

https://onlinelibrary.wiley.com/page/journal/23301619/homepage/mdc312861-sup-v001.htm
Hypermanganesemia with dystonia 1 (HMNDYT1, OMIM #613280) is caused by mutations in the manganese (Mn) transporter SLC30A10 and characterized by Mn deposition in the liver and brain.1, 2, 3 Affected individuals present with dystonia–parkinsonism, liver disease, polycythemia, and markers of iron depletion. Brain magnetic resonance imaging (MRI) appearances are pathognomonic, and diagnosis is confirmed by genetic testing. The mainstay of treatment is chelation with disodium calcium edetate combined with iron supplementation.1, 2, 4 In this report, we describe the favorable response of an individual with HMNDYT1 to sole iron treatment.
Case Report
A 28‐year‐old female was followed up in our clinic for generalized dystonia. She was born at term to healthy, consanguineous parents. Her initial developmental milestones were normal. She was able to sit at 7 months and walk and use first words at 13 months. At 18 months she developed difficulty with walking and lost the ability to walk at 2 years.
At the age of 7, she presented to us with moderate generalized dystonia, particularly prominent in the lower extremities. She was moderately dysarthric and had mild oromandibular dystonia. Her skin tone was darker than that of her parents and healthy sibling. She was polycythemic, had severe hypermanganesemia and markers of iron depletion, and liver function tests were deranged (Table 1). Liver ultrasound was normal, whereas liver biopsy showed fibrosis and mononuclear cell infiltration. Brain MRI revealed T1‐hyperintensity of the globus pallidus consistent with Mn deposition.
Table 1.
Result of laboratory investigations prior to and during treatment with oral iron
| Blood results | Prior treatment (at 7 years) | Prior treatment (at 23 years) | During treatment (at 24 years) |
|---|---|---|---|
| Whole blood manganese (<273 nmol/L) | 1946 ↑ | 3528 ↑ | 263 |
| Erythrocytes (3.8–5.0 106/μL) | 7.63 ↑ | 7.11 ↑ | 4.209 |
| Hemoglobin (11.7–15.5 g/dL) | 23 ↑ | 17.9 ↑ | 12.5 |
| Hematocrit (36%–46%) | 62.6 ↑ | 57.7 ↑ | 37.7 |
| TIBC (250–410 μg/dL) | 539 ↑ | 673 ↑ | 291 |
| Ferritin (13–150 ng/mL) | 10 ↓ | 10.65 ↓ | 339.2 ↑ |
| sGOT (5–42 U/L) | 78 ↑ | 35 | |
| sGPT (5–45 U/L) | 130 ↑ | 30 | |
| Indirect blirubin (0.1–0.5 mg/dL) | 1.1 ↑ | 0.37 | |
| Total bilirubin (0.2–1.2 mg/dL) | 1.8 ↑ | 0.66 |
Abnormal values in bold. ↑ above reference range, ↓ below reference range.
TIBC, Total iron binding capacity; sGOT, serum Glutamate Oxaloacetate Transaminase; sGPT, serum Glutamic‐pyruvic transaminase.
At the age of 23 years, she was unable to sit unsupported as a result of severe generalized dystonia with prominent oromandibular dystonia and dyskinetic movements in the upper extremities. Features of parkinsonism were not present. She scored 111 on the Fahn Marsden Rating Scale, including both the motor and the disability subscales. She experienced irritability, insomnia, logorrhea, emotional instability, and auditory and visual hallucinations. A diagnosis of organic psychosis was made, which responded to risperidone. Intelligence quotient (IQ) score was 70. Brain MRI showed worsening of the basal ganglia and white matter T1‐hyperintensities (Fig. 1). Sanger sequencing of the solute carrier family 30 member 10 (SLC30A10) gene according to published protocols1 revealed a homozygous sequence variant (c.1188 dup [p.Leu397Thrfs*15]) that has not been previously reported (Fig. 1). Both parents were found to be heterozygous carriers.
Figure 1.
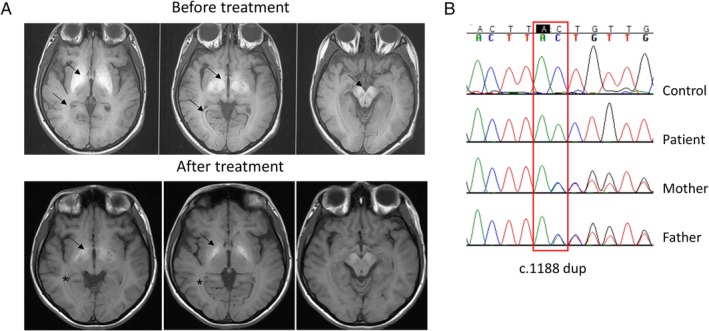
A: Axial, T1‐weighted magnetic resonance imaging brain imaging showing marked hyperintensity of bilateral gobus pallidus, putamen, substantia nigra, and white matter (arrows) before treatment initiation. At 2 years after iron treatment, the globus pallidus hyperintensity has improved (arrows), and the white matter appearance normalized (*). B: Sequencing chromatogram showing a homozygous duplication of an adenine residue at position 1188 (c.1188 dup [p.Leu397Thrfs*15]) of the SLC30A10 gene (NM_018713.2) in the patient. Both parents are heterozygous for the identified variant.
The family decided against chelation with disodium calcium edetate because of its possible side effects and added disease burden. Therefore, the patient was commenced on oral iron alone with a standard dose of ferrous fumarate at 200 mg elemental iron per day. At 10 months after treatment initiation, her speech was clearer and echolalia was no longer present. Fine motor movements improved, and she was able to use her hands to eat solid food. Generalized dystonia was less severe; however, her sitting posture remained affected by axial dystonia, and she was unable to stand without support. After 2 years, dystonic contractions decreased, her skin color normalized, and T1‐hyperintensities on MRI decreased (Fig. 1). Laboratory parameters including hemoglobin, iron parameters, liver function tests, and blood Mn normalized (Table 1). A 3‐month treatment pause initiated by the parents led to the worsening of dystonia and a reoccurrence of psychotic symptoms. Restarting of iron stabilized symptoms within 3 weeks. Currently, 5 years after treatment initiation, she is able to speak clearly, though mildly dysarthric, eat solid food by herself, brush her teeth, and sit more comfortably without severe dystonic postures. Her latest Fahn Marsden Rating Scale score is 54.
Discussion
The patient reported here presented with characteristic clinical, radiological, and biochemical features of HMNDYT1, including generalized dystonia, liver disease, severe hypermanganesemia, and polycythemia. In addition, she developed psychosis during adolescence. To date, psychotic symptoms have not been described in HMNDYT1.1, 2, 3, 4 However, they are a well‐characterized feature of manganism caused by environmental Mn overexposure.5 MRI brain appearances were pathognomonic as expected for an inherited Mn transporter defect associated with Mn overload.1 Other causes of symmetric T1 hyperintensities of the basal ganglia including acquired Mn overload through environmental overexposure, parenteral nutrition, and end‐stage liver disease as well as Wilson's and Fahr disease were excluded.
Mn and iron homeostasis are interdependent as both metals share transporters such as the divalent metal transporter 1, transferrin/transferrin receptor complex, and ferroportin.6 Because chelation therapy was declined, treatment was initiated with iron alone with a view to limiting intestinal Mn absorption. This led to significant improvement of dystonia, normalization of biochemical parameters, and a reduction in Mn deposition on MRI. The literature only reports 1 patient with HMNDYT1 who was treated with iron alone and who, similar to our case, experienced partial neurological improvement and moderate reduction in serum Mn.7 Iron supplementation also normalized blood Mn levels in a patient on chelation therapy to an extent that could not be achieved with chelation alone.8 Given that HMNDYT1 causes neurodegeneration,9 it is surprising that in individuals with significant disease progression, treatment with either chelator or iron leads to the improvement of neurological disease. This suggests that, in addition to neuronal loss, metal toxicity reversibly affects neuronal function similar to what is observed in Wilson's disease.10
In conclusion, an early diagnosis of HMNDYT1 is of paramount importance as it is a treatable neurological disorder. Iron supplementation alone presents a promising alternative to chelation, particularly in patients who do not tolerate or have no access to chelation therapy.
Author Roles
(1) Research Project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing of the First Draft, B. Review and Critique.
Z.Y.: 1A, 1B, 1C, 3A, 3B
K.T.: 3A, 3B
M.E.: 3B
Disclosures
Ethical Compliance Statement: This study was approved by the West London Research Ethics Committee. Verbal and written informed consent were obtained from the patient and the patient's guardian for publication of the video S1. The authors confirm that they have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest: KT was funded by a National Institute for Health Research (NIHR), (Academic Clinical Lecturership), the Academy of Medical Sciences, Action Medical Research and Great Ormond Street Hospital Children's Charity (UK) for this research project. This publication presents independent research funded by the NIHR. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months: The authors declare that there are no additional disclosures to report.
Supporting information
Video S1. Segment 1 shows the affected individual prior to treatment initiation at the age of 23 years. She is unable to sit in the wheelchair without support as a result of severe generalized dystonia at rest. Segment 2 shows the patient 13 months after the start of iron supplementation. She is able to sit in the wheelchair more comfortably although axial dystonia remains. On request, she is able to use her hands to eat and brush her hair and teeth. Her speech is clear.
Acknowledgments
We thank Cüneyt Bademcioğlu, Dr. Alper Alnak, Dr. Peter Clayton, Dr. Philippa Mills, Dr. Steve Wilson, Dr. Corinne Houart, and Dr. Aysegul Gunduz for their input and fruitful discussions.
Zuhal Yapici and Karin Tuschl contributed equally to this work.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Tuschl K, Clayton PT, Gospe SM, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet 2012;90:457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Quadri M, Federico A, Zhao T, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet 2012;90:467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zaki MS, Issa MY, Elbendary HM, et al. Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: Six novel SLC30A10 mutations and further phenotype delineation. Clin Genet 2018;93:905–912. [DOI] [PubMed] [Google Scholar]
- 4. Tuschl K, Clayton PT, Gospe SM Jr, Mills PB. Dystonia/parkinsonism, hypermanganesemia, polycythemia, and chronic liver disease In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993–2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK100241/ [Google Scholar]
- 5. Bowler RM, Koller W, Schulz PE. Parkinsonism due to manganism in a welder: neurological and neuropsychological sequelae. Neurotoxicology 2006;27:327–332. [DOI] [PubMed] [Google Scholar]
- 6. Fitsanakis VA, Zhang N, Garcia S, Aschner M. Manganese (Mn) and iron (Fe): interdependency of transport and regulation. Neurotox Res 2010;18:124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Avelino MA, Fusao EF, Pedroso JL, et al. Inherited manganism: the “cock‐walk” gait and typical neuroimaging features. J Neurol Sci 2014;341:150–152. [DOI] [PubMed] [Google Scholar]
- 8. Tuschl K, Mills PB, Parsons H, et al. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia—a new metabolic disorder. J Inherit Metab Dis 2008;31:151–163. [DOI] [PubMed] [Google Scholar]
- 9. Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW, Gospe SM Jr. Pathology of inherited manganese transporter deficiency. Ann Neurol 2014;75:608–612. [DOI] [PubMed] [Google Scholar]
- 10. Aggarwal A, Bhatt M. Advances in treatment of Wilson disease. Tremor Other Hyperkinet Mov 2018;8:525. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Segment 1 shows the affected individual prior to treatment initiation at the age of 23 years. She is unable to sit in the wheelchair without support as a result of severe generalized dystonia at rest. Segment 2 shows the patient 13 months after the start of iron supplementation. She is able to sit in the wheelchair more comfortably although axial dystonia remains. On request, she is able to use her hands to eat and brush her hair and teeth. Her speech is clear.