

Abstract
Blood supply returned to infracted tissue causes tissue damage. Therefore, ischemia/reperfusion (I/R) injuries are usually accompanied by synapse formation, but the exact cause is still unknown. To address this question, we established a middle cerebral artery occlusion (MCAO) rat model with different reperfusion times, and we examined neurological deficit scores and brain infarct size. Subsequently, thrombospondin (TSP)-1 and TSP-2 expression levels in the cingulated cortex, striate cortex, aud cortex, and piriform cortex at different time points after I/R were examined using immunohistochemistry (IHC). In addition, synaptophysin expression in the hippocampus was examined using IHC. As expected, after ischemia with different reperfusion times, higher neurological deficits scores were observed in MCAO rats compared to sham-operated rats. Brain infarct sizes were increased in different sections, and brain sections exhibited obvious necrosis in the right cerebra. In addition, TSP-1 and TSP-2 expression levels in the cingulated cortex, striate cortex, aud cortex, and piriform cortex significantly increased with increasing reperfusion times. Similarly, synaptophysin expression levels in the hippocampus significantly increased with increasing reperfusion times. Our results indicate that altered TSP-1 and TSP-2 expression in cortical areas may contribute to synapse formation. Our model not only allowed us to observe the time-related expression of TSP-1, TSP-2, and synaptophysin after I/R injury but also provides a potential tool for studying synapse formation.
Keywords: Ischemia/reperfusion, middle cerebral artery occlusion, thrombospondin-1, thrombospondin-2, synaptophysin, immunohistochemistry
Introduction
Ischemia/reperfusion (I/R) injury is a prevalent condition induced by sub-lethal ischemia. I/R leads to adaptive responses that protect against future severe ischemic injuries [1-3]. I/R occurs when an excessive excitatory amino acid release leads to neurodegenerative processes such as excessive calcium influx, brain edema, electrophysiological and metabolic dysfunction, lipid peroxidation, cellular apoptosis, oxidation, and inflammatory stress [3-6]. To date, focal cerebral I/R injury is becoming a leading cause of the morbidity and mortality worldwide [7-11]. Lacking a sufficient blood supply, large primary and secondary neuronal areas die, and denervated regions stimulate post-ischemic angiogenesis and synaptogenesis [12-17].
Thrombospondins (TSPs) are a family of secreted glycoproteins with antiangiogenic functions, and they include TSP-1 and TSP-2 [18-20]. TSP-1 and TSP-2 are homotrimers, consisting of three identical subunits [19,21-24]. Previous studies demonstrated that the expression of TSP-1 and TSP-2 significantly increases after focal cerebral I/R. Furthermore, the expression of TSP-2 occurs much later than TSP-1, peaking 2 weeks after I/R injury. However, the expression sites of TSP-1 and TSP-2 in different cortical areas have not been reported [24-26]. In addition, recent studies have shown that TSP-1 and TSP-2 might promote the development of new synapses [19]. However, the underlying mechanisms triggering the formation of synapses via TSP-1/TSP-2 upon focal cerebral I/R have not yet been investigated [19]. Therefore, we developed the rat middle cerebral artery occlusion (MCAO) model to evaluate neurological behavior and brain infarct size. Furthermore, we examined the expression levels of TSP-1/TSP-2 in different cortical areas, and the expression of synaptophysin in the hippocampus using immunohistochemistry (IHC). Our model provides a significant reference for the time-related expression of TSP-1 and TSP-2 in different brain regions after I/R injury. Moreover, increased expression levels of TSP-1 and TSP-2 may be associated with the formation of new synapses.
Materials and methods
Experimental animals and groups
A total of 25 adult specific pathogen-free Sprague Dawley rats (male, 250-330 g) were purchased and raised in the Laboratory Animal Center of the Academy of Military Medical Sciences (room temperature: 25±2°C, Humidity: 40-60%, 12 h light/dark cycle). They were randomly divided into the following three groups: sham group, ischemia group (only ligation of the right carotid artery communis by line, labeled as I-2 h), and I/R (ligation of the right carotid artery communis by line, and the line was removed for reperfusion at a different time) group. The I/R group was further divided into three groups, 2 h ischemia/1 d reperfusion (labeled as I/R-2 h/1 d), 2 h ischemia/7 d reperfusion (labeled as I/R-2 h/7 d), and 2 h ischemia/14 d reperfusion (labeled as I/R-2 h/14 d). Each group comprised 5 rats. All animals and experimental procedures were approved by the Ethics Committee of the Academy of Military Medical Sciences of the Chinese People’s Liberation Army (Beijing, China).
Establishment of the rat middle cerebral artery occlusion (MCAO) model
The above-mentioned Sprague Dawley rats were anesthetized with sodium pentobarbital (25 mg/kg), and the four limbs were fixed before an incision was made along the neck midline. The right external carotid artery and the right common carotid artery were ligated with a 10-0 suture. The thyroid, vein, and nerve tissue were stripped to expose the right carotid artery communis, which was ligated with a 10-0 surgical line for 2 h ischemia. Subsequently, the line was removed to allow reperfusion for 1 d, 7 d, and 14 d. After MCAO modeling, the rats were kept separately in an air-ventilated room with water and food. The sham group rats were subjected to vascular surgeries without I/R. After the experiment, animals were sacrificed by dislocation of the cervical spine to prepare brain tissue for slicing and further examination.
Neurological deficits score
After MCAO modeling, the rats were tested for neurological deficits. Neurological deficit scores were given by a blinded observer as described by Bederson (27), with the following minor modification: 0 indicated no observable deficit (normal); 1 indicated failure to extend the right forepaw (mild); 2 indicated circling to the contralateral side (moderate); 3 indicated loss of walking or righting reflex (severe). Animals with no neurological deficit after MCAO modeling, or with significant symptoms of subarachnoid brain hemorrhage, were excluded from the analysis.
2,3,5-triphenyl four azole nitrogen chloride (TTC) staining
The rats’ brain tissue was collected, washed with saline for 5 min, and frozen for 20 min at -20°C. Then, their brains were transferred to a brain sectioning mold and cut into 5 slices (labeled as P1, P2, P3, P4, and P5). The sections were transferred to 1% TTC at 37°C for 30 min, and washed three times with ddH2O. Images were taken for further analysis.
Immunohistochemistry (IHC)
The rat brain tissue was cut into thin slices (1 μm), and sections were deparaffinized, rehydrated, and post-fixed with 4% paraformaldehyde for 10 min. Then slices were washed three times with 0.01 M phosphate-buffered saline (PBS). The endogenous peroxidase was inactivated by incubating sections in 3% hydrogen peroxide (H2O2) for 30 min. The sections were subjected to sequential incubations with 10% normal goat serum in 0.01 M PBS for 30 min at room temperature. Slices were then incubated in rabbit anti-TSP-1/-2 polyclonal antibody (1:100, Santa Cruz, USA) and rabbit anti-synaptophysin (1:100, Santa Cruz, USA) in PBS containing 0.3% Triton X-100 overnight at 4°C. Sections were washed three times for 5 min with PBS, and then incubated in peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) (1:200; Zymed, South San Francisco, CA, USA) for 1 h at room temperature. Finally, the sections were developed with diaminobenzidine (Sigma, St. Louis, MO, USA) in 0.1 M Tris-buffered saline (TBS) containing 0.001% H2O2 for 30 min. The cingulated cortex, striate cortex, aud cortex, and piriform cortex were observed under a microscope (Olympus, Tokyo, Japan), and 5 specific areas in each region were captured. The integrated optical density (IOD) values of TSP-1 and TSP-2 labeling in the cingulated cortex, striate cortex, aud cortex, and piriform cortex were measured by Image-Pro Plus 7.0 software (http://www.mediacy.com/index.aspx?page=IP_Premier; Media Cybernetics, Bethesda, MD, USA) and a histogram analysis was performed using Origin 9.0 software (http://www.originlab.com/; Northampton, MA, USA). Similarly, the hippocampus was observed under a microscope (Olympus, Tokyo, Japan), and five specific areas in each region were captured. The integrated optical density (IOD) values of synaptophysin labeling were measured. Each assessment was repeated at least three times.
Statistical analysis
All data are expressed as the mean ± standard deviation (SD). The statistical analysis was performed with a one-way analysis of variance using Statistical Package for Social Sciences software (version 21.0, http://spss.en.softonic.com/; Chicago, IL, USA). Student’s t-tests were performed for two sample groups. The threshold for statistically significant differences and highly statistically significant differences were defined as P<0.05 and P<0.01, respectively.
Results
Rat MCAO model showed higher neurological deficits score
Table 1 shows neurological deficits scores for the MCAO rats and the sham rats. For the MCAO rats, the neurological deficits score was increased to 1 (indicating the failure to extend the right forepaw) and increased to 2 and/or 3 after different reperfusion times, indicating that reperfusion could enhance the injury caused by ischemia. Taken together, these results suggest that the rat MCAO model was correctly established.
Table 1.
Neurological deficit score increased with increasing reperfusion times in the rat MCAO model
| Groups | Score |
|---|---|
| Sham | 0 |
| I-2 h | 1 |
| I/R-2 h/1 d | 2 |
| I/R-2 h/7 d | 3 |
| I/R-2 h/14 d | 3 |
Infarct size was significantly large in the right cerebra of P1 to P5 sections
As shown in Figure 1A and 1B, when compared to P1 sections, infarct size significantly increased in the right cerebra of P2 to P4 in the group of I-2 h, I/R-2 h/1 day, I/R-2 h/7 day, and I/R-2 h/14 day (*: P<0.05; **: P<0.01), indicating that the rat MCAO model was correctly established.
Figure 1.
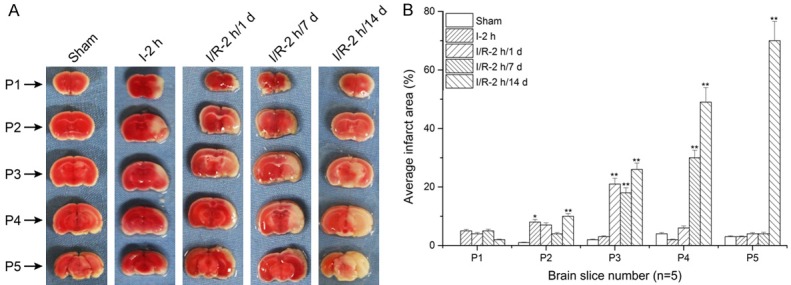
2,3,5-Triphenyl four azole nitrogen chloride (TTC) staining of rat brain tissue and histogram analysis. Images show that infarct size significantly increased in P2-P4 sections compared to P1 (*: P<0.05; **: P<0.01, compared to P1).
Thrombospondin (TSP)-1 and TSP-2 expression levels in cingulate cortex significantly increased with prolonged reperfusion times
As shown in Figure 2A and 2B, when compared to the sham and ischemia groups, TSP-1 expression levels in the cingulated cortex significantly increased with prolonged reperfusion times (**: P<0.01). Similarly, the TSP-2 expression level in the cingulated cortex significantly increased with prolonged reperfusion times (**: P<0.01) and reached a peak after 7 d of reperfusion (Figure 2A and 2C).
Figure 2.

Thrombospondin (TSP)-1 and TSP-2 expression level assays in the cingulated cortex using immunohistochemistry and histogram analysis. A. Immunohistochemistry assay of TSP-1 and TSP-2 in the cingulated cortex; B. Histogram analysis of the TSP-1 expression level in the cingulated cortex; C. Histogram analysis of the TSP-2 expression level in the cingulated cortex. The images indicate that TSP-1/TSP-2 expression levels in the cingulated cortex significantly increased with increasing reperfusion times (**: P<0.01, compared to sham).
TSP-1 and TSP-2 expression levels in striate cortex significantly increased with prolonged reperfusion times
As shown in Figure 3A and 3B, when compared to the sham and ischemia groups, TSP-1 expression levels in the striate cortex significantly increased with prolonged reperfusion times (**: P<0.01) and reached a peak after 7 d reperfusion. Similarly, TSP-2 expression levels in the striate cortex significantly increased with prolonged reperfusion times (**: P<0.01) and reached a peak after 7 d of reperfusion (Figure 3A and 3C).
Figure 3.
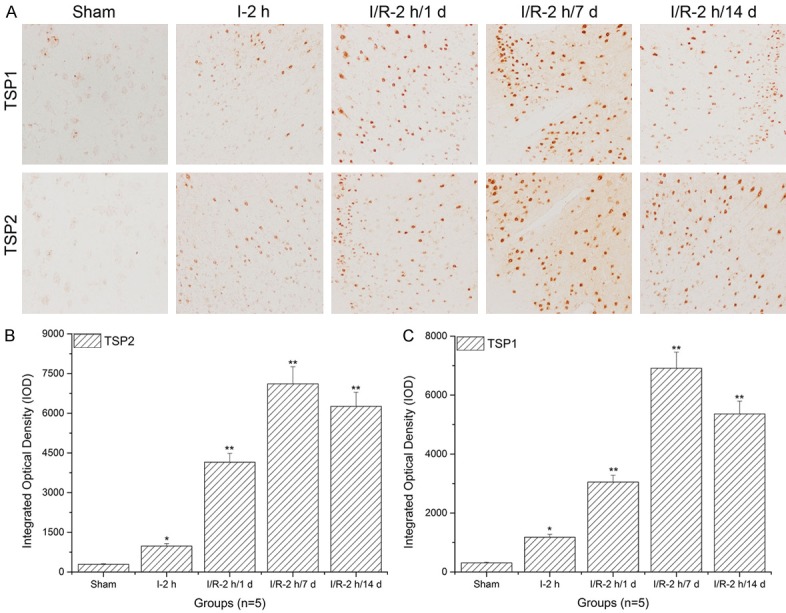
Thrombospondin (TSP)-1 and TSP-2 expression level assays in the striate cortex using immunohistochemistry and histogram analysis. A. Immunohistochemistry assay of TSP-1 and TSP-2 in the striate cortex; B. Histogram analysis of TSP-1 expression levels in the striate cortex; C. Histogram analysis of TSP-2 expression levels in the striate cortex. Images indicate that the TSP-1/TSP-2 expression levels in the striate cortex significantly increased with increasing reperfusion times (**: P<0.01, compared to sham).
TSP-1 and TSP-2 expression levels in the aud cortex significantly increased with prolonged reperfusion times
As shown in Figure 4A and 4B, when compared to the sham and ischemia groups, TSP-1 expression levels in the aud cortex significantly increased with prolonged reperfusion times (**: P<0.01). Similarly, TSP-2 expression levels in the aud cortex significantly increased with prolonged reperfusion times (**: P<0.01) and reached a peak after 7 d reperfusion (Figure 4A and 4C).
Figure 4.
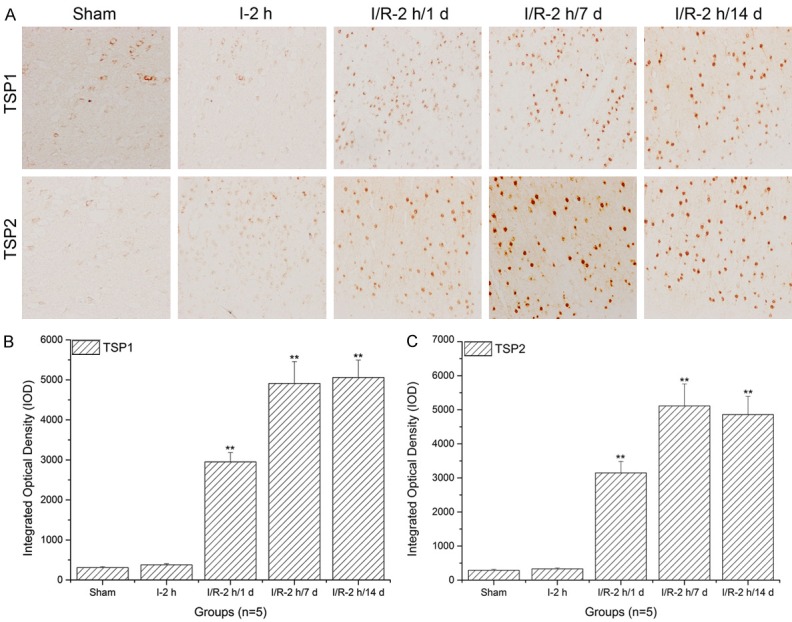
Thrombospondin (TSP)-1 and TSP-2 expression level assays in the aud cortex using immunohistochemistry and histogram analysis. A. Immunohistochemistry assay of TSP-1 and TSP-2 in aud cortex; B. Histogram analysis of TSP-1 expression levels in the aud cortex; C. Histogram analysis of TSP-2 expression levels in the aud cortex. The images indicate that TSP-1/TSP-2 expression levels in the aud cortex significantly increased with increasing reperfusion times (**: P<0.01, compared to sham).
TSP-1 and TSP-2 expression levels in the piriform cortex significantly increased with prolonged reperfusion times
As shown in Figure 5A, when compared to the sham and ischemia groups, TSP-1 expression levels in the piriform cortex significantly increased with prolonged reperfusion times (**: P<0.01) and reached a peak after 7 d of reperfusion. Similarly, TSP-2 expression levels in the piriform cortex significantly increased with prolonged reperfusion times (**: P<0.01) and reached a peak after 7 d of reperfusion (Figure 5A and 5C).
Figure 5.
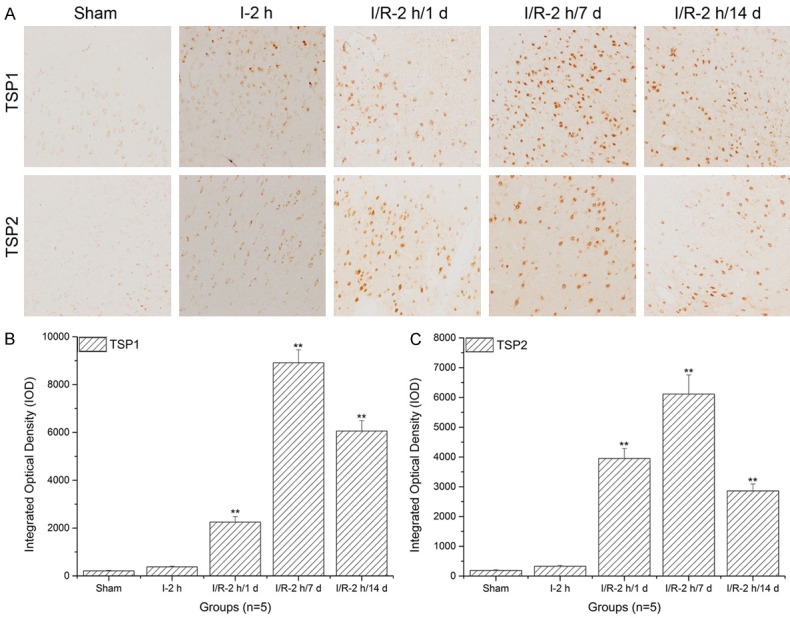
Thrombospondin (TSP)-1 and TSP-2 expression level assays in the piriform cortex using immunohistochemistry and histogram analysis. A. The immunohistochemistry assays of TSP-1 and TSP-2 in the piriform cortex; B. Histogram analysis of TSP-1 expression levels in the piriform cortex; C. Histogram analysis of TSP-2 expression levels in the piriform cortex. Images indicate that the TSP-1/TSP-2 expression levels in the piriform cortex significantly increased with increasing reperfusion times (**: P<0.01, compared to sham).
Synaptophysin expression levels in the hippocampus significantly increased with prolonged reperfusion times
As shown in Figure 6, when compared to the sham and ischemia groups, synaptophysin expression levels in the hippocampus significantly increased with prolonged reperfusion times (**: P<0.01) and reached a peak after 7 d of reperfusion.
Figure 6.
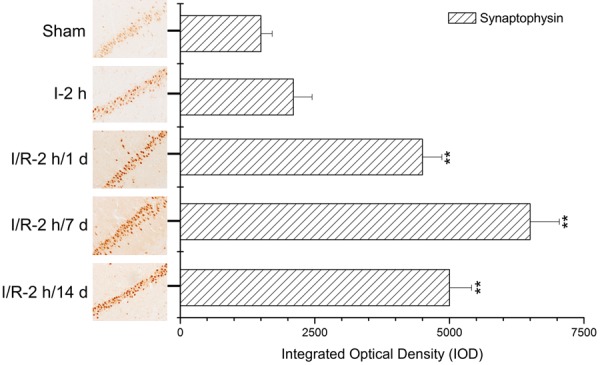
Synaptophysin expression level assays in the hippocampus using immunohistochemistry and histogram analysis. Images indicate that synaptophysin expression levels in the hippocampus significantly increased with increasing reperfusion times (**: P<0.01, compared to sham).
Discussion
In the present study, we show that our rat MCAO model was correctly established. The MCAO rats showed higher neurological deficits scores and obvious necrosis. Moreover, TSP-1 and TSP-2 expression levels significantly increased with prolonged reperfusion times in different cortical areas (cingulated cortex, striate cortex, aud cortex, and piriform cortex). Similarly, synaptophysin expression levels in hippocampus significantly increased with prolonged reperfusion times. Our results indicate that altered expression levels of TSP-1 and TSP-2 may contribute to the regulation of synaptophysin in the hippocampus and may regulate synapse formation.
MCAO models have been created by injecting particles (such as blood clots or artificial spheres) into the carotid arteries of experimental animals [28-30]. Reperfusion of ischemic tissue (transient focal cerebral ischemia) and ischemia without reperfusion (permanent focal cerebral ischemia) have also been modeled [29,31]. The area of injury is typically concentrated in the periventricular regions of the brain, especially the cortical and hippocampal areas [28,32,33]. To evaluate different models of MCAO, several neurological deficit score criteria have been used to evaluate the severity and curative effects [27]. In the present study, rat MCAO models were correctly established and further tested to obtain neurological deficit scores by a blinded observer as described by Bederson [27]. After 2 h of ischemia, the neurological deficit score increased to 1 and reperfusion for up to 14 d led to increases in neurological deficit scores to 2 or 3. These results indicate that the MCAO model was correctly established.
TTC is a redox indicator commonly used in biochemical experiments to indicate cellular respiration. In particular, TTC is often used to differentiate between metabolically active and inactive tissue [34-37]. In living tissue, the white compound is reduced to red owing to the activity of various dehydrogenases. In areas of necrosis, TTC remains white, because dehydrogenases have been either denatured or degraded [35,36,38,39]. Because of these properties, TTC staining has been widely used to identify I/R injury. Healthy tissue appears red and infarcted tissue appears white. In order to determine the MCAO model was correctly established, rat brain tissues with 14 d reperfusion were chosen, and as expected, after MCAO modeling, the necrosis manifested itself as white areas in the brain sections. The extent of the white areas significantly increased in the P2-P4 sections, indicating that the MCAO model had been correctly established.
Proteins from the TSP family, including TSP-1 and TSP-2, are matricellular proteins that inhibit angiogenesis and cause apoptosis in vivo and in vitro in several cancerous cells and tissues [25,40-44]. Previous studies have revealed that proteins from the TSP family act as a mediators of cell injury in kidney ischemia and brain ischemia [42,45-47]. The striate cortex, cingulate cortex, aud cortex, and piriform cortex were affected by the reduction of the blood supply. In this study, we examined the expression levels of TSP-1 and TSP-2 in different cortical areas (striate cortex, cingulate cortex, aud cortex, and piriform cortex) at 1 d, 7 d, and 14 d after reperfusion. The expression levels of TSP-1 and TSP-2 were significantly higher at various time points tested after I/R injury in the I/R groups as compared with the sham group. Additionally, synaptophysin expression levels in the hippocampus significantly increased after I/R injury and exhibited a similar trend as TSP-1 and TSP-2. Therefore, we hypothesize that the altered expression levels of TSP-1 and TSP-2 may have contributed to the up regulation of synaptophysin in the hippocampus and may regulate synapse formation. This interpretation is in line with a previous report on TSPs as astrocyte-secreted proteins that promote the formation of ultrastructurally normal central nervous system synapses [48]. However, we only assessed the expression levels of TSP-1 and TSP-2 in the cortex and the expression levels of synaptophysin in the hippocampus. Therefore, our conclusion that TSP-1 and TSP-2 were closely associated with synaptophysin in the hippocampus needs to be validated more directly with experiments specifically designed to address this issue in the future.
In summary, in our MCAO model with different reperfusion times, TSP-1 and TSP-2 expression in different cortical areas and synaptophysin expression in the hippocampus exhibited similar trends. Our results suggest that increased TSP-1 and TSP-2 expression is closely associated with synaptophysin expression in the hippocampus and might mediate the formation of new synapses. Therefore, this study not only provides a reference for TSP-1 and TSP-2 and synaptophysin expression in MCAO models, but also reveals a potential mechanism of synapse formation.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 30772244).
Disclosure of conflict of interest
None.
References
- 1.Hou S, Zhao MM, Shen PP, Liu XP, Sun Y, Feng JC. Neuroprotective effect of salvianolic acids against cerebral ischemia/reperfusion injury. Int J Mol Sci. 2016:17. doi: 10.3390/ijms17071190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farag MM, Khalifa AA, Elhadidy WF, Rashad RM. Hepatorenal protection in renal ischemia/reperfusion by celecoxib and pentoxifylline. J Surg Res. 2016;204:183–191. doi: 10.1016/j.jss.2016.04.064. [DOI] [PubMed] [Google Scholar]
- 3.Hussein AM, Barakat N, Awadalla A, Gabr MM, Khater S, Harraz AM, Shokeir AA. Modulation of renal ischemia/reperfusion in rats by a combination of ischemic preconditioning and adipose-derived mesenchymal stem cells (ADMSCs) Can J Physiol Pharmacol. 2016;94:936–46. doi: 10.1139/cjpp-2016-0018. [DOI] [PubMed] [Google Scholar]
- 4.Messner F, Grahammer J, Hautz T, Brandacher G, Schneeberger S. Ischemia/reperfusion injury in vascularized tissue allotransplantation: tissue damage and clinical relevance. Curr Opin Organ Transplant. 2016;21:503–9. doi: 10.1097/MOT.0000000000000343. [DOI] [PubMed] [Google Scholar]
- 5.Hussein AM, Sakr HF, Alenzi FQ. Possible underlying mechanisms of the renoprotective effect of remote limb ischemic preconditioning against renal ischemia/reperfusion injury: a role of osteopontin, transforming growth factor-beta and survivin. Nephron. 2016;134:117–129. doi: 10.1159/000447953. [DOI] [PubMed] [Google Scholar]
- 6.Tanaka S, Chen-Yoshikawa TF, Kajiwara M, Menju T, Ohata K, Takahashi M, Kondo T, Hijiya K, Motoyama H, Aoyama A, Masuda S, Date H. Protective effects of imatinib on ischemia/reperfusion injury in rat lung. Ann Thorac Surg. 2016;102:1717–1724. doi: 10.1016/j.athoracsur.2016.05.037. [DOI] [PubMed] [Google Scholar]
- 7.Han J, Xiao Q, Lin YH, Zheng ZZ, He ZD, Hu J, Chen LD. Neuroprotective effects of salidroside on focal cerebral ischemia/reperfusion injury involve the nuclear erythroid 2-related factor 2 pathway. Neural Regen Res. 2015;10:1989–1996. doi: 10.4103/1673-5374.172317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li XJ, Wei LY, Li CK, Li DL. [Neuroprotective effect of progesterone on focal cerebral ischemia/reperfusion injury in rats and its mechanism] . Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2015;31:231–234. [PubMed] [Google Scholar]
- 9.Li XJ, Li CK, Wei LY, Lu N, Wang GH, Zhao HG, Li DL. Hydrogen sulfide intervention in focal cerebral ischemia/reperfusion injury in rats. Neural Regen Res. 2015;10:932–937. doi: 10.4103/1673-5374.158353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Awooda HA, Lutfi MF, Sharara GG, Saeed AM. Oxidative/nitrosative stress in rats subjected to focal cerebral ischemia/reperfusion. Int J Health Sci (Qassim) 2015;9:17–24. doi: 10.12816/0024679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gong G, Xiang L, Yuan L, Wu W, Cai L, Yin L, Dong H. Protective effect of glycyrrhizin, a direct HMGB1 inhibitor, on focal cerebral ischemia/reperfusion-induced inflammation, oxidative stress, and apoptosis in rats. PLoS One. 2014;9:e89450. doi: 10.1371/journal.pone.0089450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang Y, Liu P, Chen L, Liu Z, Zhang H, Wang J, Sun X, Zhong W, Wang N, Tian K, Zhao J. Therapeutic effect of Ginkgo biloba polysaccharide in rats with focal cerebral ischemia/reperfusion (I/R) injury. Carbohydr Polym. 2013;98:1383–1388. doi: 10.1016/j.carbpol.2013.07.045. [DOI] [PubMed] [Google Scholar]
- 13.Zheng YQ, Yao MJ, Liu JX, Song WT, Li L, Liu SB, Hu Y, Si JX. [Effect and mechanism of huatuo zaizao extractum on focal cerebral ischemia/reperfusion-induced blood-brain barrier injury in rats] . Zhongguo Zhong Yao Za Zhi. 2013;38:585–590. [PubMed] [Google Scholar]
- 14.Ma M, Uekawa K, Hasegawa Y, Nakagawa T, Katayama T, Sueta D, Toyama K, Kataoka K, Koibuchi N, Kuratsu J, Kim-Mitsuyama S. Pretreatment with rosuvastatin protects against focal cerebral ischemia/reperfusion injury in rats through attenuation of oxidative stress and inflammation. Brain Res. 2013;1519:87–94. doi: 10.1016/j.brainres.2013.04.040. [DOI] [PubMed] [Google Scholar]
- 15.Qian L, Shen M, Tang H, Tang Y, Zhang L, Fu Y, Shi Q, Li NG. Synthesis and protective effect of scutellarein on focal cerebral ischemia/reperfusion in rats. Molecules. 2012;17:10667–10674. doi: 10.3390/molecules170910667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao LD, Wang JH, Jin GR, Zhao Y, Zhang HJ. Neuroprotective effect of Buyang Huanwu decoction against focal cerebral ischemia/reperfusion injury in rats--time window and mechanism. J Ethnopharmacol. 2012;140:339–344. doi: 10.1016/j.jep.2012.01.026. [DOI] [PubMed] [Google Scholar]
- 17.Ye N, Liu S, Lin Y, Rao P. Protective effects of intraperitoneal injection of TAT-SOD against focal cerebral ischemia/reperfusion injury in rats. Life Sci. 2011;89:868–874. doi: 10.1016/j.lfs.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 18.Kirk JA, Cingolani OH. Thrombospondins in the transition from myocardial infarction to heart failure. J Mol Cell Cardiol. 2016;90:102–110. doi: 10.1016/j.yjmcc.2015.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mendus D, Sundaresan S, Grillet N, Wangsawihardja F, Leu R, Müller U, Jones SM, Mustapha M. Thrombospondins 1 and 2 are important for afferent synapse formation and function in the inner ear. Eur J Neurosci. 2014;39:1256–1267. doi: 10.1111/ejn.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stenina-Adognravi O. Thrombospondins: old players, new games. Curr Opin Lipidol. 2013;24:401–409. doi: 10.1097/MOL.0b013e3283642912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Li L, Wang B, Qian DM, Song XM, Hu M. Human cytomegalovirus infection modulates thrombospondins 1 and 2 in primary fetal astrocytes. Neuroreport. 2013;24:526–535. doi: 10.1097/WNR.0b013e32836206d1. [DOI] [PubMed] [Google Scholar]
- 22.Huang J, Zhou L, Wang H, Luo J, Zeng L, Xiong K, Chen D. Distribution of thrombospondins and their neuronal receptor alpha2delta1 in the rat retina. Exp Eye Res. 2013;111:36–49. doi: 10.1016/j.exer.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Liauw J, Hoang S, Choi M, Eroglu C, Choi M, Sun GH, Percy M, Wildman-Tobriner B, Bliss T, Guzman RG, Barres BA, Steinberg GK. Thrombospondins 1 and 2 are necessary for synaptic plasticity and functional recovery after stroke. J Cereb Blood Flow Metab. 2008;28:1722–1732. doi: 10.1038/jcbfm.2008.65. [DOI] [PubMed] [Google Scholar]
- 24.Armstrong LC, Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22:63–71. doi: 10.1016/s0945-053x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 25.Bornstein P, Agah A, Kyriakides TR. The role of thrombospondins 1 and 2 in the regulation of cell-matrix interactions, collagen fibril formation, and the response to injury. Int J Biochem Cell Biol. 2004;36:1115–1125. doi: 10.1016/j.biocel.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Bornstein P, Sage EH. Thrombospondins. Methods Enzymol. 1994;245:62–85. doi: 10.1016/0076-6879(94)45006-4. [DOI] [PubMed] [Google Scholar]
- 27.Chen J, Zhang ZG, Li Y, Wang Y, Wang L, Jiang H, Zhang C, Lu M, Katakowski M, Feldkamp CS, Chopp M. Statins induce angiogenesis, neurogenesis, and synaptogenesis after stroke. Ann Neurol. 2003;53:743–751. doi: 10.1002/ana.10555. [DOI] [PubMed] [Google Scholar]
- 28.Wang LX, Huang HH, Chen YF, Cai HC, Qian JQ. [The effects of prenatal stress on the cell apoptosis after MCAO in adult offspring rats] . Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2015;31:427–430. 436. [PubMed] [Google Scholar]
- 29.Zuo XL, Deng HL, Wu P, Xu E. Do different reperfusion methods affect the outcomes of stroke induced by MCAO in adult rats? Int J Neurosci. 2016;126:850–855. doi: 10.3109/00207454.2015.1074903. [DOI] [PubMed] [Google Scholar]
- 30.El Amki M, Clavier T, Perzo N, Bernard R, Guichet PO, Castel H. Hypothalamic, thalamic and hippocampal lesions in the mouse MCAO model: potential involvement of deep cerebral arteries? J Neurosci Methods. 2015;254:80–85. doi: 10.1016/j.jneumeth.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 31.Pramila B, Kalaivani P, Anita A, Saravana Babu C. L-NAME combats excitotoxicity and recuperates neurological deficits in MCAO/R rats. Pharmacol Biochem Behav. 2015;135:246–253. doi: 10.1016/j.pbb.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 32.Gupta S, Gupta YK. Combination of Zizyphus jujuba and silymarin showed better neuroprotective effect as compared to single agent in MCAo-induced focal cerebral ischemia in rats. J Ethnopharmacol. 2017;197:118–127. doi: 10.1016/j.jep.2016.07.060. [DOI] [PubMed] [Google Scholar]
- 33.Gu J, Chen J, Yang N, Hou X, Wang J, Tan X, Feng L, Jia X. Combination of ligusticum chuanxiong and radix paeoniae ameliorate focal cerebral ischemic in MCAO rats via endoplasmic reticulum stress-dependent apoptotic signaling pathway. J Ethnopharmacol. 2016;187:313–324. doi: 10.1016/j.jep.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 34.Sun YY, Yang D, Kuan CY. Mannitol-facilitated perfusion staining with 2,3,5-triphenyltetrazolium chloride (TTC) for detection of experimental cerebral infarction and biochemical analysis. J Neurosci Methods. 2012;203:122–129. doi: 10.1016/j.jneumeth.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng C, Fan X, Zhang C, Shi X. [On the quantitative analysis of focal ischemic cerebral infarction by TTC staining] . Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2009;26:1363–1366. [PubMed] [Google Scholar]
- 36.Kramer M, Dang J, Baertling F, Denecke B, Clarner T, Kirsch C, Beyer C, Kipp M. TTC staining of damaged brain areas after MCA occlusion in the rat does not constrict quantitative gene and protein analyses. J Neurosci Methods. 2010;187:84–89. doi: 10.1016/j.jneumeth.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 37.Okuno S, Nakase H, Sakaki T. Comparative study of 2,3,5-triphenyltetrazolium chloride (TTC) and hematoxylin-eosin staining for quantification of early brain ischemic injury in cats. Neurol Res. 2001;23:657–661. doi: 10.1179/016164101101198983. [DOI] [PubMed] [Google Scholar]
- 38.Dettmers C, Hartmann A, Rommel T, Krämer S, Pappata S, Young A, Hartmann S, Zierz S, MacKenzie ET, Baron JC. Immersion and perfusion staining with 2,3,5-triphenyltetrazolium chloride (TTC) compared to mitochondrial enzymes 6 hours after MCA-occlusion in primates. Neurol Res. 1994;16:205–208. doi: 10.1080/01616412.1994.11740228. [DOI] [PubMed] [Google Scholar]
- 39.Holmbom B, Naslund U, Eriksson A, Virtanen I, Thornell LE. Comparison of triphenyltetrazolium chloride (TTC) staining versus detection of fibronectin in experimental myocardial infarction. Histochemistry. 1993;99:265–275. doi: 10.1007/BF00269099. [DOI] [PubMed] [Google Scholar]
- 40.Tan K, Lawler J. The interaction of thrombospondins with extracellular matrix proteins. J Cell Commun Signal. 2009;3:177–187. doi: 10.1007/s12079-009-0074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Calzada MJ, Roberts DD. Novel integrin antagonists derived from thrombospondins. Curr Pharm Des. 2005;11:849–866. doi: 10.2174/1381612053381792. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Turpin CP, Wang S. Role of thrombospondin 1 in liver diseases. Hepatol Res. 2016;47:186–193. doi: 10.1111/hepr.12787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frazier WA. Thrombospondins. Curr Opin Cell Biol. 1991;3:792–799. doi: 10.1016/0955-0674(91)90052-z. [DOI] [PubMed] [Google Scholar]
- 44.Lawler J. Thrombospondins. Curr Drug Targets. 2008;9:820–821. doi: 10.2174/138945008785909266. [DOI] [PubMed] [Google Scholar]
- 45.Chatila K, Ren G, Xia Y, Huebener P, Bujak M, Frangogiannis NG. The role of the thrombospondins in healing myocardial infarcts. Cardiovasc Hematol Agents Med Chem. 2007;5:21–27. doi: 10.2174/187152507779315813. [DOI] [PubMed] [Google Scholar]
- 46.Teraoku H, Morine Y, Ikemoto T, Saito Y, Yamada S, Yoshikawa M, Takasu C, Higashijima J, Imura S, Shimada M. Role of thrombospondin-1 expression in colorectal liver metastasis and its molecular mechanism. J Hepatobiliary Pancreat Sci. 2016;23:565–73. doi: 10.1002/jhbp.376. [DOI] [PubMed] [Google Scholar]
- 47.Kyriakides TR, Maclauchlan S. The role of thrombospondins in wound healing, ischemia, and the foreign body reaction. J Cell Commun Signal. 2009;3:215–225. doi: 10.1007/s12079-009-0077-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jayakumar AR, Tong XY, Curtis KM, Ruiz-Cordero R, Shamaladevi N, Abuzamel M, Johnstone J, Gaidosh G, Rama Rao KV, Norenberg MD. Decreased astrocytic thrombospondin-1 secretion after chronic ammonia treatment reduces the level of synaptic proteins: in vitro and in vivo studies. J Neurochem. 2014;131:333–347. doi: 10.1111/jnc.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]