610-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rady I., Siddiqui I.A., Rady M., Mukhtar H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett. 2017;402:16–31. doi: 10.1016/j.canlet.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shin J.M., Jeong Y.J., Cho H.J., Park K.K., Chung I.K., Lee I.K.…Chang Y.C. Melittin suppresses HIF-1 alpha/VEGF expression through inhibition of ERK and mTOR/p70S6K pathway in human cervical carcinoma cells. PLoS One. 2013;8(7) doi: 10.1371/journal.pone.0069380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zhang M., He Y., Sun X., Li Q., Wang W., Zhao A., Di W. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 2014;7:19. doi: 10.1186/1757-2215-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gluzman-Poltorak Z., Cohen T., Herzog Y., Neufeld G. Neuropilin-2 is a receptor for the vascular endothelial growth factor (VEGF) forms VEGF-145 and VEGF-165 [corrected] J. Biol. Chem. 2000;275(24):18040–18045. doi: 10.1074/jbc.M909259199. [DOI] [PubMed] [Google Scholar]
- 143.Delgoffe G.M., Woo S.R., Turnis M.E., Gravano D.M., Guy C., Overacre A.E.…Vignali D.A. Stability and function of regulatory T cells is maintained by a neuropilin-1-semaphorin-4a axis. Nature. 2013;501(7466):252–256. doi: 10.1038/nature12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hansen W., Hutzler M., Abel S., Alter C., Stockmann C., Kliche S.…Helfrich I. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J. Exp. Med. 2012;209(11):2001–2016. doi: 10.1084/jem.20111497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Roth L., Agemy L., Kotamraju V.R., Braun G., Teesalu T., Sugahara K.N.…Ruoslahti E. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012;31(33):3754–3763. doi: 10.1038/onc.2011.537. [DOI] [PubMed] [Google Scholar]
- 146.Ou W., Thapa R.K., Jiang L., Soe Z.C., Gautam M., Chang J.H.…Kim J.O. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J. Control. Release. 2018;281:84–96. doi: 10.1016/j.jconrel.2018.05.018. [DOI] [PubMed] [Google Scholar]
- 147.Pastor F., Soldevilla M.M., Villanueva H., Kolonias D., Inoges S., de Cerio A.L.…Bendandi M. CD28 aptamers as powerful immune response modulators. Mol. Ther. Nucleic Acids. 2013;2:e98. doi: 10.1038/mtna.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lozano T., Soldevilla M.M., Casares N., Villanueva H., Bendandi M., Lasarte J.J., Pastor F. Targeting inhibition of Foxp3 by a CD28 2'-Fluro oligonucleotide aptamer conjugated to P60-peptide enhances active cancer immunotherapy. Biomaterials. 2016;91:73–80. doi: 10.1016/j.biomaterials.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 149.Qin H., Lerman B., Sakamaki I., Wei G., Cha S.C., Rao S.S.…Kwak L.W. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 2014;20(6):676–681. doi: 10.1038/nm.3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang Y., Zhang X., Yang L., Xue J., Hu G. Blockade of CCL2 enhances immunotherapeutic effect of anti-PD1 in lung cancer. J Bone Oncol. 2018;11:27–32. doi: 10.1016/j.jbo.2018.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chang A.L., Miska J., Wainwright D.A., Dey M., Rivetta C.V., Yu D.…Lesniak M.S. CCL2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. 2016;76(19):5671–5682. doi: 10.1158/0008-5472.CAN-16-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sawanobori Y., Ueha S., Kurachi M., Shimaoka T., Talmadge J.E., Abe J.…Matsushima K. Chemokine-mediated rapid turnover of myeloid-derived suppressor cells in tumor-bearing mice. Blood. 2008;111(12):5457–5466. doi: 10.1182/blood-2008-01-136895. [DOI] [PubMed] [Google Scholar]
- 153.Shen L., Tenzer S., Storck W., Hobernik D., Raker V.K., Fischer K.…Bros M. Protein corona-mediated targeting of nanocarriers to B cells allows redirection of allergic immune responses. J. Allergy Clin. Immunol. 2018;142(5):1558–1570. doi: 10.1016/j.jaci.2017.08.049. [DOI] [PubMed] [Google Scholar]
- 154.Zanganeh S., Spitler R., Erfanzadeh M., Alkilany A.M., Mahmoudi M. Protein corona: opportunities and challenges. Int. J. Biochem. Cell Biol. 2016;75:143–147. doi: 10.1016/j.biocel.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Darvin P., Toor S.M., Sasidharan Nair V., Elkord E. Immune checkpoint inhibitors: recent progress and potential biomarkers. Exp. Mol. Med. 2018;50(12):165. doi: 10.1038/s12276-018-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bridge J.A., Lee J.C., Daud A., Wells J.W., Bluestone J.A. Cytokines, chemokines, and other biomarkers of response for checkpoint inhibitor therapy in skin cancer. Front. Med. 2018;5:351. doi: 10.3389/fmed.2018.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]