

Abstract
Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder. The abnormal accumulation and deposition of amyloid-beta peptide (Aβ) in senile plaques and cerebral vasculature is widely recognized to be the most likely culprit in the pathogenesis of AD. Long non-coding RNAs (lncRNAs), a kind of evolutionarily conserved non-coding RNAs with over 200 nucleotides in length, have introduced a novel field of biology, and are involved in various human diseases, including neurological diseases. Recently, lncRNA X-inactive specific transcript (XIST) is reported to be upregulated in the rat spinal cord injury (a neurological disease) model and XIST knockdown has a prominent protective effect on the recovery of spinal cord injury. However, little is known about the expression and function of XIST in AD. Here, we showed that Aβ25-35 treatment increased XIST expression in hippocampal neurons. XIST knockdown ameliorated toxicity, oxidative stress, and apoptosis induced by Aβ25-35 treatment in hippocampal neurons. We further identified and confirmed that miR-132 was the target of XIST, and XIST functioned by targeting miR-132. Collectively, these data show that knockdown of XIST relieves Aβ25-35-induced toxicity, oxidative stress, and apoptosis in primary cultured rat hippocampal neurons by upregulation of miR-132. These findings encourage continued investigation of the potential of manipulating XIST in the treatment of AD.
Keywords: Alzheimer’s disease, lncRNA X-inactive specific transcript, miR-132, amyloid-beta peptide, oxidative stress, apoptosis
Introduction
Alzheimer’s disease (AD) is one of the most prevalent neurodegenerative diseases with a global prevalence of up to 15 million people, and is characterized by slowly progressive dementia and brain atrophy [1]. The hippocampus, one of the most important brain structures, plays a central role in spatial learning and memory, and is the most affected area of pathological alteration in AD. It is widely accepted that the accumulation and deposition of amyloid-beta peptide (Aβ), the main constituent of the amyloid core of senile plaques, is an important event in the pathogenesis of AD [2]. Much research has proven the toxicity of Aβ on neurons both in vitro and in vivo [3,4]. In addition, the transgenic mice with the overexpression of human amyloid precursor protein develop neuritic plaques, which are similar to those seen in brains of AD patients [5]. In fact, increasing evidence points to Aβ as the most likely culprit in the pathogenesis of AD, and it plays a key role in AD [6-8]. In recent years, the molecular mechanisms whereby Aβ exerts its role in AD are widely studied, but have not yet been fully elucidated.
Recent evidence suggests that oxidative stress is a critical event in the pathogenesis of AD, and exerts a key role in AD development. For example, Michel et al. [9] reported that activity of mitochondrial aldehyde dehydrogenase (ALDH) increases in the putamen of individuals with AD. Pratico et al. [10] reported that individuals with mild cognitive impairment have increased brain oxidative damage before the onset of symptomatic dementia. Oxidative stress can cause cell death by damaging cellular macromolecules, including proteins, lipids, DNA, and RNA [11]. The brain is particularly susceptible to oxidative stress, due to its high concentration of easily oxidized fatty acids, and low level of antioxidant enzymes [12]. In AD, oxidative stress has been suggested to be induced by Aβ, and crucially involved in Aβ toxicity [13-15]. In recent years, many compounds have been reported to play neuroprotective effects against Aβ-induced oxidative stress and neuronal apoptosis, which may be promising therapeutics against AD [16-19].
Long non-coding RNAs (lncRNAs) are a kind of evolutionarily conserved non-coding RNAs with over 200 nucleotides in length, and lack significant protein-coding capacity. There is growing evidence that lncRNAs can regulate multiple biologic processes and are involved in various human diseases, including neurological diseases [20-22]. In addition, the deregulation of many lncRNAs has been confirmed to be associated with the occurrence and development of diseases, and restoration of aberrantly expressed lncRNA is favorable for curing the diseases [23-25]. LncRNA X-inactive specific transcript (XIST), first discovered by searching cDNA libraries for clones in 1980s and 1990s, is dysregulated in some cancers and is correlated with tumor progression and poor prognosis. Recently, XIST is reported to be upregulated in rat spinal cord injury (a neurological disease) model and XIST knockdown has a prominent protective effect on the recovery of spinal cord injury by suppressing apoptosis [26]. However, little is known about the expression and function of XIST in AD.
In this study, we examined XIST expression in Aβ25-35-insulted primary cultured hippocampal neurons, and investigated the effects of XIST expression on toxicity, oxidative stress, and apoptosis induced by Aβ25-35. In addition, we also investigated the molecular mechanism whereby XIST exerts its role.
Materials and methods
Primary hippocampal neuron culture
Use of animals was approved by the Animal Care and Use Committee of Xinxiang Medical University. Primary dissociated hippocampal neurons were prepared from pregnant 16 to 18-day Sprague-Dawley rat embryos. In brief, hippocampus was dissected and placed in 35-mm petri dishes containing calcium, magnesium-free Hanks’ balanced salt solution (HBSS). Then the hippocampus tissue was minced into small pieces by using crossed scalpels and trypsinized with 0.25% trypsin (Sigma, St. Louis, MO, USA) for 25 min at 37°C with 5% CO2. Trypsinization was terminated and tissue was triturated by aspirating 8 times with a flame-narrowed Pasteur pipette to produce cell suspension. Cell suspension was centrifuged and the supernatant was discarded. The cell pellet was resuspended in Neurobasal/B27 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 2 mM-glutamine. Hippocampal neurons were manually counted via hematocytometer, and then seeded into Petri dishes coated with poly-L-lysine at a density of 2×105/cm2. Cultures were maintained in a humidified chamber filled with 5% CO2 at 37°C. After 3 days, these cells were used for further experiments.
Aβ25-35 treatment
Three days after culturing as mentioned previously, hippocampal neurons were transfected as described below, and then cultured in the fresh medium with a concentration of 25 μM Aβ25-35 (Sigma) for 24 h at 37°C. The morphologic changes of hippocampal neurons were observed under the microscope (Olympus, Japan).
RNA extraction and qRT-PCR
After Aβ25-35 treatment, total RNA samples were extracted using Trizol reagent (Invitrogen), and cDNA was synthesized using a Reverse Transcription Kit according to the manufacturer’s instructions (Takara, Dalian, China). The expression of miR-132 was determined by the TaqMan miRNA assay (Applied Biosystems, Foster City, CA, USA), and U6 snRNA was used as an internal standard. The expression of lncRNA XIST was determined on ABI 7500 real-time PCR system (Applied Biosystems), and GAPDH was used as a loading control. The average fold-change of genes was calculated using the 2-ΔΔCt method.
Cell transfection
A plasmid that carries complementary DNA encoding XIST (pcDNA-XIST) was synthesized by introducing the XIST cDNA sequence into the pcDNA3.1 expression vector (Invitrogen). Small interfering RNAs (siRNA) targeting XIST (si-XIST), miR-132 mimic, and their respective controls were obtained from Dharmacon (Chicago, IL, USA). All transfection reactions were performed using Lipofectamine 2000 Reagent following the manufacturer’s protocol (Invitrogen).
MTT assay
Cell viability was assessed by MTT assay. Briefly, hippocampal neurons were cultured at a density of 2.5×103 cells per well in fresh medium with 25 μM Aβ25-35 for 24 h in 96-well plates. Then 20 μl of MTT solution (5 mg/ml) was added to each well, and the cells were continued to culture. After 4 h incubation, the medium was gently removed from the wells, and 100 μl of dimethyl sulfoxide (DMSO, Sigma) was added to each well to dissolve the formazan product. The absorbance was recorded at 570 nm with a microplate reader Dynatech MR 4000 (Dynex, Denkendorf, Germany).
Lactate dehydrogenase (LDH) release assay
Cell viability was also evaluated by measurement of LDH released into the medium. After Aβ25-35 treatment, LDH activity was determined by LDH-cytotoxicity assay kit (BioVision, Mountain View, CA, USA) in accordance with the manufacturer’s instructions. The absorbance was measured at 570 nm with a microplate reader Dynatech MR 4000 (Dynex).
Level of potassium ions (K+) leakage
The permeability of neuron membranes was assessed by measurement of level of K+ leakage. Briefly, after Aβ25-35 treatment, cell culture supernatants were collected and centrifuged at 10,000 g for 10 min. Then 0.5 ml of supernatant was transferred to a 1.5 ml Eppendorf tube, and normal culture medium was used as control. The absorbance was measured with a flame spectrophotometer (Beckman Coulter, Fullerton, CA, USA).
The activity levels of superoxide dismutase (SOD), glutathione peroxidase (GSH-Px) and malonaldehyde (MDA)
After Aβ25-35 treatment, the activities of SOD and GSH-Px, and the level of MDA in hippocampal neurons were measured by using the assay kits from Nanjing Jiancheng Bioengineering Institute (Nanjing, China) according to manufacturer’s protocols.
TdT-mediated dUTP nick-end labeling (TUNEL)
Hippocampal neurons were washed in 0.01 M PBS, and fixed with 4% (w/v) paraformaldehyde for 30 min. Then the fixed cells were permeated in 0.1% Triton X-100 in 0.1% sodium citrate for 10 min. TUNEL was carried out using an in situ cell death detection kit (Roche, Mannheim, Germany) in accordance with manufacturer’s inductions. The neuronal nuclei were counterstained with Hoechst 33342. The TUNEL-positive cells were observed through a fluorescent microscope (Olympus BX51 microscope, Tokyo, Japan), and were counted manually.
Western blot analysis
Western blot assay was performed as previously described [16]. The primary antibodies against Bax (Bioss Inc., Beijing, China), Bcl-2 (Bioss Inc.), cytochrome c release (cytC, CST, Danvers, MA, USA), cleaved caspase-3 (Abcam, Cambridge, MA, USA) and β-actin (Abcam), and peroxidase-conjugated secondary antibody (Abcam) were employed in this assay.
Luciferase reporter assay
The reporter construct was generated by inserting six fragments of XIST mRNA containing predicted miR-132 binding site 1, 2, 3, 4, 5, or 6 into a modified pcDNA3.1 vector containing a luciferase gene. Mutant reporter plasmid was obtained by Mutagenesis Kit according to manufacturer’s protocols (Stratagene, Heidelberg, Germany). Cells were cotransfected with the WT or MUT reporter plasmid and miR-132 mimic or miR-control. At 48 h post-transfection, luciferase activity assay was conducted with the dual luciferase reporter assay system (Promega, Madison, WI, USA).
Statistical analysis
All data were expressed as mean ± standard deviation (SD), and analyzed using the SPSS-17.0 software (SPSS, Chicago, IL, USA). Stu-dent’s t test or ANOVA test was used to the comparisons between groups. The statistical significances were achieved when P < 0.05.
Results
Aβ25-35 induces the increase of XIST expression in hippocampal neurons
After exposure to 25 μM Aβ25-35 for 24 h, hippocampal neurons were collected, and XIST expression was detected by qRT-PCR. As shown in Figure 1, the cells treated with Aβ25-35 had significantly higher XIST expression compared with the control group, suggesting that Aβ induces an increase in XIST expression in hippocampal neurons.
Figure 1.
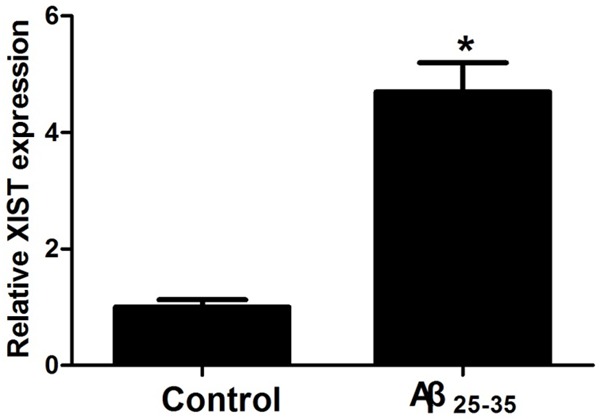
Aβ25-35 treatment increases XIST expression in hippocampal neurons. After exposure to 25 μM Aβ25-35 for 24 h, hippocampal neurons were collected for XIST expression detection. *P < 0.05.
LncRNA XIST knockdown ameliorates Aβ25-35-induced toxicity in hippocampal neurons
The toxicity of Aβ25-35 in hippocampal neurons was assessed by morphologic observation, cell viability, and membrane permeability. As shown in Figure 2A, hippocampal neurons treated with Aβ25-35 had irregular shapes, and less arborization, synaptic connections and inter-cellular associations than the hippocampal neurons in the control group, while XIST knockdown prevented the morphologic changes induced by Aβ25-35. MTT and LDH release assays were used to detect cell viability. Level of K+ leakage was used to assess the permeability of neuron membranes. The results of MTT assay showed that after Aβ25-35 insults, cell viability had a significant decrease, but the decrease was attenuated prominently by knockdown of XIST (Figure 2B). The results of LDH release and K+ leakage assays showed that Aβ25-35 treatment significantly increased LDH release and K+ leakage, while XIST knockdown alleviated the effects induced by Aβ25-35 insults (Figure 2C, 2D). Taken together, these data suggested that XIST knockdown ameliorates Aβ25-35-induced toxicity in hippocampal neurons.
Figure 2.
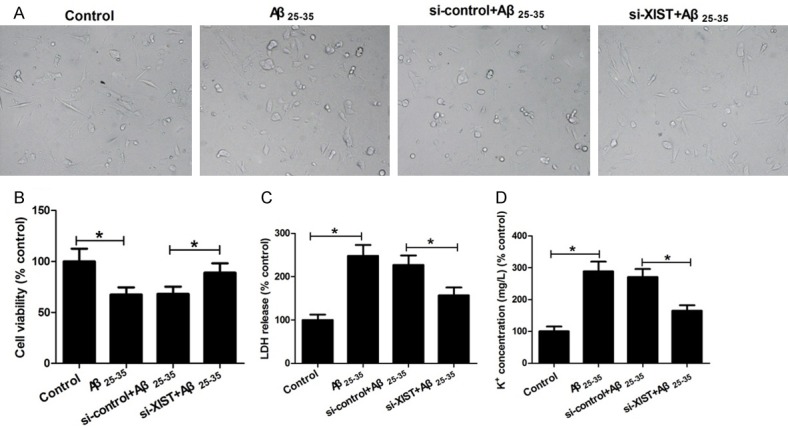
XIST knockdown ameliorates Aβ25-35-induced toxicity in hippocampal neurons. Hippocampal neurons were treated with control, 25 μM Aβ25-35, si-control+25 μM Aβ25-35, and si-XIST+25 μM Aβ25-35, and then used for morphologic observation (A), cell viability (B), LDH release (C), and K+ leakage (D) determination. *P < 0.05.
LncRNA XIST knockdown inhibits Aβ25-35-induced oxidative stress and apoptosis in hippocampal neurons
Oxidative stress causes cell apoptosis and plays an important role in Aβ25-35-induced toxicity. To investigate whether XIST knockdown suppressed Aβ25-35-induced oxidative stress, the activities of two anti-oxidases SOD and GSH-Px, and the level of oxidation product MDA were determined. As shown in Figure 3A-C, Aβ25-35 treatment significantly decreased the activities of SOD and GSH-Px, and increased MDA level compared to controls, while XIST knockdown alleviated the effects of Aβ25-35 treatment on the activities of SOD and GSH-Px, and MDA level. We subsequently performed TUNEL assay to investigate whether XIST knockdown suppressed Aβ25-35-induced apoptosis in hippocampal neurons. The results showed that Aβ25-35 treatment significantly increased the number of TUNEL-positive and apoptotic neurons, but the increase was attenuated by knockdown of XIST (Figure 3D). Bax and Bcl-2 are two important regulators of mitochondria-dependent apoptotic pathway. cytC and caspase-3 are biomarkers of oxidative stress-induced cell apoptosis that are regulated through the mitochondria-dependent apoptotic pathway. To further investigate whether XIST knockdown suppressed Aβ25-35-induced apoptosis via inhibition of oxidative stress and the activation of mitochondrial apoptotic pathway, Bax, Bcl-2, cytC and caspase-3 expression were detected by western blot. As shown in Figure 3E, Aβ25-35 treatment significantly increased the expression of Bax, cytC and caspase-3, and decreased Bcl-2 expression compared to control, while XIST knockdown obviously alleviated the effects of Aβ25-35 treatment on the expression of Bax, cytC, caspase-3 and Bcl-2. Collectively, these data indicated that XIST knockdown inhibits Aβ25-35-induced oxidative stress and mitochondria-mediated apoptosis in hippocampal neurons.
Figure 3.
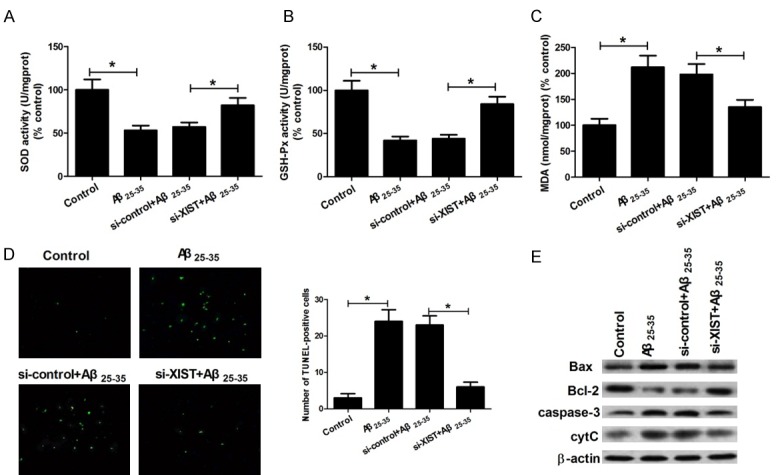
LncRNA XIST knockdown inhibits Aβ25-35-induced oxidative stress and apoptosis in hippocampal neurons. Hippocampal neurons were treated with control, 25 μM Aβ25-35, si-control+25 μM Aβ25-35, and si-XIST+25 μM Aβ25-35. Then the activities of SOD (A) and GSH-Px (B), MDA level (C), the number of TUNEL-positive neurons (D), and the protein expression of Bax, Bcl-2, caspase-3 activity and cytochrome c release (E) were determined. *P < 0.05.
miR-132 is a direct target of lncRNA XIST
Increasing evidence shows that lncRNAs contain a motif with sequence complementary to miRNAs and competitively inhibit miRNA expression and function. To test whether XIST exerts its role via such mechanisms in hippocampal neurons, bioinformatics tools were used to search for potential miRNAs interacted with XIST. The results showed that XIST contained six conserved binding sites for miR-132, and the binding sites were presented in Figure 4A. Then we determined miR-132 expression in Aβ25-35-treated hippocampal neurons. The results showed that Aβ25-35 treatment decreased miR-132 expression, which was opposite to the effects of Aβ25-35 treatment on XIST expression (Figure 4B). To further confirm that XIST could directly target miR-132, we performed the dual-luciferase reporter assay. It is worth noting that XIST harbors six binding sites for miR-132 and XIST mutant sequence (XIST-MUT) is constructed by mutation of the six predicted binding sites. The results showed that overexpression of miR-132 significantly inhibited the luciferase activity of wild-type reporter, but had little effect on the luciferase activity of mutant reporter (Figure 4C). In addition, we explored whether XIST could regulate miR-132 expression. The results showed that miR-132 expression was reduced after overexpression of XIST, whereas it was enhanced after knockdown of XIST (Figure 4D). These data suggested that XIST could negatively regulate miR-132 expression by binding to complementary miR-132 seed regions.
Figure 4.
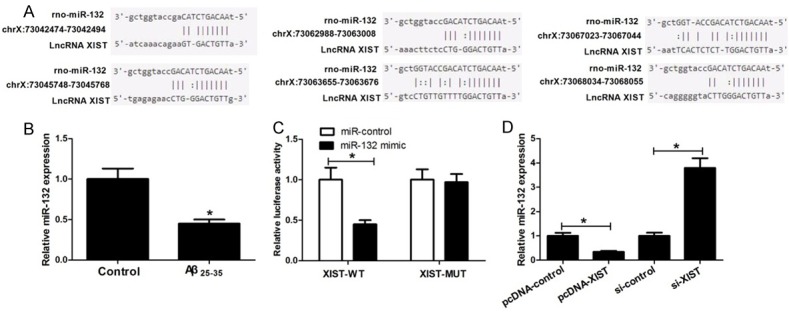
miR-132 is a direct target of lncRNA XIST. A. The binding sites for miR-132 in XIST. B. Relative miR-132 expression in hippocampal neurons exposed to 25 μM Aβ25-35 for 24 h. C. Relative luciferase activities of luciferase reporters bearing wild-type or mutant XIST 48 h following transfection with the indicated miR-132 mimics or miR-control in hippocampal neurons. D. Relative miR-132 expression in the cells with overexpression or knockdown of XIST. *P < 0.05.
miR-132 inhibits the toxicity, oxidative stress and apoptosis caused by Aβ25-35 treatment in hippocampal neurons
To further explore whether XIST exerted its role by targeting miR-132, hippocampal neurons were transfected with miR-132 mimic or miR-control and then cultured in the medium with or without 25 μM Aβ25-35 for cell toxicity, oxidative stress and apoptosis detection. Results showed that an increase of miR-132 could relieve the toxicity, oxidative stress, and apoptosis caused by Aβ25-35 treatment (Figure 5A-I), which played a similar role to XIST knockdown. Collectively, these data suggested that XIST knockdown attenuates Aβ25-35-induced toxicity, oxidative stress, and apoptosis in hippocampal neurons by targeting miR-132.
Figure 5.
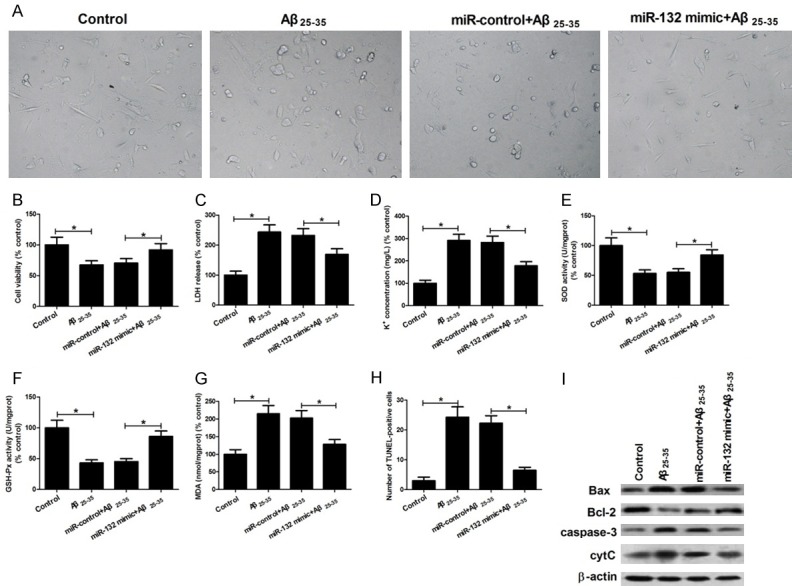
miR-132 inhibits the toxicity, oxidative stress and apoptosis caused by Aβ25-35 treatment in hippocampal neurons. Hippocampal neurons were treated with control, 25 μM Aβ25-35, miR-control+25 μM Aβ25-35, and miR-132 mimic+25 μM Aβ25-35. Then the morphologic observation (A), cell viability (B), LDH release (C), K+ leakage (D), activities of SOD (E) and GSH-Px (F), MDA level (G), the number of TUNEL-positive neurons (H), and the protein expression of Bax, Bcl-2, caspase-3 activity and cytochrome c release (I) were determined. *P < 0.05.
Discussion
AD is a progressive and irreversible neurodegenerative disorder. Mounting evidence shows that Aβ plays a key role in the pathogenic mechanism of AD. When AD occurs,abnormal accumulation and deposition of Aβ forms amyloid fibrils and plaques that are toxic to neurons [27]. Recent evidence suggests that oxidative stress is induced by Aβ and involved in Aβ toxicity, playing an important role in the development of AD [9,10,13-15]. In addition, a large body of evidence suggests that oxidative stress is involved in Aβ-induced neuronal apoptosis [28,29]. In fact, in recent years, many Chinese herb extracts have been reported to exert neuroprotective effects against Aβ-induced oxidative stress and neuronal apoptosis, and are promising drugs for the management of AD. For example, ginkgo biloba extract decreases cell toxicity and oxidative stress induced by Aβ25-35 via blocking mitochondria-mediated apoptosis signaling; baicalin effectively improves Aβ25-35-induced learning and memory deficit, hippocampal injury, and neuron apoptosis; and ginsenoside Rd attenuates Aβ25-35-induced oxidative stress and apoptosis in primary cultured hippocampal neurons [16-18].
LncRNAs have been formed as a novel field of biology, and regulate multiple biologic processes, involved in various human diseases, such as cancer, cardiovascular disease, and neurologic disease. Increasing evidence suggests that deregulation of many lncRNAs is related to the progress of diseases, and restoration of aberrantly expressed lncRNA might contribute to the treatment of diseases. For example, Yang et al. [23] reported that lncRNA PVT1 expression is significantly upregulated in non-small cell lung cancer tissues, and is closely correlated with histologic grade and lymph node metastasis, while knockdown of PVT1 inhibits cell proliferation, migration, and invasion in vitro. Braconi et al. [24] reported that lncRNA MEG3 expression is very weak in human hepatocellular cancer tissues compared with paired normal tissues, while enforced expression of MEG3 in hepatocellular cancer cells significantly decreases both anchorage-dependent and -independent cell growth, and induces apoptosis. Chen et al. [25] reported that lncRNA TUG1 is significantly upregulated in brain ischemic penumbra from rat middle carotid artery occlusion model, while TUG1 knockdown decreases the ratio of apoptotic cells and promotes cell survival in vitro. LncRNA XIST, expressed among many tissues, is associated with 12 disease classes and 50 diseases, including neurological diseases. For example, recently, Gu et al. [26] reported that XIST is one of the most upregulated lncRNAs in the rat spinal cord injury model, and its expression is increased continually after spinal cord injury, reaching a peak at three days. XIST knockdown has a protective effect and significantly improves spinal cord injury recovery by suppressing apoptosis through reactivation of the PI3K/AKT signaling pathway. However, the expression and function of XIST in AD were uncerain. Here, we first investigated the effects of Aβ25-35 treatment on XIST expression in hippocampal neurons, and found that Aβ25-35 treatment increased XIST expression. Then we investigated the effects of XIST on Aβ25-35-induced toxicity, oxidative stress, and apoptosis in hippocampal neurons, and found that XIST knockdown ameliorated toxicity, oxidative stress, and apoptosis induced by Aβ25-35 treatment. We further investigated the underlying mechanism by which XIST functioned. Mounting evidence suggests that lncRNAs exert regulatory functions by binding to miRNAs and inhibiting miRNA expression and function. For example, lncRNA UCA1 contributes to progression of hepatocellular carcinoma through inhibition of miR-216b; lncRNA XIST knockdown exerts tumor-suppressive functions in human glioblastoma stem cells by upregulating miR-152; lncRNA MALAT1 exerts oncogenic functions in lung adenocarcinoma by targeting miR-204; and lncRNA GAS5 inhibits proliferation and progression of prostate cancer by targeting miR-103 through the AKT/mTOR signaling pathway [30-33]. Likewise, we performed bioinformatics analysis of miRNA recognition sequences on XIST, and found that miR-132 was a potential XIST-binding miRNA. miR-132 has been widely reported to be involved in AD. For example, Wong et al. [34] reported that miR-132 is downregulated in temporal cortical areas and CA1 hippocampal neurons of human AD brains, and sequence specific inhibition of miR-132 induces apoptosis in cultured primary neurons, whereas miR-132 overexpression plays a neuroprotective role against oxidative stress. Smith et al. [35] reported that miR-132 is downregulated in tauopathies such as AD, and treatment of AD mice with miR-132 mimics restores in part memory function and tau metabolism. Salta et al. [36] reported that miR-132 loss exacerbates both amyloid and TAU pathology via inositol ITPKB upregulation in an AD mouse model. Based on these findings, it was tempting to speculate that XIST functioned by targeting miR-132. To test this speculation, we first performed a luciferase assay and validated the direct binding of miR-132 to XIST. We also detected the effects of Aβ25-35 treatment on miR-132 expression in hippocampal neurons, and found that Aβ25-35 treatment significantly decreased miR-132 expression, which was opposite to the effects of Aβ25-35 treatment on XIST expression. In addition, miR-132 expression was enhanced after knockdown of XIST, whereas it was reduced after overexpression of XIST. Functional assays showed that miR-132 inhibited the toxicity, oxidative stress, and apoptosis caused by Aβ25-35 treatment in hippocampal neurons. Collectively, these data show that XIST knockdown inhibits Aβ25-35-induced toxicity, oxidative stress, and apoptosis in hippocampal neurons by targeting miR-132.
In summary, our study suggested that knockdown of XIST relieved Aβ25-35-induced toxicity, oxidative stress, and apoptosis in primary cultured rat hippocampal neurons by upregulation of miR-132. These findings encourage continued investigation of the potential of manipulating XIST in the treatment of AD.
Acknowledgements
This work is supported by National Natural Science Foundation of China (grant No. U1504809 and 31571106), and key research project plan of colleges and universities in Henan province (grant No. 17A310016).
Disclosure of conflict of interest
None.
References
- 1.Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, De Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of alzheimer’s disease. Nature. 2008;451:720–4. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. In vitro aging of ß-amyloid protein causes peptide aggregation and neurotoxicity. Brain Res. 1991;563:311–314. doi: 10.1016/0006-8993(91)91553-d. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelaein alzheimer’s disease brain contribute to neuronal death. Neurobiol Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 5.Paulson JB, Ramsden M, Forster C, Sherman MA, McGowan E, Ashe KH. Amyloid plaque and neurofibrillary tangle pathology in a regulatable mouse model of alzheimer’s disease. Am J Pathol. 2008;173:762–772. doi: 10.2353/ajpath.2008.080175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hardy J, Selkoe DJ. The amyloid hypothesis of alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 7.Tanzi RE, Bertram L. Twenty years of the alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 8.Huebbe P, Wagner AE, Boesch-Saadatmandi C, Sellmer F, Wolffram S, Rimbach G. Effect of dietary quercetin on brain quercetin levels and the expression of antioxidant and alzheimer’s disease relevant genes in mice. Pharmacol Res. 2010;61:242–246. doi: 10.1016/j.phrs.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 9.Michel TM, Gsell W, Käsbauer L, Tatschner T, Sheldrick AJ, Neuner I, Schneider F, Grünblatt E, Riederer P. Increased mitochondrial aldehydedehydrogenase in the putamen of individuals with alzheimer’s disease. J Alzheimers Dis. 2010;19:1295–1301. doi: 10.3233/JAD-2010-1326. [DOI] [PubMed] [Google Scholar]
- 10.Pratico D, Clark CM, Liun F, Lee VY, Trojanowski JQ. Increase of brain oxidative stress in mild cognitive impairment: a possible predictor of alzheimer disease. Arch Neurol. 2002;59:972–976. doi: 10.1001/archneur.59.6.972. [DOI] [PubMed] [Google Scholar]
- 11.Echeverria V, Clerman A, Doré S. Stimulation of PGE2 receptors EP2 and EP4 protects cultured neurons against oxidative stress and cell death following β-amyloid exposure. Eur J Neurosci. 2005;22:2199–2206. doi: 10.1111/j.1460-9568.2005.04427.x. [DOI] [PubMed] [Google Scholar]
- 12.Butterfield DA, Galvan V, Lange MB, Tang H, Sowell RA, Spilman P, Fombonne J, Gorostiza O, Zhang J, Sultana R. In vivo oxidative stress in brain of alzheimer disease transgenic mice: requirement for methionine 35 in amyloid β-peptide of APP. Free Radic Biol Med. 2010;48:136–144. doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turunc Bayrakdar E, Uyanikgil Y, Kanit L, Koylu E, Yalcin A. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Aβ (1-42)-induced rat model of alzheimer’s disease. Free Radic Res. 2014;48:146–158. doi: 10.3109/10715762.2013.857018. [DOI] [PubMed] [Google Scholar]
- 14.Celsi F, Svedberg M, Unger C, Cotman C, Carri M, Ottersen O, Nordberg A, Torp R. Betaamyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol Dis. 2007;26:342–352. doi: 10.1016/j.nbd.2006.12.022. [DOI] [PubMed] [Google Scholar]
- 15.Carrillo-Mora P, Luna R, Colín-Barenque L. Amyloid beta: multiple mechanisms of toxicity and only some protective effects? Oxid Med Cellul Longev. 2014;2014:795375. doi: 10.1155/2014/795375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian X, Zhang L, Wang J, Dai J, Shen S, Yang L, Huang P. The protective effect of hyperbaric oxygen and Ginkgo biloba extract on Aβ25-35-induced oxidative stress and neuronal apoptosis in rats. Behav Brain Res. 2013;242:1–8. doi: 10.1016/j.bbr.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 17.Ding H, Wang H, Zhao Y, Sun D, Zhai X. Protective effects of baicalin on Aβ1-42-induced learning and memory deficit, oxidative stress, and apoptosis in rat. Cell Mol Neurobiol. 2015;35:623–632. doi: 10.1007/s10571-015-0156-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu JF, Yan XD, Qi LS, Li L, Hu GY, Li P, Zhao G. Ginsenoside Rd attenuates Aβ25-35-induced oxidative stress and apoptosis in primary cultured hippocampal neurons. Chem Biol Interact. 2015;239:12–18. doi: 10.1016/j.cbi.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 19.Ghasemi R, Zarifkar A, Rastegar K, Moosavi M. Insulin protects against Aβ-induced spatial memory impairment, hippocampal apoptosis and MAPKs signaling disruption. Neuropharmacology. 2014;85:113–120. doi: 10.1016/j.neuropharm.2014.01.036. [DOI] [PubMed] [Google Scholar]
- 20.Zhou X, Xu J. Identification of alzheimer’s disease-associated long noncoding RNAs. Neurobiol Aging. 2015;36:2925–2931. doi: 10.1016/j.neurobiolaging.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 21.Roberts TC, Morris KV, Wood MJ. The role of long non-coding RNAs in neurodevelopment, brain function and neurological disease. Philos Trans R Soc Lond B Biol Sci. 2014:369. doi: 10.1098/rstb.2013.0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qureshi IA, Mattick JS, Mehler MF. Long non-coding RNAs in nervous system function and disease. Brain Res. 2010;1338:20–35. doi: 10.1016/j.brainres.2010.03.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang YR, Zang SZ, Zhong CL, Li YX, Zhao SS, Feng XJ. Increased expression of the lncRNA PVT1 promotes tumorigenesis in nonsmall cell lung cancer. Int J Clin Exp Pathol. 2014;7:6929–35. [PMC free article] [PubMed] [Google Scholar]
- 24.Braconi C, Kogure T, Valeri N, Huang N, Nuovo G, Costinean S, Negrini M, Miotto E, Croce C, Patel T. microRNA-29 can regulate expression of the long non-coding RNA gene MEG3 in hepatocellular cancer. Oncogene. 2011;30:4750–6. doi: 10.1038/onc.2011.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen S, Wang M, Yang H, Mao L, He Q, Jin H, Ye ZM, Luo XY, Xia YP, Hu B. LncRNA TUG1 sponges microRNA-9 to promote neurons apoptosis by up-regulated Bcl2l11 under ischemia. Biochem Biophys Res Commun. 2017;485:167–173. doi: 10.1016/j.bbrc.2017.02.043. [DOI] [PubMed] [Google Scholar]
- 26.Gu S, Xie R, Liu X, Shou J, Gu W, Che X. Long coding RNA XIST contributes to neuronal apoptosis through the downregulation of AKT phosphorylation and is negatively regulated by miR-494 in rat spinal cord injury. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18040732. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Irvine GB, El-Agnaf OM, Shankar GM, Walsh DM. Protein aggregation in the brain: the molecular basis for alzheimer’s and Parkinson’s diseases. Mol Med. 2008;14:451–64. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tamagno E, Parola M, Guglielmotto M, Santoro G, Bardini P, Marra L, Tabaton M, Danni O. Multiple signaling events in amyloid β-induced, oxidative stress-dependent neuronal apoptosis. Free Radic Biol Med. 2003;35:45–58. doi: 10.1016/s0891-5849(03)00244-2. [DOI] [PubMed] [Google Scholar]
- 29.Ho PI, Collins SC, Dhitavat S, Ortiz D, Ashline D, Rogers E, Shea TB. Homocysteine potentiates β-amyloid neurotoxicity: role of oxidative stress. J Neurochem. 2001;78:249–253. doi: 10.1046/j.1471-4159.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- 30.Wang F, Ying HQ, He BS, Pan YQ, Deng QW, Sun HL, Chen J, Liu X, Wang SK. Upregulated lncRNA-UCA1 contributes to progression of hepatocellular carcinoma through inhibition of miR-216b and activation of FGFR1/ERK signaling pathway. Oncotarget. 2015;6:7899–917. doi: 10.18632/oncotarget.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao Y, Ma J, Xue Y, Wang P, Li Z, Liu J, Chen L, Xi Z, Teng H, Wang Z. Knockdown of long non-coding RNA XIST exerts tumor-suppressive functions in human glioblastoma stem cells by up-regulating miR-152. Cancer Lett. 2015;359:75–86. doi: 10.1016/j.canlet.2014.12.051. [DOI] [PubMed] [Google Scholar]
- 32.Li J, Wang J, Chen Y, Li S, Jin M, Wang H, Chen Z, Yu W. LncRNA MALAT1 exerts oncogenic functions in lung adenocarcinoma by targeting miR-204. Am J Cancer Res. 2016;6:1099–107. [PMC free article] [PubMed] [Google Scholar]
- 33.Xue D, Zhou C, Lu H, Xu R, Xu X, He X. LncRNA GAS5 inhibits proliferation and progression of prostate cancer by targeting miR-103 through AKT/mTOR signaling pathway. Tumor Biol. 2016 doi: 10.1007/s13277-016-5429-8. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 34.Wong HK, Veremeyko T, Patel N, Lemere CA, Walsh DM, Esau C, Vanderburg C, Krichevsky AM. De-repression of FOXO3a death axis by microRNA-132 and-212 causes neuronal apoptosis in alzheimer’s disease. Hum Mol Genet. 2013;22:3077–3092. doi: 10.1093/hmg/ddt164. [DOI] [PubMed] [Google Scholar]
- 35.Smith PY, Hernandez-Rapp J, Jolivette F, Lecours C, Bisht K, Goupil C, Dorval V, Parsi S, Morin F, Planel E. miR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum Mol Genet. 2015;24:6721–6735. doi: 10.1093/hmg/ddv377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salta E, Sierksma A, Eynden EV, De Strooper B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in alzheimer’s brain. EMBO Mol Med. 2016;8:1005–1018. doi: 10.15252/emmm.201606520. [DOI] [PMC free article] [PubMed] [Google Scholar]