

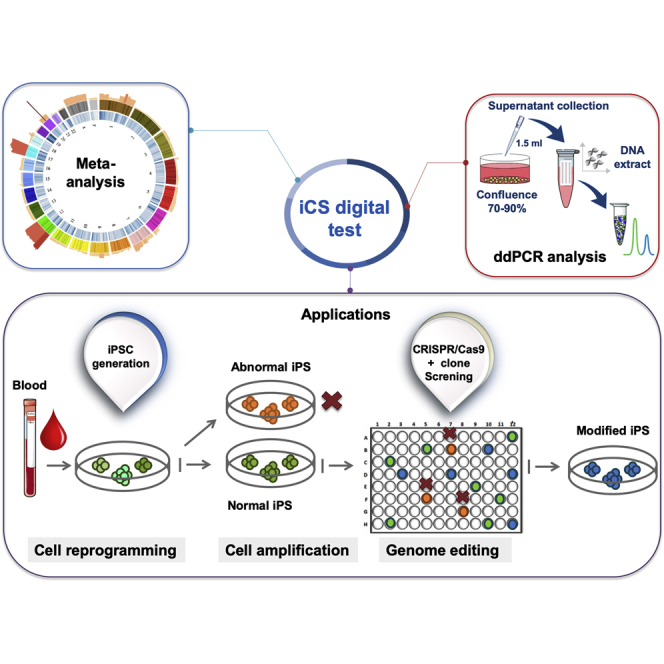
Summary
Genomic integrity of human pluripotent stem cells (hPSCs) is essential for research and clinical applications. However, genetic abnormalities can accumulate during hPSC generation and routine culture and following gene editing. Their occurrence should be regularly monitored, but the current assays to assess hPSC genomic integrity are not fully suitable for such regular screening. To address this issue, we first carried out a large meta-analysis of all hPSC genetic abnormalities reported in more than 100 publications and identified 738 recurrent genetic abnormalities (i.e., overlapping abnormalities found in at least five distinct scientific publications). We then developed a test based on the droplet digital PCR technology that can potentially detect more than 90% of these hPSC recurrent genetic abnormalities in DNA extracted from culture supernatant samples. This test can be used to routinely screen genomic integrity in hPSCs.
Keywords: pluripotent stem cells, induced pluripotent stem cells, genetic abnormalities, pluripotency, chromosome instability, genetic integrity, quality control, cell-free DNA, ddPCR
Graphical Abstract
Highlights
-
•
A meta-analysis was carried out to list genetic abnormalities detected in hPSCs
-
•
Recurrent genetic abnormalities in hPSCs were precisely defined
-
•
ddPCR is a robust and sensitive approach to screen these recurrent abnormalities
-
•
In culture supernatant digital test can be used to rapidly screen iPSC clones
In this article, De Vos and colleagues performed a comprehensive meta-analysis of genetic abnormalities detected in hPSC lines. They then used this information to devise a ddPCR test targeting the majority of recurrent abnormalities and show that this test can be applied to the cell culture supernatant. Finally, they illustrate the interest of this test for routine hPSC screening.
Introduction
Human pluripotent stem cells (hPSCs) are a physiologically relevant cell material for research (in vitro modeling of human development and diseases) and regenerative medicine/cell therapies. Therefore, it is crucial that the hPSC genome remains the faithful genetic copy of the cells from which they were derived. However, genetic abnormalities (e.g., karyotype abnormalities) can arise in hPSCs, for example, during cell reprogramming, cell culture, or genome editing (Assou et al., 2018). Many of these genetic abnormalities are often recurrent. For example, gains of chromosome 12 (most frequently 12p), 17 (particularly 17q), 20, or X have been often detected using standard cytogenetic procedures (G-banding) (Lefort et al., 2009). Sub-chromosomal abnormalities, such as 20q11.21 amplification, also can be recurrent (Lefort et al., 2008). The biological significance of such recurrent abnormalities is still discussed, but they might result in a strong selective growth advantage for cultured cells, as already demonstrated for the 20q11.21 amplification (Zhang et al., 2019). Therefore, it is crucial to carefully catalog all genetic alterations found in hPSCs and identify the recurrent ones. To this aim, we carried out a meta-analysis of published genetic abnormalities found in hPSCs. We could give a precise definition of recurrent genetic abnormality and then listed all of them in a large dataset. As these recurrent genetic abnormalities are found in specific genomic regions, we developed a focused droplet digital PCR (ddPCR) approach that allows screening more than 90% of these recurrent abnormalities in DNA isolated from cell culture supernatant. This method greatly simplifies and therefore encourages the regular and systematic hPSCs screening.
Results
Meta-Analysis of hPSC Genetic Abnormalities and Identification of a Recurrence Pattern
To catalog all genetic abnormalities previously detected in hPSCs using various techniques (karyotyping, fluorescence in situ hybridization, comparative genomic hybridization, microarray-based comparative genomic hybridization, and next-generation sequencing [NGS]), we selected primary research articles that reported genetic abnormalities in hESCs and human induced PSCs (hiPSCs), and extracted the DNA abnormality genomic coordinates as well as the experimental data to characterize these abnormalities. We collected data on 942 cell samples and on 415,750 variants and abnormalities from 107 different studies published between 2004 and 2016 (Figures 1A and 1B; Table S1). The dataset included the major publications on genetic abnormalities in hPSCs during culture and also articles that identified one or several abnormal clones in new hPSC lines. A first global analysis of all listed mutations allowed identifying genome locations where these abnormalities were more frequently localized: trisomy 12 and 12p amplification, 20q11.21 amplification, trisomy 17 and 17q amplification, chromosome 1 amplification, and trisomy X (female cell lines) (Figure S1A). Abnormalities that accumulated at a specific genome location (i.e., recurrent abnormalities) were mostly aneuploidy or copy-number variations (CNV), in agreement with previous reports. No abnormality smaller than 10 base pairs (bp) displayed a recurrent profile in this large dataset (Figure S1B). We also reported 93 translocations and 20 inversions, involving all chromosomes with the exception of chromosome 12. They were mainly at chromosomes 1 and 17, but without a clear recurrent pattern (Figure 1C).
Figure 1.

Description of the Genetic Abnormalities Dataset
(A) Countries contributing to the articles included in the analysis.
(B) Number of genetic abnormalities and variations collected, according to their length.
(C) Circos plot representing all translocations in this study. Numbers, chromosome; green, balanced translocations; gray, unbalanced translocations.
In summary, this large meta-analysis of genetic abnormalities in hPSCs confirms the recurrence of large CNVs and chromosomal abnormalities, and provides a large dataset of recurrent abnormalities.
Definition and Analysis of Recurrent Genetic Abnormalities
As no quantitative definition of a recurrent hPSC genetic abnormality exists in the literature, we wanted to establish a clear threshold for such events. Therefore, we defined a recurrent genetic abnormality as a non-polymorphic variant that overlaps with abnormalities found in other hPSC lines. The recurrence pattern most likely reflects a common functional cause that occurs in different laboratories and in different cell lines (Assou et al., 2018). We hypothesized that the abnormalities with the strongest functional impact on hPSC growth would be those that are (1) common to different hPSC lines and (2) found in different culture conditions. We estimated that a genetic abnormality met these two criteria if all/part of the altered sequence overlapped with that of other genetic abnormalities that were described in at least four other distinct scientific publications (thus, at least five articles in total) (Figure 2A). To identify recurrent genetic abnormalities based on these criteria, we analyzed all variants >10-bp-long variants that were not polymorphisms (n = 8,284). We found that recurrent abnormalities were only CNVs (including chromosomal gain or losses) (Figure 2B). By plotting the genomic coordinates of these 738 recurrent abnormalities, we found that they were mainly localized in known hotspots, such as chromosome 1, 12, 17q, 20q11.21, or X (Figure 2C). Conversely, there were no recurrent abnormalities, as defined above, in chromosome 2, 4, 10, or 21.
Figure 2.

Definition and Analysis of Recurrent Genetic Abnormalities in hPSC
(A) Graphic representation of eight genetic abnormalities (or variants that are not polymorphic) (#1 to #8), from five different articles (A–E), one color for each article. Abnormalities in orange are recurrent genetic abnormalities because they overlapped and were from five different articles.
(B) Dot plot showing the length of all genetic abnormalities larger than 10 bp and that were not polymorphisms (n = 8,284) (x axis) versus the number of different articles that described these overlapping abnormalities (y axis).
(C) Bar plot showing the 738 recurrent genetic abnormalities according to their genomic coordinates (one color per chromosome). y axis, number of recurrent abnormalities.
(D) Percentage of cumulated recurrent abnormalities found in the 24 common abnormal regions with the most recurrent genetic abnormalities.
(E) Comparison of the recurrent abnormalities reported in this work and in a selection of other major original papers or reviews. Colors, chromosome; large bubble, high recurrence; average bubble, intermediate recurrence or no quantitative information; small bubble, low recurrence; no bubble, no recurrence. References found in Supplemental Information section.
We then investigated the nature of these hotspot regions (Figure 2A) (Table 1 lists the 20 more frequent common regions). Specifically, four regions included more than 50% of all reported abnormal genetic abnormalities. Moreover, a limited set of common abnormal regions comprised the most recurrent genetic abnormalities. Indeed, more than 90% of recurrent abnormalities were restricted to 20 common regions. A set of probes designed to cover these regions would detect all genomic abnormalities of these regions, except balanced translocations (Figure 2D).
Table 1.
Common Abnormal Regions
| Rank | Chromosome | Start | End | n | % | Cumulated % |
|---|---|---|---|---|---|---|
| 1 | 20 | 29 848 383 | 30 754 613 | 169 | 22.9% | 22.9% |
| 2 | 12 | 11 937 418 | 25 556 120 | 116 | 15.7% | 38.6% |
| 3 | 17 | 51 281 495 | 52 293 893 | 75 | 10.2% | 48.8% |
| 4 | X | 1 | 60 600 000 | 39 | 5.3% | 54.1% |
| 5 | 1 | 172 900 000 | 185 800 000 | 34 | 4.6% | 58.7% |
| 6 | 5 | 104 500 000 | 117 404 202 | 26 | 3.5% | 62.2% |
| 7 | 18 | 56 200 000 | 61 600 000 | 24 | 3.3% | 65.4% |
| 8 | 17 | 7 211 004 | 8 044 174 | 21 | 2.8% | 68.3% |
| 9 | 7 | 132 600 000 | 133 785 759 | 18 | 2.4% | 70.7% |
| 10 | 9 | 68 700 000 | 114 900 000 | 17 | 2.3% | 73.0% |
| 11 | 11 | 2 800 000 | 10 700 000 | 17 | 2.3% | 75.3% |
| 12 | 13 | 87 700 000 | 101 700 000 | 16 | 2.2% | 77.5% |
| 13 | 16 | 1 | 90 354 753 | 16 | 2.2% | 79.7% |
| 14 | 1 | 16 200 000 | 17 074 942 | 14 | 1.9% | 81.6% |
| 15 | 8 | 93 300 000 | 127 230 818 | 13 | 1.8% | 83.3% |
| 16 | 14 | 19 152 018 | 107 349 540 | 13 | 1.8% | 85.1% |
| 17 | 6 | 130 300 000 | 139 000 000 | 12 | 1.6% | 86.7% |
| 18 | 15 | 67 200 000 | 67 300 000 | 11 | 1.5% | 88.2% |
| 19 | 3 | 60 799 | 26 400 000 | 9 | 1.2% | 89.4% |
| 20 | 22 | 24 268 025 | 49 570 503 | 9 | 1.2% | 90.7% |
Cell Culture Supernatant as a DNA Source to Evaluate hPSC Genomic Integrity by ddPCR
We then decided to take advantage of this highly biased recurrence profile of hPSC genetic abnormalities to develop a rapid PCR-based approach to detect the most common recurrent abnormalities, including those that cannot be detected by karyotyping due to its resolution limits. We analyzed DNA extracted from different hPSC lines (cell-DNA) without (HY03, UHOMi001-A, iCOPD2, and iCOPD9) and with genetic abnormalities (RSP4: chromosome 20 triploidy), using ddPCR and primer pairs that target chromosome X or chromosome 20. We could observe two chromosome 20 copies in DNA samples from normal hPSC lines, and three copies in the RSP4 cell-DNA sample (Figures 3A and S2). Second, to test the sensitivity of our approach, we prepared cell-DNA from UHOMi001-A cells (diploid line) mixed with increasing percentages (0%–100%) of HD291 cells (chromosome 12q trisomy). Our ddPCR approach could detect the presence of the chromosome abnormality, starting from the sample containing 10% of HD291 cells (Figure 3B).
Figure 3.

Detecting Recurrent Genetic Abnormalities by Focused ddPCR
Each ddPCR data point is obtained from one sample using Poisson statistics and error bars indicate the Poisson distribution at 95% confidence intervals.
(A) Copy-number variation analysis using droplet digital PCR and DNA extracted from different hPSC lines in culture.
(B) Sensitivity of the droplet digital PCR method for detecting increasing percentages (from 0% to 100%) of hPSCs harboring a trisomy 12 within a sample of euploid hPSCs. The panels represent three independent experiments (p < 0.05, Student's t test).
(C) Quantification of DNA in supernatant samples from one hESC line (HS291) and two hiPS lines (HY03 and UHOMi001 cells) cultured in E8 medium on Geltrex matrix. Supernatant was collected at the indicated days (D) after seeding (75,000 cells/well in a 35-mm plate). DNA was extracted from 300 μL of supernatant and quantified by ALU-qPCR with ALU115 primers.
(D) The percentage of apoptotic, necrotic, and viable cells in supernatant samples collected at day 5 was evaluated by flow cytometric analysis after staining with annexin V and 7-amino-actinomycin (7-AAD). Every dot corresponds to a single cell. ScEP, single-cell enzymatic passaging; MP, mechanical passaging.
(E) Copy number of chromosome 20q measured by ddPCR using genomic DNA from cells and supernatant as template. The error bar varies in function of the DNA source (cells or supernatant).
(F) The iCS-digital test using six probes for chromosomes 20q, 12, X, 17, 1, and 5 can identify aneuploidy. The hPSC lines HD129 (chromosome 20 triploidy), HD291 (chromosome 12 triploidy), and UHOMi001-A (euploid) were analyzed by karyotyping (classical G-banding) and with the iCS-digital test using probes targeting common abnormal regions on chromosomes 20q, 12, X, 17, 1, and 5. Karyotype images are reprinted from Stem Cells Dev. 24(5):653-62, 2015 and Stem Cell Res. 33:15-19, 2018 with permission from Elsevier.
(G) Identification of genome modifications associated with culture conditions using the iCS-digital test. After 15 passages using single-cell enzymatic passaging, the HY03 hiPSC line displayed a copy-number gain on chromosome 1 (2.66), suggesting a mosaic cell population that comprises an abnormal clone with at least three genome copies at the probe location.
(H) Analysis of genome stability using the iCS-digital test in the male hiPSC line HY03 before (mechanical passage M53, clumps passage Cl2, single-cell passage SC11) and seven passages after genome editing.
(I) CNV characterization of chromosome 20q, 12, X, 17, 1, and 5 in four CCDC40_KO HY03 clones obtained using CRISPR/Cas9 technology. No abnormality was detected in any of the four clones.
Another major constraint to hPSC genome integrity analysis is the need to dedicate a significant part of the cell culture for this purpose. Therefore, we investigated whether genome integrity could be assessed using DNA extracted from hPSC culture supernatants. We found that when using supernatant-derived DNA (supernatant-DNA), the concentration of DNA fragments, estimated by qPCR, increased progressively during hPSC culture and was at a highest after 7 days in culture, when cell confluence was higher than 70% (Figure 3C). To help elucidating the origin of the supernatant-DNA, we analyzed floating cells by staining with annexin V and 7-amino-actinomycin followed by flow cytometry (Figure 3D). This analysis showed that 74.5% and 56.5% of floating cells were apoptotic, 12.2% and 6.6% were dead, and 13.4% and 36.9% were viable after single-cell and mechanical passaging, respectively. We also measured the DNA integrity index by qPCR using the ALU115 primers that amplify both short (apoptotic) and long (non-apoptotic) DNA fragments, and the ALU247 primers that amplify only long non-apoptotic DNA fragments (Umetani et al., 2006). The ALU115/ALU247 ratio was higher than 2, showing that the supernatant contained a majority of apoptotic cells (Figure S3A). We also investigated the influence of the interval before DNA extraction and of the number of freeze-thaw cycles on the quantity of DNA extracted from cell culture supernatant samples (Figures S3B and S3C). These results demonstrated that stable and significant quantities of DNA can be extracted from cell culture supernatant samples.
Finally, we observed an excellent agreement between the results obtained using cell-DNA and the corresponding supernatant-DNA (supernatant collected at days 5–7 of culture) (Figure 3E). This shows that ddPCR offers the sensitivity required for evaluating genomic integrity in supernatant-DNA, and that supernatant-DNA could be used instead of cell-DNA.
We then evaluated whether supernatant-DNA could be used to perform focused ddPCR (in culture supernatant digital PCR test, iCS-digital test) using a panel of six commercial pre-designed probes that correspond to or are close to the six most common abnormal regions (chromosomes 20q, 12, 17, X, 1, and 5). These six probes target genome regions that comprise more than 50% of all recurrent genetic abnormalities found in hPSCs (61% cumulated coverage of recurrent abnormalities). We analyzed supernatant-DNA from two hPSC lines with abnormal karyotype (HD129 and HD291: chromosome 20 and 12 triploidy, respectively) (Bai et al., 2015) and from one diploid line (UHOMi001-A). The iCS-digital test results overlapped with those obtained by karyotyping (Figure 3F). In conclusion, targeted ddPCR can efficiently detect CNVs and can be carried out using supernatant-DNA.
Routine Screening of hPSC Lines during Cell Culture and after CRISPR Gene Editing Using the iCS-Digital Test
The simplicity of the iCS-digital test could allow the routine screening of the most recurrent genetic abnormalities in hPSC lines, particularly when using single-cell or small-clump passaging, a major cause of genomic alterations (Bai et al., 2015). The iCS-digital test revealed that the HY03 hiPSC line, which was euploid at passage 5, harbored a chromosome 1 gain at passage 15 after single-cell passaging, but not after mechanical passaging (Figure 3G).
Cell reprogramming and genome editing using CRISPR/Cas9 require clonal expansion/selection that favors the emergence of abnormal clones. The iCS-digital test did not detect any alteration at chromosomes 20q, 12, 17, X, 1, and 5 in the hiPSC line HY03 before genome editing (Figure 3H). Conversely, after introduction of the mCherry cassette at the 3′ of the FOXJ1 gene, or knockout (KO) of the CCDC40 gene using the CRISPR/Cas9 methodology, the iCS-digital test showed that the single correctly edited clone (passage 7 after editing) harbored a CNV in the long arm of chromosome 5 (copy number = 1.3) (Figure 3H), while the four CCDC40-KO clones analyzed appeared euploid (six regions checked) (Figure 3I). Taken together, our results show that focused ddPCR can be used to rapidly screen iPSCs after derivation, during cell culture or amplification, and after cell cloning in settings, such as gene editing.
Discussion
The present study is, to our knowledge, the largest meta-analysis of hPSC genetic abnormalities in more than 100 different research articles from many different laboratories and cell lines. This allowed us to propose a quantitative threshold to define recurrent genetic abnormalities in hPSCs and to test this threshold. Hence, a recurrent genetic abnormality is an abnormality that shares part of its abnormal sequence with other abnormalities that have been reported in at least five different publications. Our threshold should favor the detection of abnormalities that are common to different hPSC lines and different laboratories, as opposed to abnormalities that are specific to a unique cell line, a hiPSC donor cell, or to the culture conditions used in one laboratory. As expected, many regions were already reported, but some are new. Figure 2E compares our recurrent abnormalities with those reported in a selection of other major original papers or reviews. Our study adds to previous studies a rigorous definition of recurrence, and the analysis of a larger number of different samples compared with all previous works. Our analysis confirmed that structural variants, including CNVs of various lengths, are among the main recurrent genetic abnormalities in hPSC lines. It has been proposed that the high hPSC susceptibility to mitotic division errors and to the upregulation of anti-apoptotic proteins contributes to the high frequency of aneuploidy and CNVs at specific genomic locations (Zhang et al., 2019). For instance, chromosome 20q11.21 gain leads to upregulation of the anti-apoptotic protein BCL-XL (Zhang et al., 2019). Noteworthy, with the exception of the BCL2L1 locus, we did not find convergence on any specific chromosome locus, even by using the large dataset collected for our meta-analysis. As illustrated by Figure 2C, most recurrent zones are large parts of chromosome arms. Although regions with more recurrences than others can be identified, only the BCL2L1 locus has a pattern pointing to a precise zone. This suggests that recurrence is the consequence of several genes dispersed on a large chromosome section that must be simultaneously upregulated through copy-number gains (or downregulated for the rare cases of recurrent deletions) to promote in vitro culture fitness. For example, the most recurrent part of chromosome X (X:1, 60,600,000) contains several genes that are annotated as involved in regulation of cell cycle, DNA repair, apoptosis, stemness, or cancer. Consequently, two or more of these genes could be central players in the recurrence of copy-number gains at this locus (Figure S4). Recent analyses of hPSC NGS data identified recurrent small mutations in oncogenes and tumor suppressor genes, such as TP53, with a frequency still to be determined (Avior et al., 2019, Merkle et al., 2017). We expect that additional recurrent abnormalities will be uncovered, but this requires collecting many genome sequencing data, especially whole genome data. This will allow drawing the complete map of genetic abnormalities of hPSCs. In addition, one pitfall of our current analysis is that samples used in our work were analyzed using methods which have biases, and this could have influenced the recurrence score. Whole-genome NGS would provide the unbiased data necessary for the comprehensive identification of anomaly recurrence in PSC. Finally, the comparison with parental samples (in the case of iPSC lines), and the comparison with data from a large number of healthy volunteer DNA samples is necessary to rigorously distinguish recurrence from polymorphisms because the databases routinely used to identify polymorphisms are neither comprehensive nor error-free.
Nonetheless, the recurrence distribution is biased, and this allows studying common abnormal regions by targeted PCR. For example, by targeting only the four most common abnormal regions, more than 50% of all recurrent genetic abnormalities are covered, and more than 90% by targeting 20 regions (Table 1). Screening of recurrent genetic abnormalities is paramount to claim that hPSCs are normal (Bai et al., 2013, International Stem Cell Initiative et al., 2011). Although a targeted approach is by definition not exhaustive, it is an effective strategy to rule out the most frequent and functionally damaging abnormalities found in hPSC lines. We demonstrated that this strategy can be carried out simply by using DNA from culture supernatant using ddPCR. Other competitive technologies exist, but none can meet all the following requirements: range, resolution, sensitivity, low cost, data analysis workload, and complex result interpretation. For example, DNA microarrays, and whole-exome and whole-genome sequencing can be used for large-scale hPSC genome integrity analyses, but are limited by the long time required for sample processing and their high cost. Karyotyping is still the gold standard for hPSC analysis, but it is time-consuming. Rapid tests for the routine screening of cultured cells are mostly provided by PCR-based technologies (Baker et al., 2016). The advantages and disadvantages of the techniques used to assess hPSC genome integrity are listed in Table S2. Ultimately, we anticipate that NGS will become the primary technique for assessing hPSC quality because sequencing costs continue to decrease.
Genomic integrity of hPSCs in culture should be frequently assessed. We recently noted in a series of 25 consecutive studies on hiPSCs that the current genomic screening practices were unsatisfactory because no genomic integrity follow-up was carried out for any of the hiPSC lines (Assou et al., 2018). This could be explained by the labor and costs involved in the implementation of classical screening techniques, such as karyotyping (see also Table S2). Therefore, a simple test that can rapidly rule out the most frequent recurrent genomic abnormalities might promote adhesion to good practices for hPSC genomic integrity screening. Moreover, karyotyping can miss abnormalities that are smaller than 5–10 Mb. For instance, we found that among all 170 recurrent genomic abnormalities on chromosome 20, 168 overlapped with 20q11.21, and among them 135 were smaller than 5 Mb. By contrast, the probes of the iCS-digital test can detect all 168 20q11.21 abnormalities. Therefore, PCR-based tests are appropriate for early detection of genetic abnormalities that arise during cell culture to avoid performing experiments with compromised hPSC lines.
In conclusion, we used a large dataset of hPSC genomic abnormalities based on more than 100 publications to define recurrent genetic abnormalities, including a set of common abnormal genomic regions that involve more than 90% of all recurrent abnormalities. This offered the opportunity to develop a targeted ddPCR approach that could be performed on culture supernatant. Therefore, we propose a simple test based on supernatant-DNA (iCS-digital test) that could be used to routinely screen cultured hPSC lines for excluding the presence of common CNVs.
Experimental Procedures
Recurrence Analysis
All genetic abnormalities were converted using their GRCh37A genome coordinates. The complete pipeline can be found in Supplemental Experimental Procedures. In brief, to define recurrent genetic abnormalities, each abnormality >10 bp and that was not a polymorphism (n = 8,284 abnormalities in total) was compared with all the others, and abnormalities with a reciprocal overlap >0.33 and larger than 0.2 Mb, and the number of publications from which these abnormalities originated were counted, using R. Abnormalities that overlapped with other abnormalities that came from at least four other publications (number of total publications ≥5) were defined as recurrent and kept. This process was carried out iteratively and rapidly converged. Loss and gain were considered indiscriminately because the aim was to identify common abnormal regions.
Cell Culture
All PSC were derived with the ethical oversight of a Comité de protection des Personnes (CPP). PSC lines were maintained on Geltrex matrix (Thermo Fisher Scientific) in Essential 8 Medium (Thermo Fisher Scientific) or on Matrigel (BD Biosciences) in mTeSR1medium (STEMCELL Technologies), according to the manufacturer's instructions. Cells were grown in 35-mm dishes at 37°C and were passaged either mechanically or dissociated into single cells or small clumps every week. More details for cell reprogramming and cell passaging can be found in Supplemental Experimental Procedures.
Culture Medium Collection, Nucleic Acid Extraction, Quantification of Supernatant-DNA, Flow Cytometry, ddPCR, and Genome Editing using CRISPR/Cas9
See Supplemental Experimental Procedures for details.
Statistical Analysis
For ddPCR, absolute quantification was based on the number of positive droplets and Poisson sampling statistics, as follows: λ = −ln (1 − k/n), where k is the number of positive droplets, n is the total number of droplets, and λ is the mean copies per droplet. To test the statistical difference of the ddPCR results between samples with different percentages of trisomic cells, first the values were normalized to those of the 0% sample within each biological replicate. Poisson statistics and Student's t test were applied; p < 0.05 defined statistical significance.
Author Contributions
S.A., N.G., and J.D.V. designed the study and analyzed data. S.A., M.C., J.M., and E.A., performed the experiments. M.P., J.B., C.S., C.B., M.F., and E.A., collected and analyzed data. T.C. and A.B. analyzed and interpreted the data. S.A. and J.D.V. wrote the paper. J.-M.L. revised the manuscript. All authors approved the final version prior to submission.
Conflict of Interests
S.A. and J.D.V. filed a patent entitled “Non-invasive methods for assessing genetic integrity of pluripotent stem cells” (priority number: EP20150306389) and are co-founder, shareholder and scientific advisor of Stem Genomics SAS that acquired the exploitation rights of this patent.
Acknowledgments
We thank Camille Novo for contributing to the collection of PSC genomic abnormalities, Elena Hauser for technical help and Guilhem Requirand for his expertise in flow cytometry. This study was funded by INSERM, France, ANR (INGESTEM), France and the SATT AxLR, France.
Published: January 02, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2019.12.004.
Supplemental Information
References
- Assou S., Bouckenheimer J., De Vos J. Concise review: assessing the genome integrity of human induced pluripotent stem cells: what quality control metrics? Stem Cells. 2018;36:814–821. doi: 10.1002/stem.2797. [DOI] [PubMed] [Google Scholar]
- Avior Y., Eggan K., Benvenisty N. Cancer-related mutations identified in primed and naive human pluripotent stem cells. Cell Stem Cell. 2019;25:456–461. doi: 10.1016/j.stem.2019.09.001. [DOI] [PubMed] [Google Scholar]
- Bai Q., Desprat R., Klein B., Lemaître J.-M., De Vos J. Embryonic stem cells or induced pluripotent stem cells? A DNA integrity perspective. Curr. Gene Ther. 2013;13:93–98. doi: 10.2174/1566523211313020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai Q., Ramirez J.-M., Becker F., Pantesco V., Lavabre-Bertrand T., Hovatta O., Lemaître J.-M., Pellestor F., De Vos J. Temporal analysis of genome alterations induced by single-cell passaging in human embryonic stem cells. Stem Cells Dev. 2015;24:653–662. doi: 10.1089/scd.2014.0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker D., Hirst A.J., Gokhale P.J., Juarez M.A., Williams S., Wheeler M., Bean K., Allison T.F., Moore H.D., Andrews P.W. Detecting genetic mosaicism in cultures of human pluripotent stem cells. Stem Cell Reports. 2016;7:998–1012. doi: 10.1016/j.stemcr.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Stem Cell Initiative. Amps K., Andrews P.W., Anyfantis G., Armstrong L., Avery S., Baharvand H., Baker J., Baker D., Munoz M.B., Beil S. Screening ethnically diverse human embryonic stem cells identifies a chromosome 20 minimal amplicon conferring growth advantage. Nat. Biotechnol. 2011;29:1132–1144. doi: 10.1038/nbt.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort N., Feyeux M., Bas C., Féraud O., Bennaceur-Griscelli A., Tachdjian G., Peschanski M., Perrier A.L. Human embryonic stem cells reveal recurrent genomic instability at 20q11.21. Nat. Biotechnol. 2008;26:1364–1366. doi: 10.1038/nbt.1509. [DOI] [PubMed] [Google Scholar]
- Lefort N., Perrier A.L., Laâbi Y., Varela C., Peschanski M. Human embryonic stem cells and genomic instability. Regen. Med. 2009;4:899–909. doi: 10.2217/rme.09.63. [DOI] [PubMed] [Google Scholar]
- Merkle F.T., Ghosh S., Kamitaki N., Mitchell J., Avior Y., Mello C., Kashin S., Mekhoubad S., Ilic D., Charlton M. Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature. 2017;545:229–233. doi: 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umetani N., Kim J., Hiramatsu S., Reber H.A., Hines O.J., Bilchik A.J., Hoon D.S.B. Increased integrity of free circulating DNA in sera of patients with colorectal or periampullary cancer: direct quantitative PCR for ALU repeats. Clin. Chem. 2006;52:1062–1069. doi: 10.1373/clinchem.2006.068577. [DOI] [PubMed] [Google Scholar]
- Zhang J., Hirst A.J., Duan F., Qiu H., Huang R., Ji Y., Bai L., Zhang F., Robinson D., Jones M. Anti-apoptotic mutations desensitize human pluripotent stem cells to mitotic stress and enable aneuploid cell survival. Stem Cell Reports. 2019;12:557–571. doi: 10.1016/j.stemcr.2019.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.