

https://onlinelibrary.wiley.com/page/journal/23301619/homepage/mdc312858-sup-v001.htm
X‐linked adrenoleukodystrophy (X‐ALD, Online Mendelian Inheritance in Man #300100) is a genetic peroxisomal disorder caused by mutations in the adenosine triphosphate (ATP)‐binding cassette subfamily D member 1 gene. Impaired peroxisomal beta‐oxidation of very long chain fatty acids (VLCFA) results in VLCFA accumulation in all tissues. However, it mainly affects the adrenal cortex and the central nervous system. This condition exhibits multiple phenotypes and its pathophysiological role remains uncertain. Some patients may present with either an inflammatory demyelination (cerebral form) or a progressive axonopathy of the spinal cord (myeloneuropathy and rarely spastic paresis). Herein we report a case of a patient with late‐onset X‐ALD who presented with progressive spastic paraplegia resembling hereditary spastic paraplegia (HSP).
A 24‐year‐old male patient was referred to our service with a history of lower limb pain, progressive gait impairment, and low blood pressure for 1 year. He did not show any behavioral changes, cognitive symptoms, or incontinence. His family history was remarkable: mother and grandmother with mild symptoms and 2 maternal uncles with same symptoms. Examination of the proband disclosed proximal weakness of lower limbs, moderate to severe spasticity, and bilateral Babinski and Hoffman signs (Video S1). No sensory or cranial nerve abnormalities were found. The presence of alopecia and skin and mucosal hyperpigmentation (Fig. 1) led to a suspected diagnosis of adrenal insufficiency. Low serum cortisol levels and sustained low cortisol response in the adrenocorticotropic hormone stimulation test confirmed Addison's disease.
Figure 1.
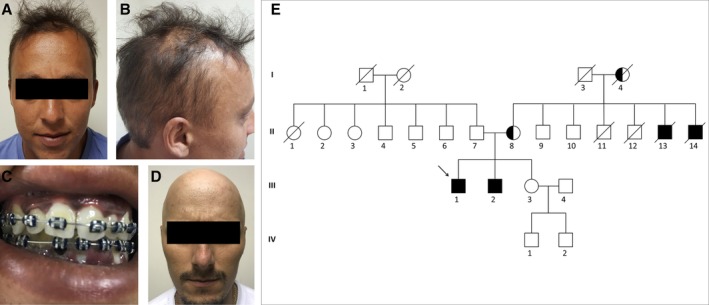
Index patient with X‐linked adrenoleukodystrophy and adrenal dysfunction presenting with skin hyperpigmentation (A) and alopecia (B) associated with gingival hyperpigmentation (C). Sibling of the index patient presenting with alopecia (D). Family pedigree of the index patient (black arrow; E).
Magnetic resonance imaging of the brain and spine was normal. Acquired causes of myelopathy were ruled out. The measurement of plasma VLCFA levels showed increased hexacosanoic acid levels and a high ratio of hexacosanoic to docosanoic acid levels. Therefore, the diagnosis of X‐ALD was established, and he has been on prednisone and fludrocortisone since then. Six years later, his 28‐year‐old brother presented with mild similar neurologic complaints and alopecia (Fig. 1D).
The defect in the ATP‐binding cassette subfamily D member 1 gene maps to Xq28 and has almost 100% penetrance. X‐ALD has a wide spectrum of clinical manifestations that cannot be predicted with currently available genetic tests or VLCFA levels. The 3 most common clinical phenotypes are (1) cerebral ALD that often manifests during childhood at a mean age of 7 years; (2) adrenomyeloneuropathy, which is the most frequent form in adults; and (3) isolated Addison's disease.1, 2
Late‐onset forms of X‐ALD may mimic HSP, particularly when family history is remarkable. Whether this presentation is a subtype of adrenomyeloneuropathy or another phenotypical manifestation of the disease is not yet clear. The fact there is no sphincter dysfunction, or peripheral neuropathy may suggest otherwise.3 The HSP‐like cases reported show similar baseline characteristics with symptom onset in the third decade of life and normal brain magnetic resonance imaging.3, 4, 5 An X‐linked inheritance or signs of adrenal dysfunction should guide genetic testing or measurement of VLCFA levels for X‐ALD investigation. These considerations should not be limited to male patients.3, 4, 5
In conclusion, X‐ALD should be biochemically and genetically investigated in patients with progressive spastic paraplegia resembling HSP. We suggest measuring plasma VLCFA levels in all of these patients.
Author Roles
(1) Research project: A. Conception, B. Organization, C. Execution; (2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; (3) Manuscript: A. Writing of the first draft, B. Review and Critique.
V.B.C.: 1A, 1B, 1C, 3A, 3B
J.L.F.: 1A, 1B, 1C, 3B
O.G.P.B.: 1A, 1B, 1C, 3B
J.L.P.: 1A, 1B, 1C, 3A, 3B
Disclosures
Ethical Compliance Statement
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines. The authors confirm that the approval of an institutional review board was not required for this work. Oral and written informed consent was obtained from the patient.
Funding Sources and Conflict of Interest
The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the Previous 12 Months
The authors declare that there are no additional disclosures to report.
Supporting information
Video S1. Segment 1: index case with X‐linked adrenoleukodystrophy presenting with spastic gait and increased deep tendon reflexes similar to patients with hereditary spastic paraplegia. Segment 2: sibling of the index patient presenting with spastic gait.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Moser HW, Mahmood A, Raymond GV. X‐linked adrenoleukodystrophy. Nat Clin Pract Neurol 2007;3(3):140–151. 10.1038/ncpneuro0421 [DOI] [PubMed] [Google Scholar]
- 2. Kemp S, Berger J, Aubourg P. X‐linked adrenoleukodystrophy: clinical, metabolic, genetic and pathophysiological aspects. Biochimica et Biophysica Acta 2012;1822(9):1465–1474. 10.1016/j.bbadis.2012.03.012 [DOI] [PubMed] [Google Scholar]
- 3. Maris T, Androulidakis EJ, Tzagournissakis M, Papavassiliou S, Moser H, Plaitakis A. X‐linked adrenoleukodystrophy presenting as neurologically pure familial spastic paraparesis. Neurology 1995;45:1101–1104. 10.1212/WNL.45.6.1101 [DOI] [PubMed] [Google Scholar]
- 4. Zhan ZX, Liao XX, Du J, et al. Exome sequencing released a case of X‐linked adrenoleukodystrophy mimicking recessive hereditary spastic paraplegia. Eur J Med Genet 2013;56(7):375–378. 10.1016/j.ejmg.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 5. Kim A, Kumar KR, Davis RL, et al. Increased diagnostic yield of spastic paraplegia with or without cerebellar ataxia through whole‐genome sequencing. Cerebellum 2009;18(4):781–790. 10.1007/s12311-019-01038-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video S1. Segment 1: index case with X‐linked adrenoleukodystrophy presenting with spastic gait and increased deep tendon reflexes similar to patients with hereditary spastic paraplegia. Segment 2: sibling of the index patient presenting with spastic gait.