

ABSTRACT
Background
Adenylate cyclase 5 (ADCY5)‐related dyskinesia is a childhood‐onset movement disorder. Manifestations vary in frequency and severity and may include chorea, tremor, dystonia, facial twitches, myoclonus, axial hypotonia, and limb hypertonia. Psychosis is likely part of the broader spectrum. ADCY5 is widely expressed in the brain, especially in the striatum. Previous reports of brain autopsies of 2 subjects with likely ADCY5‐dyskinesia were limited by the absence of a molecular diagnosis. In 1 case, normal gross pathology was reported. In the other case, ADCY5 expression was not examined and neuropathological findings were confounded by age and comorbidities.
Objectives
To examine ADCY5 expression and neuropathological changes in ADCY5‐dyskinesia.
Methods
An extensive brain autopsy, including immunohistochemical analyses with antibodies to paired helical filament tau, α‐synuclein, amyloid‐β, microtubule‐associated protein 2, and ADCY5, was performed.
Results
The patient, with a p.M1029K ADCY5 variant, had severe dyskinesias from early childhood, later recurrent episodes of psychosis, and died at age 46. Gross pathology was unremarkable, but we detected increased immunoreactivity for ADCY5 in neurons in multiple brain regions. Despite no history of brain trauma to suggest chronic traumatic encephalopathy, we found tau deposits in the deep cortical sulci, midbrain, and hippocampus with minimal amyloid pathology and no Lewy bodies.
Conclusions
We present the first brain autopsy findings in a molecularly proven case of ADCY5‐dyskinesia, showing increased ADCY5 immunoreactivity in neurons and evidence of tau deposition. Additional patients will need to be studied to determine whether increased immunoreactivity for ADCY5 is a signature for ADCY5‐dyskinesia and whether this disease has a tauopathy component.
Keywords: ADCY5‐dyskinesia, tau, neurogenetics, neuropathology
Adenylate cyclase 5 (ADCY5) dyskinesia is a childhood‐onset movement disorder. It manifests primarily with a wide range of adventitious movements, including dystonia, tremor, chorea, spasticity, facial twitches, and myoclonus that vary in severity and periodicity.1, 2 Associated abnormalities include axial hypotonia, limb hypertonia, mild‐to‐moderate cognitive impairment, and likely other neurobehavioral problems such as attention deficit hyperactivity disorder, depression, and psychosis.1, 3 The disease is usually transmitted as an autosomal‐dominant trait, but two families with a recessive pattern of inheritance have been reported.4, 5 Many sporadic cases and some progenitors in multigenerational families are found to be somatic mosaics for the ADCY5 variant.1
Adenylate cyclases are a family of enzymes that convert adenosine‐5′‐triphosphate to 3′,5′‐cyclic adenosine monophosphate (cAMP), a second messenger in signal transduction.6 The nine closely related membrane‐bound mammalian isoforms are classified into six distinct groups based on tissue specificity and patterns of regulation by stimulators and inhibitors. ADCY5 is widely expressed in the brain, most highly in medium spiny neurons of the striatum, a region involved in modulating movement. At least 15 pathogenic variants in ADCY5 have been published. All five missense and in‐frame deletion variants that we studied in vitro confer increased ability to generate cAMP in response to beta‐adrenergic or Gαs‐mediated stimulation and significantly reduce inhibition following D2 dopamine receptor activation.7, 8 The homozygous missense variants reported in two families4, 5 and a splice site variant in another9 are thought to act through loss of function.
With the exception of Huntington's and Parkinson's diseases, there is a dearth of reports on neuropathological correlates of the heterogenous group of inherited dystonias, given the difficulty in obtaining brain tissues from patients.10 Characterization of the brain findings may be helpful in distinguishing among the dystonias and giving clues to the pathogeneses. Brain autopsies of 2 individuals who likely carried pathogenic variants in ADCY5 have been reported, but the molecular diagnoses were not known before death and ADCY5 expression was not studied. One of these cases is a man, described as II.3 by McCann and colleagues,11 and subject 3 in pedigree K5 by Chang and colleagues,2 with a personal history consistent with mild ADCY5‐dyskinesia, and descendants with the recurrent p.R418G variant. His autopsy reportedly revealed abundant α‐synuclein Lewy bodies and low Alzheimer's disease (AD) neuropathological change (A1B2C1), characterized by mild amyloid‐β (Aβ) plaque deposition in most cortical regions and rare neurofibrillary tangles confined to the hippocampus. His age at death of 84 years and comorbidities of emphysema, angina, pituitary tumor, glucose intolerance, and celiac disease make it difficult to determine mutant ADCY5‐specific effects. Autopsy of a woman who likely carried a R418Q ADCY5 variant, based on her neurological manifestations and the presence of the variant in her son, and who died of suicide at age 26, was stated to be normal, but details of the evaluation were not provided.3 We now report brain autopsy findings in a woman with a p.M1029K mutation in the C2 domain of ADCY5 whom we followed for more than three decades.
Materials and Methods
Gross Neuropathology Evaluation
The brain‐only autopsy was performed by leadership and staff of the University of Washington (UW) Neuropathology Core. The brain was removed and immersion fixed in 10% neutral buffered formalin for at least 2 weeks, at which point the gross exam was performed with photographs and then the cerebellum and brainstem were removed from the cerebrum by axial transection of the midbrain at the level of the oculomotor nerves. The cerebrum was sectioned coronally into uniform 4‐mm slices after agarose embedding; the brainstem and cerebellum were similarly sectioned axially and parasagitally (respectively). Sections were laid out and photographed under uniform conditions and the tissue examined by neuropathologists (C.S.L., C.D.K.) to identify any developmental anomalies, focal lesions, or other abnormalities. In addition to the 22 samples routinely submitted for brain autopsy cases, composite sections of neostriatum and medial, middle, and lateral cerebellum were taken for a broader evaluation of pathology relevant to ADCY5 mutation. Samples were processed in standard fashion and embedded in paraffin for subsequent histological protocols.
Histological Evaluation
Routine and additional histological processes were performed in the UW Neurohistology Laboratory, where histological sections (4 μm thick) were cut from formalin‐fixed, paraffin‐embedded tissue blocks, mounted on charged glass microscope slides, and submitted for staining. Hematoxylin and eosin (H&E) as well as combined H&E/Luxol fast blue stains were prepared from all blocks. Bielschowsky silver stain was performed according to routine protocols.
Glial Fibrillary Acidic Protein, pTau, 3R tau, 4R tau, Aß, and α‐Synuclein Immunostaining
Using previously optimized conditions in the Neurohistology Laboratory, immunohistochemistry (IHC) was performed using a Leica Bond III Fully Automated IHC and in situ hybridization Staining System (Leica Bio‐Systems, Buffalo Grove, IL). Select brain regions were immunostained with rabbit polyclonal antibodies against glial fibrillary acidic protein (#Z033401‐2, 1:2000; Dako, Carpinteria, CA), mouse monoclonal antibodies against paired helical filament tau (PHF‐tau; AT8, 1:1000 dilution; Pierce, Rockford, IL), 4R tau (1E1/A6, 1:200 dilution; Millipore, Billerica, MA), α‐synuclein (LB509, 1:250 dilution; Invitrogen, Carlsbad, CA), and Aβ (6E10, 1:5000 dilution; Covance, Princeton, NJ). Manual IHC staining was performed for 3R tau (8E6/C11, 1:800 dilution; Millipore). Appropriate positive and negative controls were included.
ADCY5 and Microtubule‐Associated Protein 2 Immunostaining
Additional IHC and immunofluorescence staining protocols were developed for this study. Expression and localization of ADCY5 was assessed in the patient and in 4 sex‐ and age‐matched subjects who had no clinical or pathological evidence of central nervous system disease. Microtome sections (4 μm thick) were cut from paraffin‐embedded brain tissues from the frontal lobe, striatum, and cerebellum from the patient and controls. After deparaffinization with Histo‐Clear (National Diagnostics, Atlanta, GA) and rehydration with graded alcohols (100%, 95%, and 70%), antigen retrieval was carried out by boiling slides in citrate buffer (pH 6.0; Sigma‐Aldrich, St Louis, MO) in a pressure cooker for 10 minutes followed by peroxidase blocking in H2O2 for 10 minutes. To block nonspecific protein binding, 10% goat serum was applied to tissues. Immunochemical staining against ADCY5 was performed using a polymer‐HRP (horseradish peroxidase) detection kit (GBI Labs, Bothell, WA) with incubation with ADCY5 antibody at 1:100 (ABS573; Millipore) overnight and then a second antibody polymer HRP immunoglobulin G for 60 minutes. DAB (3,3‐diaminobenzidine) developing was monitored under a microscope, followed by rinsing in phosphate‐buffered saline. Images were visualized and captured on an Olympus microscope (FSX100; Olympus Corporation, Tokyo, Japan).
For immunofluorescence double staining of ADCY5 and microtubule‐associated protein 2 (MAP2), deparaffinization and antigen retrieval were performed as above. The staining procedure utilized the MaxDouble kit (MaxVision Biosciences, Kenmore, WA), which includes steps of autofluorescence reduction and antibody signal amplification. Antibodies to rabbit‐ADCY5 (ABS573, 1:100; Millipore) and mouse‐MAP2 (AP20, 1:1600; Thermo Fisher Scientific) were applied to the slides for overnight incubation, followed by incubation with MaxFluor 550 conjugated antirabbit and MaxFluor 488 conjugated antimouse antibodies from the MaxDouble kit for 60 minutes. Stained slides were mounted with antifading mounting medium with 4',6‐diamidino‐2‐phenylindole (DAPI; Prolong Gold; Thermo Fisher Scientific), and images were captured on an Olympus fluorescence microscope with appropriate filter settings. The captured images were processed and analyzed with ImageJ software (NIH, Bethesda, MD).
With anti‐ADCY5 chromogenic IHC staining, we evaluated ADCY5 signal intensity in the frontal cortex (cortical layers II–V), cerebellum, putamen, and caudate. After color deconvolution to separate color panels, and calibration, a DAB‐only image was selected. For each subject, we randomly selected and outlined 20 cells as the region of interest (ROI). Mean DAB intensity in each selected cell was measured by ImageJ, and the final DAB intensity density was calculated as maximum intensity minus mean intensity over the ROI in each cell (IntDen), where maximum intensity is 255 in 8‐bit images.
Statistical Analysis
Statistical analyses were done with Prism software (GraphPad Software Inc., La Jolla, CA), and scatter and box‐whiskers plots were generated. To be cautious regarding the results in a single case, we did not conduct P‐value analysis.
Results
Patient Information
A longitudinal clinical history of the patient and her family was previously described. She is subject III‐6 in Figure 2B of Chen and colleagues.1 Briefly, her motor milestones were delayed, by age 4 years she developed dysarthria and marked chorea of all extremities, face, and tongue, and by age 8 years she had severe jerky and spasmodic movements, poor head control, contractures of knees and ankles, and required a wheelchair. Tendon reflexes were brisk with ankle clonus. From her mid‐thirties, she had recurrent episodes of psychosis with delusions and visual and auditory hallucinations. She graduated from high school in special education, but at age 46 years her Mini‐Mental State Examination score was 16/26. A noncontrast brain CT scan performed at age 41 years for paranoia and delusions suggested mildly enlarged lateral ventricles. Despite progressive dyskinesia and spasticity, she had no documented falls or other incidents of head trauma. At age 46 years, without recent worsening of her dystonia or intercurrent illness, she was found deceased in her bed by her attendant.
Figure 2.
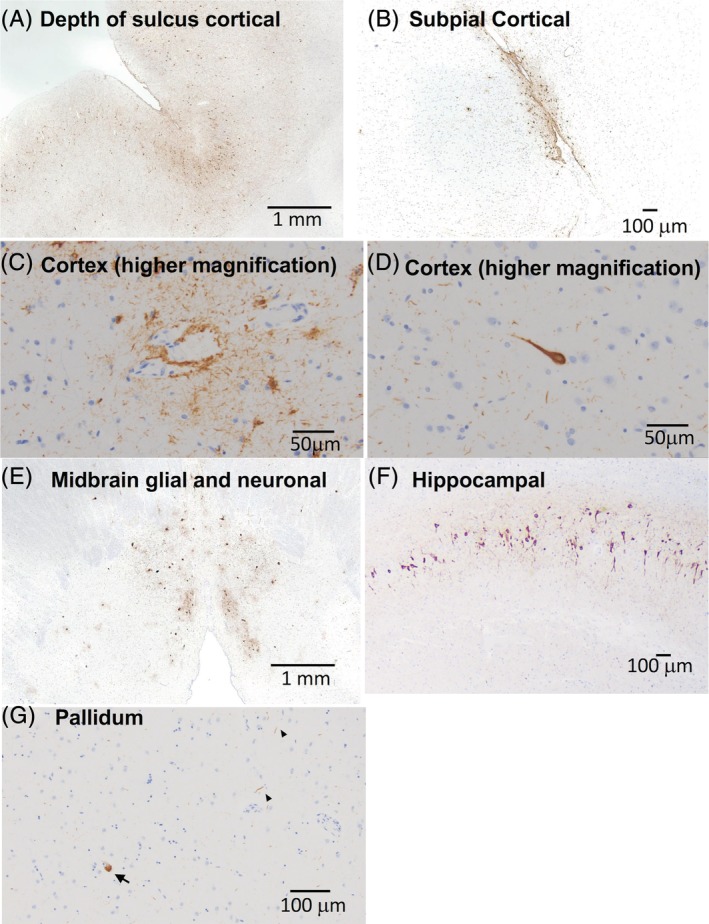
Microscopic neuropathology in a patient with ADCY5‐dyskinesia. PHF‐tau immunostained slides demonstrate patchy, but widespread, pTau pathology. (A) Sections of neocortex demonstrate neurofibrillary tangles along gyri as well as at the depths of sulci. (B) Cortical tau also consisted of subpial, astroglial tau inclusions. (C) Perivascular glial and neuritic tau pathology in the cortex. (D) High magnification of a neurofibrillary tangle. (E) Section of midbrain shows glial and neuronal tau in periaqueductal gray matter. (F) Section of hippocampus demonstrates neurofibrillary tangles in the CA1 subfield and (G) scattered tau neurites (arrowheads) and pretangles (arrow) of uncertain significance in the pallidum.
Neuropathology
At autopsy, there were no significant gross pathological findings with the exception of mild pallor of the substantia nigra (SN). Notably, the neostriatum was well developed without gross atrophy or diffuse or focal lesions (Fig. 1). Cerebral cortex, subcortical white matter, deep cerebral nuclei, brainstem, and cerebellum were well developed with no focal lesions. On histological sections, nigral pallor corresponded to mild loss of pigmented neurons, normal for age, without significant gliosis or neuron loss in the other brainstem nuclei, cerebral cortex, deep cerebral nuclei, or cerebellum. Surprisingly, comprehensive neurodegenerative disease IHC workup revealed widespread tau pathology (Fig. 2), including neurofibrillary tangles, neuritic tau, and glial tau in multiple forms in cerebral cortex, midbrain, thalamus, and hippocampus. Specifically, paired helical filament (PHF)‐tau was identified in the form of astroglial inclusions, particularly in the depths of sulci and sometimes in a perivascular distribution, in frontal, parietal, and temporal cortex, including scattered thorn‐shaped astrocytes. PHF‐tau positive neurofibrillary tangles and neuritic tau were present along gyri. Hippocampal tau was present in a typical Braak progression pattern, involving the transentorhinal, cornu ammonis subfields, subiculum, and entorhinal cortex. Rare tangles were also identified in the neocortex, often at the depths of sulci; therefore, a Braak stage V/VI could be assigned in this case. However, Braak staging is confounded by the presence of features that overlap an underlying tauopathy. Tau pathology was also identified in brainstem nuclei in the form of neurofibrillary and globose tangles, and there were scattered tau neurites and pretangles of uncertain significance in the basal ganglia, including the striatum. In addition to PHF‐tau, immunostains for 3R and 4R tau isoforms were performed in select regions of the hippocampus, frontal cortex, and striatum (data not shown). Similar amounts of both isoforms were identified in the hippocampus and frontal cortex, indicating that this process is a mixed 3R/4R tauopathy. In the striatum, 4R tau was identified, but definite 3R tau was not appreciated; however, the tau pathology in this region was very sparse and therefore harder to interpret than in the hippocampus and frontal cortex. AD pathology was very low with rare diffuse amyloid plaques limited to the neocortex (data not shown). Numerous pale bodies were noted within the remaining pigmented neurons of the SN, but Lewy bodies were not identified, and the pale bodies were not immunoreactive for α‐synuclein (data not shown).
Figure 1.
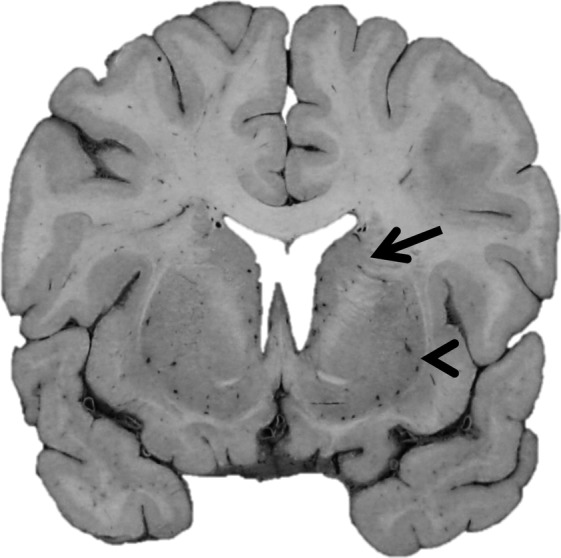
Gross pathology in a patient with ADCY5‐dyskinesia. Coronal section of the brain at the level of the anterior commissure demonstrating well‐formed caudate nucleus (arrow) and putamen (arrowhead) as well as no significant cortical atrophy or ventriculomegaly.
Expression and Localization of ADCY5
Based on its role in coordination of movement and on the high neostriatal expression of ADCY5, the neostriatum is suspected to be the primary brain region involved in ADCY5‐dyskinesia. However, no gross or cytomorphological abnormalities were detected in the caudate nucleus, putamen, or globus pallidus of the patient. Recognizing the possibility that occult pathological changes undetectable by usual approaches could be at work, we investigated the possibility that expression and/or localization of ADCY5 might be altered in the patient in the putamen, caudate, cerebellum, and frontal cortex, regions where ADCY5 is highly expressed.
MAP2 staining, a marker of mature neurons, was utilized to further evaluate neuronal populations and cytoarchitecture and revealed no significant neuron loss or pathological cytomorphologies. Double immunofluorescence staining of ADCY5 and MAP2 revealed that ADCY5 was present in all MAP2‐positive cells in all tested brain regions and ADCY5 was not identified in MAP2‐negative cells (Fig. 3). These results demonstrated that ADCY5 is expressed only in neurons and that all neurons express ADCY5. We compared this patient with control cases and discovered subtle differences in cytological patterns of ADCY5 localization, primarily characterized by increased cytoplasmic granularity without obvious intranuclear, cytoplasmic, or extracellular inclusions (Fig. 3A). Compared to the control subjects, there was no loss or decrease in size of neurons in the patient. Furthermore, in ADCY5 IHC with DAB chromogen, semiquantitative analysis showed stronger intensity of ADCY5 in all tested brain regions in the patient compared to the controls (Fig. 4).
Figure 3.
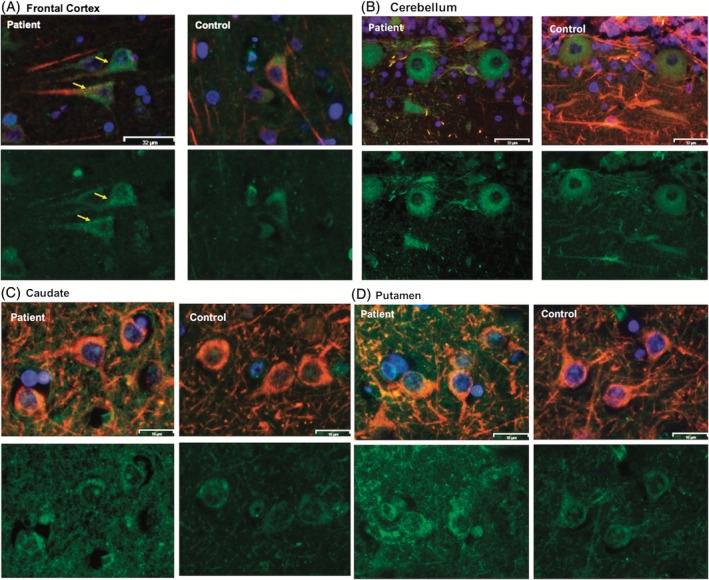
Expression and localization of ADCY5 in multiple brain areas in a patient with ADCY5‐dyskinesia. Double immunofluorescence staining of ADCY5 (in green) and the mature‐neuron marker, MAP2 (in red), in a patient with an ADCY5 p.M1029K missense variant and four age‐ and sex‐matched controls (1 representative control is shown). Nuclei are stained blue by DAPI. Multiple regions were evaluated: frontal cortex (A), cerebellum (B), caudate (C), and putamen (D). In each panel, top images show merged ADCY5 and MAP2, and the lower ones show ADCY5 only. Merged ADCY5 and MAP2 images show presence of ADCY5 in MAP2‐positive neurons, and not in MAP2‐negative cells, in all tested brain regions in the patient. Increased cytoplasmic granularity of ADCY5 is visible in the patient's cells (arrows in yellow), not seen with MAP2 staining only. Size bar: (A) and (B), 32 μm; (C) and (D), 16 μm.
Figure 4.
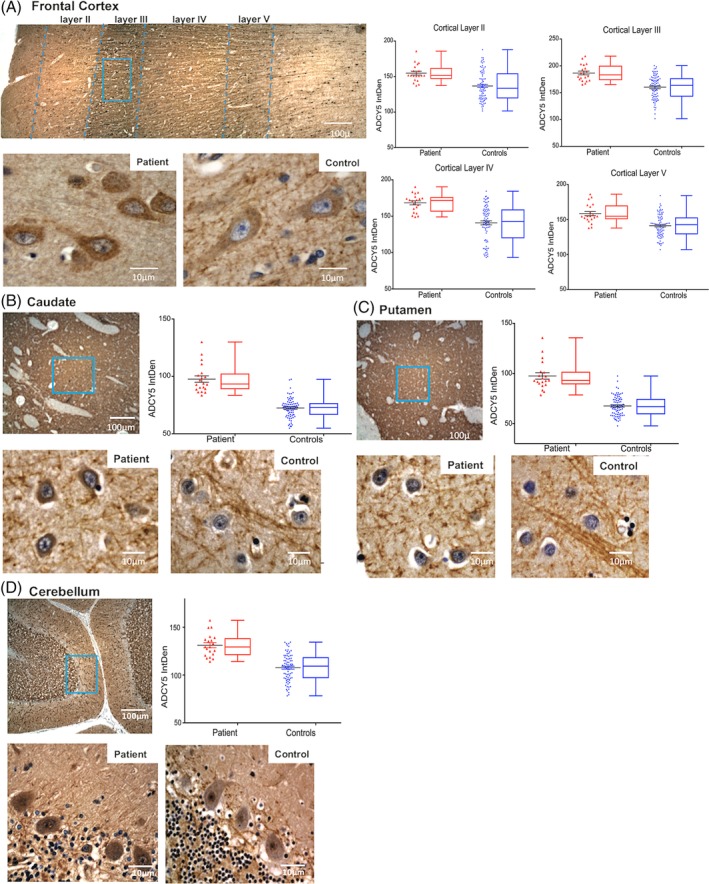
Quantitative assessment of ADCY5 staining intensity in multiple brain regions in the patient with ADCY5‐dyskinesia. DAB chromogen IHC staining for ADCY5 in the patient and 4 age‐ and sex‐matched controls. In panel (A), the top image is full thickness of gray matter in the frontal cortex from the patient showing layers I through VI. Below, the higher‐magnification images show representative sections from that area framed in blue from the patient (left) and a control (right) stained for ADCY5. Similarly, panels (B), (C), and (D) are caudate, putamen, and cerebellum, respectively. For each area, the scatter and box‐whiskers plots display quantitative measurements of the DAB chromogen intensity of ADCY5 in 20 randomly selected cells in each subject. Results from the patient (data from 20 cells) are labeled in red, and results from 4 controls (combined data from a total of 80 cells) are grouped and labeled in blue. The results demonstrate increased ADCY5 staining intensity in the patient in all brain regions studied.
Discussion
Brain autopsies from the 2 previously reported cases who likely had ADCY5‐dyskinesia and the case we report on herein showed no gross abnormalities. Specifically, we did not observe atrophy of neostriatal nuclei or other brain regions. As summarized by Sharma,10 from the limited neuropathological studies in other dystonias, there does not seem to be a consistent pattern of neuronal loss within the striatum, cerebellum, or brainstem. Similarly, we did not detect significant neuron loss or prominent astro‐ or microgliosis in association with the pathogenic ADCY5 variant, p.M1029K, in any of the regions we tested—striatum, cerebrum, and cerebellum. Our finding from pathological evaluation contrasts somewhat with those from an imaging study of 3 patients with the p.R418W variant in which significant cortical gray matter loss in multiple cortical areas and thalamus were reported.12 No significant atrophy was found in the striatum. The differences found in the cerebral cortex between our study and those of Niccolini and colleagues may reflect different effects of different genetic variants. Alternatively, they may be related to the specific manifestations in the patients. The differences may also be associated with the specificity and sensitivity of evaluation methods.
Although of unclear biological significance, we were struck by the widespread increased ADCY5 immunoreactivity in a granular pattern in neurons throughout the brain. We have previously shown that this missense variant confers gain of function in vitro with respect to generation of cAMP upon stimulation.8 The relationship of increased function to increased ADCY5 immunoreactivity is unknown. Perhaps the mutation affects ADCY5 degradation, allowing it to accumulate. This is in agreement with our observation of increased mutant M1029K‐ADCY5 protein level in comparison to wild type in cells transfected with ADCY5 in vitro (data not shown).
In addition to abnormal patterns of ADCY5 cytoplasmic staining, this case was characterized by prominent neuronal and glial pTau pathology distributed widely throughout the brain in the absence of prominent amyloid accumulation. We evaluated the brain for diagnostic features of diverse tauopathies, including PSP, corticobasal degeneration, other frontotemporal lobar degenerations with tau positive inclusions, and primary age‐related tauopathy and AD. The pattern of tau lesions, which included both 3R and 4R tau isoforms by IHC, was uncharacteristic for any particular tauopathy, but rather overlapped with many. We recognize that the findings could represent early neuropathological changes of AD,13, 14 primary age‐related tauopathy,15 or aging‐related tau astrogliopathy,16 but the patterns do not fit and these diagnoses seem unlikely in this age group. We also considered chronic traumatic encephalopathy (CTE), which is characterized by glial and neuronal pTau lesions preferentially in the depths of cortical sulci in a perivascular distribution.17 The pathognomonic neuropathological lesions of CTE were not obvious in our case. It is also notable that despite her severe movement disorder, there is no record of significant head trauma or falls. Some pTau deposition in the hippocampus found in the case described by McCann and colleagues was attributed to low AD neuropathological change.11 Because of the paucity of published ADCY5‐dyskinesia cases with autopsy data, it is unknown whether tau pathology is a component of ADCY5‐related processes. Disentangling the contribution of tau pathology from CTE versus those associated with possible early AD changes or ADCY5‐related processes is challenging and will require the neuropathological evaluation of additional cases.
Additional neuropathological findings included mild loss of pigmented neurons in the SN and numerous pale bodies, which is not necessarily abnormal for this age. Unlike the elderly case reported by McCann and colleagues,11 IHC stains for Lewy bodies were negative and the pale bodies were not immunoreactive for α‐synuclein. The significance of pale bodies is not well understood, although there are limited studies suggesting a relationship to Lewy bodies, at least in cases of Lewy body disease.18 Alternatively, these changes could be related to the tauopathy.
It is important to note that our patient inherited the variant from her mosaic mother and transmitted it to her children.1 In contrast, the relatively mild neurological manifestations of the case reported by McCann and colleagues11 and his pedigree position as the progenitor suggest that he might have been mosaic for the pathological variant.1 Mosaicism might affect the type and severity of brain pathology.
Finally, it is becoming clear that psychosis and other psychiatric disorders are likely part of the broader spectrum of ADCY5‐dyskinesia.1, 3, 5 Patients in the early stages of frontotemporal dementia, another tauopathy,19 have been given a diagnosis of schizophrenia before development of substantial cognitive impairment. Additional brain autopsies will need to be evaluated to determine whether the tau abnormalities in our case comprise a pathological signature of ADCY5‐dyskinesia and whether it is restricted to cases with psychosis.
Author Roles
(1) Research Project A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
D.‐H.C.: 1A, 1B, 1C, 2A, 2B, 3A, 3B
C.S.L.: 1A, 1C, 3A, 3B
M.S.: 1C, 2B
P.K.: 1C
M.Y.D.: 1C, 3B
L.F.G.‐C.: 3B
C.D.K.: 1A, 1B, 2C, 3B
T.D.B.: 1A, 1B, 2C, 3A, 3B
W.H.R.: 1A, 1B, 3A, 3B
Disclosures
Ethical Compliance Statement
University of Washington (UW) Institutional Review Board (IRB) approved the study, with the protocol reference STUDY00002022. The patient provided informed consent to participate in research. Consent for brain autopsy was provided by her legal next of kin, according to protocols approved by the UW IRB. We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflicts of Interest
This work was supported by grants R01NS069719 and P50AG005136 from the National Institutes of Health, Merit Review Award Number 101 CX001702 from the United States Department of Veterans Affairs, and the Nancy and Buster Alvord Endowment. The authors report no conflicts of interest.
Financial Disclosures for previous 12 months
Dong‐Hui Chen, Caitlin S. Latimer, Min Spencer, Prasanthi Karna, Luis F. Gonzalez‐Cuyar, C. Dirk Keene, and Wendy H. Raskind are employed by the University of Washington. Dong‐Hui Chen receives royalties for patent 10,036,067 (Mutations in PKCγ are the cause for spinocerebellar ataxia). Marie Y. Davis is employed by the VA Puget Sound Health Care System. Thomas D. Bird holds emeritus status at the University of Washington and receives royalties for patent 10,036,067 (Mutations in PKCγ are the cause for spinocerebellar ataxia). Wendy H. Raskind serves on the advisory boards for http://adcy5.org and the University of Washington Huntington Disease Center of Excellence, and she receives royalties for patent 10,036,067 (Mutations in PKCγ are the cause for spinocerebellar ataxia).
Acknowledgments
We thank Ms. Kim Howard and Dr. Pamela McMillan for outstanding technical support, Ms. Allison Beller for coordination of research efforts and support in case selection, Drs. Ellen Wijsman and Nicola Chapman for suggestions in biostatistics analysis, and Dr. Brian Kraemer for useful discussion.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Chen DH, Meneret A, Friedman JR, et al. ADCY5‐related dyskinesia: broader spectrum and genotype‐phenotype correlations. Neurology 2015;85:2026–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chang FC, Westenberger A, Dale RC, et al. Phenotypic insights into ADCY5‐associated disease. Mov Disord 2016;31:1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vijiaratnam N, Newby R, Kempster PA. Depression and psychosis in ADCY5‐related dyskinesia‐part of the phenotypic spectrum? J Clin Neurosci 2018;57:167–168. [DOI] [PubMed] [Google Scholar]
- 4. Barrett MJ, Williams ES, Chambers C, Dhamija R. Autosomal recessive inheritance of ADCY5‐related generalized dystonia and myoclonus. Neurol Genet 2017;3:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bohlega SA, Abou‐Al‐Shaar H, AlDakheel A, et al. Autosomal recessive ADCY5‐related dystonia and myoclonus: expanding the genetic spectrum of ADCY5‐related movement disorders. Parkinsonism Relat Disord 2019;64:145–149. [DOI] [PubMed] [Google Scholar]
- 6. Linder JU. Class III adenylyl cyclases: molecular mechanisms of catalysis and regulation. Cell Mol Life Sci 2006;63:1736–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen YZ, Friedman JR, Chen DH, et al. Gain‐of‐function ADCY5 mutations cause familial dyskinesia with facial myokymia. Ann Neurol 2014;75:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doyle TB, Hayes MP, Chen DH, Raskind WH, Watts VJ. Functional characterization of AC5 gain‐of‐function variants: impact on the molecular basis of ADCY5‐related dyskinesia. Biochem Pharmacol 2019;163:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carapito R, Paul N, Untrau M, et al. A de novo ADCY5 mutation causes early‐onset autosomal dominant chorea and dystonia. Mov Disord 2015;30:423–427. [DOI] [PubMed] [Google Scholar]
- 10. Sharma N. Neuropathology of dystonia. Tremor Other Hyperkinet Mov (N Y) 2019;9:569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. McCann H, Fung VS, Klein C, Halliday GM. Unusual alpha‐synuclein and cerebellar pathologies in a case of hereditary myoclonus‐dystonia without SGCE mutation. Neuropathol Appl Neurobiol 2015;41:837–842. [DOI] [PubMed] [Google Scholar]
- 12. Niccolini F, Mencacci NE, Yousaf T, et al. PDE10A and ADCY5 mutations linked to molecular and microstructural basal ganglia pathology. Mov Disord 2018;33:1961–1965. [DOI] [PubMed] [Google Scholar]
- 13. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 2014;128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kovacs GG, Ferrer I, Grinberg LT, et al. Aging‐related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 2016;131:87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McKee AC, Cairns NJ, Dickson DW, et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 2016;131:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dale GE, Probst A, Luthert P, Martin J, Anderton BH, Leigh PN. Relationships between Lewy bodies and pale bodies in Parkinson's disease. Acta Neuropathol 1992;83:525–529. [DOI] [PubMed] [Google Scholar]
- 19. Gambogi LB, Guimaraes HC, de Souza LC, Caramelli P. Long‐term severe mental disorders preceding behavioral variant frontotemporal dementia: frequency and clinical correlates in an outpatient sample. J Alzheimers Dis 2018;66:1577–1585. [DOI] [PubMed] [Google Scholar]