

Abstract
Hydrogen sulfide (H2S) plays an important role in diverse physiological and pathophysiological processes in cancer cells both in vitro and in vivo. We have previously shown that exogenous H2S exerts its biological effects on hepatoma, glioma, and esophageal cancer cells through the activation of NF-κB, p38-MAPK/ERK1/2-COX-2, and HSP90 pathways. However, the role of H2S and the underlying mechanism in esophageal squamous cell carcinoma remain unclear. Here we investigated whether exogenous H2S contributes to the biological behavior of esophageal squamous cancer cell line EC109, through the activation of JAK2/STAT3 signaling pathway. EC109 cells were treated with NaHS (a donor of H2S) and AG490 (a specific inhibitor of JAK2/STAT3 signaling pathway). The expression levels of p-JAK2, p-STAT3, caspase-3/9/12, Bax, Bcl-2, MMP-2/9, and VEGFR were measured by western blot analysis. Cell viability was detected by CCK-8 and quantified by direct counting of cells under a microscope. Cell migration was analyzed by the scratch-wound assay, while the level of VEGF was measured by ELISA. Cells treated with NaHS for 24 h showed significant upregulation of p-JAK2, and p-STAT3 expression, as well as increased cell viability when compared to the control cells. The expression levels of caspase-3/9/12 and Bax decreased, while those of Bcl-2, MMP-2/9, VEGFR, and VEGF increased. NaHS induced the migration of EC109 cells. However, co-treatment with NaHS and AG490 significantly inhibited these effects. Thus, JAK2/STAT3 signaling pathway may contribute to H2S-induced cell proliferation, anti-apoptosis, migration, and angiogenesis in EC109 cells.
Keywords: Hydrogen sulfide, esophageal squamous cell carcinoma, JAK2/STAT3
Introduction
Esophageal carcinoma (EC) is the sixth most common cause of cancer-related mortality worldwide, with a 5-year overall survival (OS) rate ranging from 15% to 25%. About 60% of the global EC cases occur in China, with 4,779,000 new cases reported in 2015, while 179,000 new cases were diagnosed in 2016 in the United States [1-3]. Esophageal squamous cell carcinoma (ESCC) is the major histological type of EC in China, accounting for more than 90% of all cases [4]. Multiple therapeutic approaches, including surgery, neo-/adjuvant radiation, and chemotherapy, are widely applied for ESCC, but the improvement in OS and progression-free survival (PFS), remains unsatisfactory. This observation may be attributed to the lower rate of early diagnosis, recurrence, progression, and metastasis. The effectiveness of targeted therapy is notable in many cancers such as lung cancer, hepatoma, and gastrointestinal stromal tumor (GIST), but is yet to be achieved in the case of ESCC. Thus, better therapeutic agents are required to improve patient survival. It is believed that a complicated series of molecular and biological pathways are involved in the occurrence and progression of ESCC, such as epidermal growth factor receptor (EGFR), Her-2, p53, and heat shock proteins (HSPs) signaling [5-7]. However, the detailed mechanisms underlying the process of oncogenesis in ESCC are uncharacterized.
The Janus family kinase (JAK), a type of non-receptor protein tyrosine kinases (PTK), is known to be involved in the growth, survival, development, and differentiation of a variety of cells. JAKs, including four members JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2), are selectively associated with the cytoplasmic domains of various cytokine receptors [8]. It is reported that JAKs are key intracellular mediators involved in the signaling pathways of about 60 cytokines and hormones [9]. A variety of studies demonstrated that activation of the JAK2 pathway may result in a substantial increase in proliferation, differentiation, and apoptosis of a wide variety of cancer cells [10]. Signal transducer and activator of transcription 3 (STAT3) is an important downstream target in the JAK2 pathway. Accumulating evidence suggests phosphorylation of STAT3 is associated with the oncogenic potential and anti-apoptotic activities of various tumor cells, including breast cancer [11], multiple myeloma [12], hepatocellular carcinoma, and cholangiocarcinoma [13]. However, there is a paucity of data on the relationship between the activated JAK2/STAT3 pathway and ESCC progression.
H2S is thought to be a gasotransmitter, like nitric oxide and carbon monoxide. H2S has important physiological functions such as the regulation of metabolism, cell membrane polarization,smooth muscle cell relaxation, and neuronal excitability [14]. Recent studies have demonstrated that H2S enhances the proliferation, migration, and invasion of cancer cells [15-18], and maintains cellular bioenergetics to support tumor growth and promote angiogenesis and vasorelaxation [19]. In our preliminary experiments, we found evidence that exogenous H2S strengthened carcinogenesis via the activation of NF-κB, p38-MAPK/ERK1/2-COX-2 and HSP90 pathways [20-22]. However, the relationship between H2S and the JAK2/STAT3 signaling pathway remains unclear. Here, we investigate whether exogenous H2S contributes to the biological behavior of esophageal squamous carcinoma cell line EC109, through activation of the JAK2/STAT3 signaling pathway.
Materials and methods
Cell culture and treatments
The human esophageal carcinoma cell line EC109 was purchased from Experimental Animal Center of Sun Yat-sen University (Guangzhou, China). The cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium supplemented with 10% fetal bovine serum and incubated in 5% CO2 and 95% air at 37°C. The cells were treated with 500 µmol/L sodium hydrosulfide (NaHS; a donor of H2S, purchased from Sigma-Aldrich Co., USA) and different concentrations of AG490 (specific inhibitor of JAK2; Sigma-Aldrich Co., USA).
Cell Counting Kit-8 (CCK-8) assay for the measurement of cell viability
EC109 cells were cultured at a concentration of 1 × 104/mL in 96-well plates and the cell viability was measured using the CCK-8 assay (Dojindo Lab, Japan). The cells were incubated with NaHS and AG490 for 1.5 h and absorbance was measured at 450 nm using a microplate reader (Molecular Devices, USA). The optical density (OD) values of five wells were used to evaluate cell viability as follows: Cell viability (%) = (OD treatment group/OD control group) × 100%. In addition, EC109 cells were seeded in six-well plates, and treated with either NaHS (500 μmol/L), AG490 (60 μmol/L), or both for 24 and 48 h and the number of cells was counted directly under a microscope. The experiment was performed thrice.
Western blot analysis
After different treatments, the cells were harvested and dissolved in a lysis solution. Protein concentrations were quantified using the bicinchoninic acid assay kit. Equal amounts of proteins were separated by electrophoresis on a 10% sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes. The blotted membrane was blocked with 5% nonfat dry milk (Sigma) and incubated overnight with either anti-p-JAK2 (1:1000 dilution), anti-p-STAT3 (1:1000 dilution), anti-caspase-3 (1:1000 dilution), anti-caspase-9 (1:1000 dilution), anti-caspase-12 (1:1000 dilution), anti-Bax (1:1000 dilution), anti-Bcl-2 (1:1000 dilution), anti-MMP-2 (1:1000 dilution), anti-MMP-9 (1:1000 dilution), or anti-VEGFR (1:1000 dilution) antibody at 4°C. The membranes were washed thrice in TBST and incubated for 2 h with a horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (1:2500 dilution) in TBST with 3% nonfat dry milk at room temperature. The membranes were washed thrice with TBST. Enhanced chemiluminescence (ECL) assay was used to detect the immunoreactive signals and the X-ray films were analyzed with Image J 1.47i software. Allantibodies used in the experiment were purchased from CST Co. (Boston, USA). Experiments were performed thrice.
Scratch-wound assay
EC109 cells were cultured in six-well plates. After starving the cells for 24 h in the medium without epidermal growth factor (EGF) or FBS, shaped and straight scratches were made across each well using a sterile pipette tip to create a cell-free area. Cell debris was removed and a smooth scratch edge was made by washing with phosphate-buffered saline (PBS). The cells were subsequently exposed to the following drugs: NaHS (500 μmol/L), AG490 (60 μmol/L), or a mixture of both. A scratch was also made in cells from control groups; these were washed and maintained in the culture medium supplemented with 1% FBS. The wound healing assays were performed in growth factor-free medium to exclude any differences in cell proliferation. The wound was inspected and photographed, at 200× magnification with a phase-contrast microscope at different time points. The experiment was performed thrice.
Enzyme-linked immunosorbent assay (ELISA) to assess VEGF content in the cell culture supernatant
EC109 cells were seeded in 96-well plates and subjected to different treatments. The level of VEGF in the culture media was assessed using the ELISA kit (ExCell Bio Company, Shanghai, China) according to the manufacturer’s instructions. The same experiment was performed thrice.
Statistical analysis
All the data were analyzed using the SPSS software (SPSS version 19.0; IBM SPSS Inc., Chicago, IL, USA). The experimental data are presented as the mean ± standard error of the mean (SEM). Differences between groups were analyzed by one-way analysis of variance (ANOVA), followed by least significant difference (LSD) post hoc comparison test. Statistical significance was set at P<0.05.
Results
NaHS increases the levels of both p-JAK2 and p-STAT3 in EC109 cells
To determine the effects of H2S on the levels of p-JAK2 and p-STAT3 in EC109 cells, a time-response experiment to quantify the expression of these proteins was performed. As shown in Figure 1A and 1B), the exposure of cells to NaHS (500 µmol/L) for the indicated time periods (6, 12, and 24 h) resulted in an upregulation in the expression of p-JAK2 and p-STAT3 in a time-dependent manner from 6 h and the values reached their peaks at 24 h.
Figure 1.
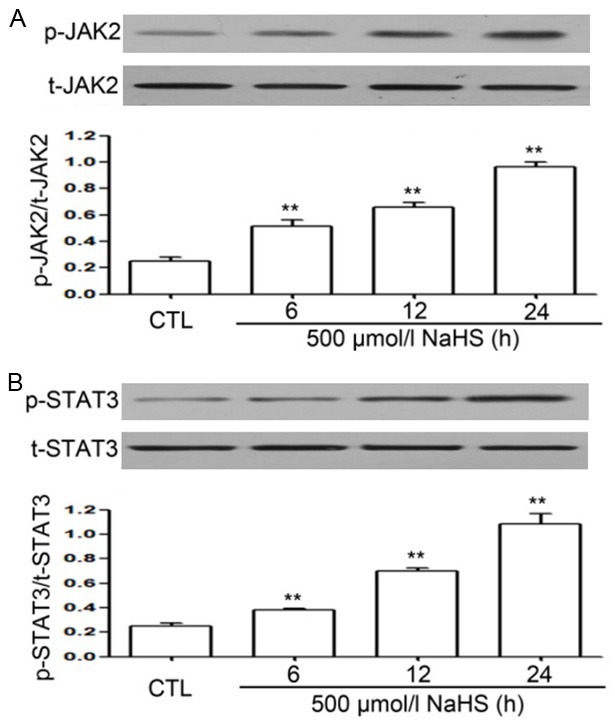
NaHS enhances the levels of phosphorylated (p)-JAK2 and p-STAT3 in EC109 cells. The expression of p-JAK2 and p-STAT3 was examined by western blot analysis (A and B) and quantified by densitometric analysis with Image J 1.47i software. To investigate the effects of NaHS on the expression of p-JAK2 and p-STAT3, EC109 cells were treated with NaHS (500 µmol/L) for indicated time periods (6, 12, and 24 h). Data are presented as mean ± SEM (n=3). **P<0.01 versus control group. CTL, control group.
Inhibition of JAK2/STAT3 signaling pathway suppresses cell proliferation in the NaHS-treated EC109 cells
As shown in Figure 2, the treatment of cells with different concentrations (70, 80 and 90 µmol/L) of AG490, an inhibitor of JAK2/STAT3 signaling pathway, resulted in significant toxicity to the cells, in a dose-dependent manner. In addition, the treatment of EC109 cells with NaHS (500 µmol/L) for 24 h resulted in a significant increase in cell viability, which was inhibited by pre-treatment with AG490 (60 µmol/L) for 2 h. To further analyze the effect of NaHS and AG490 on EC109 cell proliferation, cells were treated with either with NaHS, AG490, or the combination of NaHS and AG490 for 24 and 48 h and the cell number was counted using the fully automatic inverted microscope. Treatment with AG490 resulted in a reduction in NaHS-induced cell proliferation. These findings indicate that inhibition of the JAK2/STAT3 signaling pathway suppressed the proliferative effect of NaHS.
Figure 2.
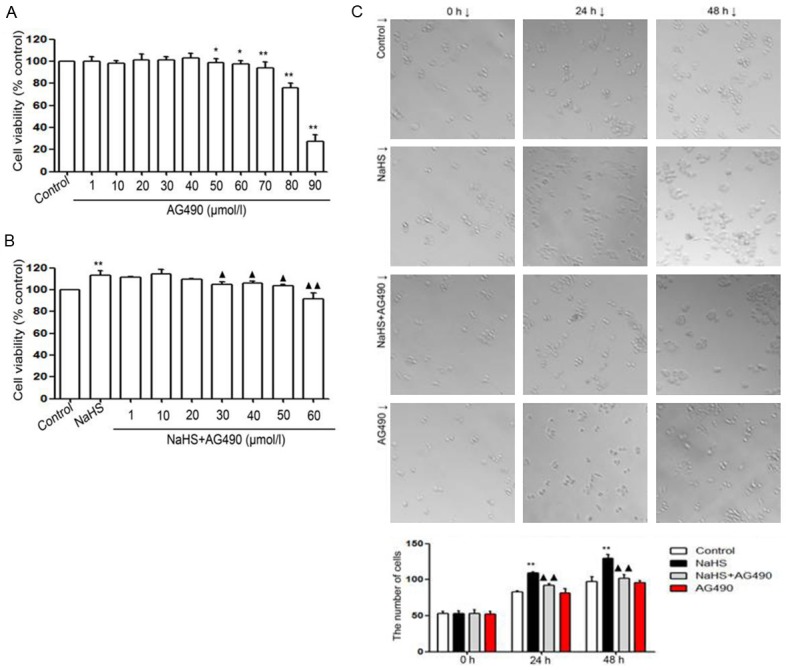
(A) AG490 reduces the proliferation of EC109 cells. EC109 cells were treated with different doses of AG490 (1, 10, 20, 30, 40, 50, 60, 70, 80, and 90 µmol/L) for 24 h. (B) AG490 reduces cell proliferation stimulated by NaHS in EC109 cells. EC109 cells were co-treated with NaHS (500 µmol/L) and different doses of AG490 (1, 10, 20, 30, 40, 50, and 60 µmol/L) for 24 h. (C) Effects of NaHS and AG490 on EC109 cell proliferation. EC109 cells were treated with NaHS, NaHS + AG490, and AG490 for 24 and 48 h. Cell number was counted directly under a microscope. AG490 reduced the NaHS-induced proliferation of EC109 cells (A and B). Data are expressed as mean ± SEM (n=3). **P<0.01 versus the control group. ++P<0.01 versus the NaHS group. C. **P<0.01 versus control group. ++P<0.01 versus NaHS group.
The effect of NaHS treatment on apoptosis regulators is suppressed by the inhibition of JAK2/STAT3 signaling pathway
To detect the effects of NaHS on cleaved caspase-3, caspase-9, caspase-12, Bax and Bcl-2 in EC109 cells, the cells were treated with NaHS (500 µmol/L) for the indicated time periods (6, 12 and 24 h). As shown in Figure 3, NaHS significantly increased the expression of Bcl-2 that reached its peak at 24 h, while the expression levels of caspase-3, caspase-9, caspase-12, and Bax were decreased. When EC109 cells were treated with NaHS (500 µmol/L) and AG490 (60 µmol/L) for 24 h, the expression of cleaved caspase-3, caspase-9, caspase-12 and Bax, that were suppressed by NaHS, were significantly enhanced, while the expression of Bcl-2, induced by NaHS, was significantly attenuated following AG490 treatment. Thus, NaHS downregulates the expression of cleaved caspase-3/9/12 and Bax, and upregulates the expression of Bcl-2 in EC109 cells. AG490 suppresses the NaHS-induced decrease in cleaved caspase-3/9/12 and Bax, and inhibits the increase in Bcl-2 expression following NaHS treatment.
Figure 3.
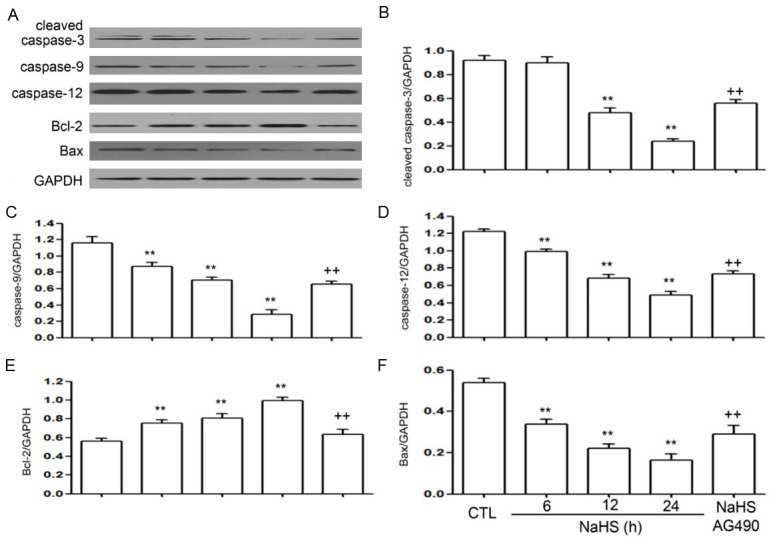
(A) NaHS downregulates the expression of cleaved caspase-3, caspase-9, caspase-12, and Bax and upregulates the expression of Bcl-2 in EC109 cells. EC109 cells were treated with NaHS (500 µmol/L) for 6, 12, and 24 h. AG490 upregulates the NaHS-induced decreased in the expression of cleaved caspase-3, caspase-9, caspase-12 and Bax (B-F) and inhibits the NaHS-induced increase in the expression of Bcl-2 in EC109 cells (E). EC109 cells were co-treated with NaHS (500 µmol/L) and AG490 (60 µmol/L) for 24 h. The expressions of cleaved caspase-3, caspase-9, caspase-12, Bax, and Bcl-2 were measured by western blot analysis followed by densitometric analysis. Data are presented as the mean ± SEM (n=3). **P<0.01 versus control group; ++P<0.01 versus NaHS group.
NaHS upregulates the level of MMP-2 and MMP-9 in EC109 cells
To observe the effects of NaHS on MMP-2 and MMP-9 expression in EC109 cells, the cells were exposed to NaHS (500 µmol/L) for the indicated time periods (6, 12, and 24 h). As shown in Figure 4, NaHS treatment significantly increased the expressions of MMP-2 and MMP-9, with maximum expression observed at 24 h. The cells treated with NaHS (500 µmol/L) and AG490 (60 µmol/L) for 24 h showed a significant decrease in NaHS-induced expression of MMP-2 and MMP-9.
Figure 4.
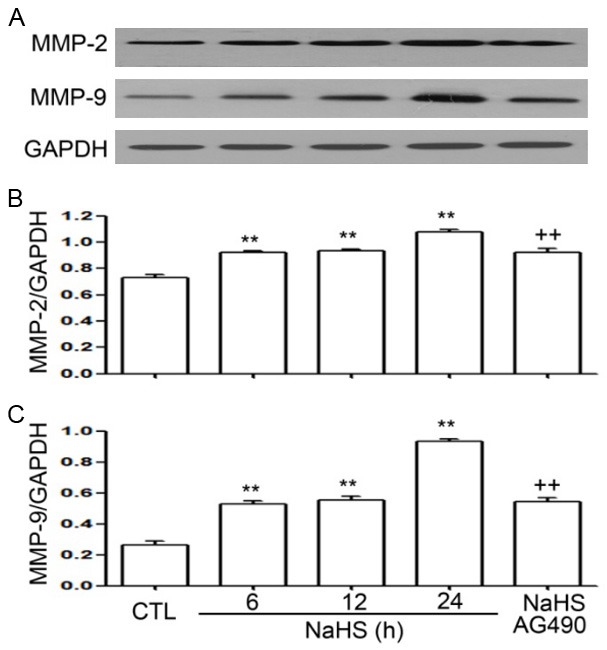
(A) NaHS promotes expression of MMP-2 and MMP-9 in EC109 cells. (B and C) EC109 cells were treated with NaHS (500 µmol/L) for 6, 12, and 24 h. AG490 suppresses the NaHS-induced increase in the expression of MMP-2 and MMP-9 in EC109 cells. EC109 cells were treated with NaHS (500 µmol/L) and AG490 (60 µmol/L) for 24 h. The protein levels of MMP-2 and MMP-9 were evaluated by western blot analysis. Data are shown as the mean ± SEM (n=3). **P<0.01 versus control group; ++P<0.01 versus NaHS group.
Wound healing was faster in cells treated with NaHS than in those treated with AG490
As shown in Figure 5, the wound closure was faster in EC109 cells cultured in the presence of NaHS for 24 h than in cells from the control group and those treated with NaHS and AG490. Furthermore, treatment with AG490 had an inhibitory effect on wound healing, indicating that inhibition of the JAK2/STAT3 signaling pathway may suppress cell migration induced by NaHS treatment.
Figure 5.

Treatment of EC109 cells with AG490 results in the suppression of cell migration-induced NaHS. NaHS-induced EC109 cells showed faster migration than cells from the control group and NaHS + AG490 group. Wound closure was monitored at 24 and 48 h, and a representative image from three independent experiments is shown (original magnification, 200×).
The compound AG490 suppresses NaHS-induced upregulation of VEGFR and VEGF in EC109 cells
As shown in Figure 6, the levels of VEGFR and VEGF were significantly increased in NaHS-treated EC109 cells, with peak values reported at 24 h. However, the increase in levels of VEGFR and VEGF was suppressed following AG490 treatment.
Figure 6.
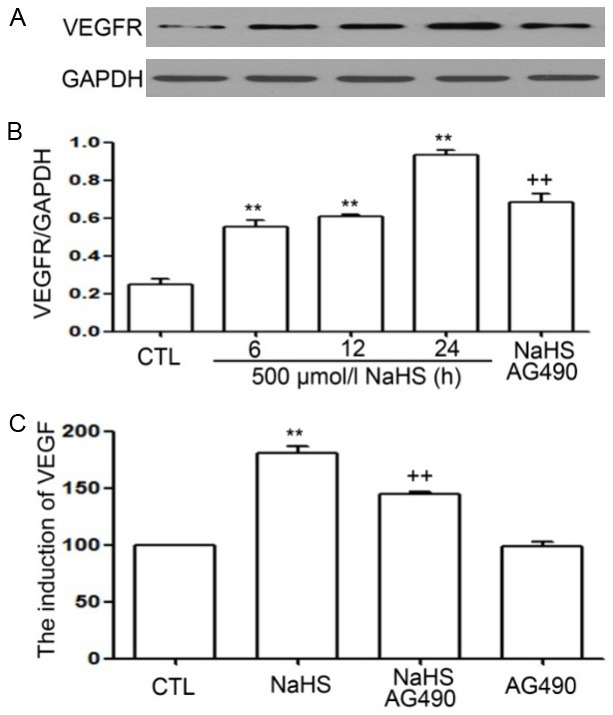
(A and C) AG490 suppresses NaHS-induced increased expression of level of VEGFR and VEGF in EC109 cells. The cells were either treated with 60 µmol/L AG490 for 30 minutes not treated with AG490, followed by incubation with 500 µmol/L NaHS for 6, 12, and 24 h. Expression of VEGFR was measured by western blot analysis. ELISA was used to determine the level of VEGF in cell supernatants. Data are shown as the mean ± SEM (n=3). **P<0.01 versus control group. ++P<0.01 versus NaHS treatment group.
Discussion
In the present study, we demonstrated that pre-treatment of EC109 cells with exogenous H2S results in proliferation, anti-apoptosis, migration, and angiogenesis through activation of the JAK2/STAT3 signaling pathway. As observed in our previous study, H2S has a positive effect on the proliferation of EC109 cells, as evident from the increase in the cell viability and number. Furthermore, the treatment of cells with NaHS resulted in a decrease in cell apoptosis, probably owing to the reduction in the level of caspase-3, caspase-9, caspase-12, and Bax, and an increase in the level of Bcl-2. NaHS-treatment stimulated cell migration and induced the expression of MMP-2 and MMP-9. On the other hand, treatment of cells with AG490 resulted in suppression of the effects induced by NaHS treatment.
The mechanism underlying the effects of H2S is complicated and involves several signaling molecules and pathways. Evidence suggests that H2S acts as a neuroprotective agent in the brain at physiological concentrations and protects neurons against oxidative stress, apoptosis, and endocytoplasmic reticulum (ER) stress damage induced by multiple drugs [23-25]. On the other hand, H2S has been shown to have tumor-promoting ability as it enhances cancer cell growth, anti-apoptosis and migration. These effects are mediated via the activation of signaling pathways such as NF-κB, p38-MAPK/ERK1/2-COX-2 and HSP90 pathways [20-22]. Additionally, the JAK2/STAT3 signaling pathway is involved in various biological processes and contributes to cell proliferation, differentiation, growth, and apoptotic and anti-apoptotic effects [26-28]. JAK2 functions as a typical kinase to mediate the phosphorylation of STAT3 and plays a crucial role in the regulation the JAK2/STAT3 signaling pathway in multiple cancer cell lines [29]. However, the roles of JAK2/STAT3 signaling pathway in NaHS-treated EC109 cells have not been studied. In order to determine the potential mechanism, we investigated the role of NaHS in JAK2/STAT3 signaling pathway activation in EC109 cells. Our data showed that the treatment of cells with NaHS markedly increased the expression levels of p-JAK2 and p-STAT3 in a time-dependent manner, suggesting that NaHS may activate the JAK2/STAT3 signaling pathway in EC109 cells. We also examined the role of the JAK2/STAT3 signaling pathway in NaHS-induced proliferation and apoptosis. The exposure of cells to NaHS for 24 h significantly decreased cell apoptosis through the upregulation of Bcl-2 and downregulation of caspase-3, caspase-9, caspase-12, and Bax, which are pro-apoptotic proteins of the Bcl-2 family. Moreover, the effects observed following NaHS treatment were suppressed in the presence of AG490. Our findings are in line with those previously reported [30].
Many studies have demonstrated that the invasion and metastasis of cancer cells is facilitated through an increase in the expression of MMPs viaremodeling of the extracellular matrix (ECM) [31]. MMP-2 and MMP-9 are two important enzymes known for their ability to degrade type IV collagen, one of the crucial elements of ECM [32]. Furthermore, MMP-2 and MMP-9 are upregulated in several types of cancer cells and this upregulation is correlated with metastasis and migration [33-36]. Our data show that NaHS stimulated the expression of MMP-2 and MMP-9 in EC109 cells, leading to their faster migration than that observed in the control group. All of these effects were suppressed following AG490 treatment, indicating that H2S promotes cancer cell invasion and migration through activation of the JAK2/STAT3 signaling pathway.
Hydrogen sulfide stimulates the expression of VEGF, which is important in the process of wound healing and tumor angiogenesis and may contribute to malignant behavior such as persistent proliferation and metastasis [37]. As shown in previous studies, NaHS upregulated the expression of VEGFR and VEGF in EC109 cells and this effect was suppressed by AG490. Hence, we hypothesize that the overexpression of JAK2/STAT3 in ESCC may be associated with upregulation of VEGFR and VEGF, resulting in tumor angiogenesis.
In conclusion, our data revealed that H2S enhances cell proliferation and migration, and exhibits anti-apoptotic and angiogenic effects in EC109 cells. These effects may be mediated through the phosphorylation of JAK2 and STAT3, which results in the overexpression of MMP-2, MMP-9, Bcl-2, VEGFR, and VEGF, and the downregulation of caspase-3, caspase-9, caspase-12, and Bax. These results provide new insight into cancer physiology and highlight a new drug target for therapy. The detailed mechanisms underlying H2S effects in EC109 esophageal cells remain unclear and need to be studied.
Acknowledgements
This study was supported by the Science and Technology Planning Project of Guangdong Province (2017ZC0038 and 2017ZC0474), Medical Scientific Research Foundation of Guangdong Province (A2015287), and the Medical Scientifc Research Foundation of Guangdong Province (2015A030313690, A2014246, A2014217 and A2016297).
Disclosure of conflict of interest
None.
References
- 1.Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381:400–412. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 2.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 4.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 5.Delektorskaya VV, Chemeris GY, Zavalishina LE, Ryazantseva AA, Grigorchuk AY, Kononets PV, Davydov MI. Squamous cell carcinoma of the esophagus: evaluation of the status of epidermal growth factor receptors (EGFR and HER-2) by immunohistochemistry and in situ hybridization. Bull Exp Biol Med. 2010;149:615–620. doi: 10.1007/s10517-010-1007-z. [DOI] [PubMed] [Google Scholar]
- 6.Langer R, Ott K, Specht K, Becker K, Lordick F, Burian M, Herrmann K, Schrattenholz A, Cahill MA, Schwaiger M, Hofler H, Wester HJ. Protein expression profiling in esophageal adenocarcinoma patients indicates association of heat-shock protein 27 expression and chemotherapy response. Clin Cancer Res. 2008;14:8279–8287. doi: 10.1158/1078-0432.CCR-08-0679. [DOI] [PubMed] [Google Scholar]
- 7.Madani K, Zhao R, Lim HJ, Casson AG. Prognostic value of p53 mutations in oesophageal adenocarcinoma: final results of a 15-year prospective study. Eur J Cardiothorac Surg. 2010;37:1427–1432. doi: 10.1016/j.ejcts.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 8.Pencik J, Pham HT, Schmoellerl J, Javaheri T, Schlederer M, Culig Z, Merkel O, Moriggl R, Grebien F, Kenner L. JAK-STAT signaling in cancer: from cytokines to non-coding genome. Cytokine. 2016;87:26–36. doi: 10.1016/j.cyto.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ward AC, Touw I, Yoshimura A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood. 2000;95:19–29. [PubMed] [Google Scholar]
- 10.Yang N, Han F, Cui H, Huang J, Wang T, Zhou Y, Zhou J. Matrine suppresses proliferation and induces apoptosis in human cholangiocarcinoma cells through suppression of JAK2/STAT3 signaling. Pharmacol Rep. 2015;67:388–393. doi: 10.1016/j.pharep.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Qiu W, Zhang G, Xu S, Gao Q, Yang Z. MicroRNA-204 targets JAK2 in breast cancer and induces cell apoptosis through the STAT3/BCl-2/survivin pathway. Int J Clin Exp Pathol. 2015;8:5017–5025. [PMC free article] [PubMed] [Google Scholar]
- 12.Hu L, Wu H, Li B, Song D, Yang G, Chen G, Xie B, Xu Z, Zhang Y, Yu D, Hou J, Xiao W, Sun X, Chang G, Zhang Y, Gao L, Dai B, Tao Y, Shi J, Zhu W. Dihydrocelastrol inhibits multiple myeloma cell proliferation and promotes apoptosis through ERK1/2 and IL-6/STAT3 pathways in vitro and in vivo. Acta Biochim Biophys Sin (Shanghai) 2017;49:420–427. doi: 10.1093/abbs/gmx021. [DOI] [PubMed] [Google Scholar]
- 13.Wu J, Guo J, Cao Q, Wang Y, Chen J, Wang Z, Yuan Z. Autophagy impacts on oxaliplatin-induced hepatocarcinoma apoptosis via the IL-17/IL-17R-JAK2/STAT3 signaling pathway. Oncol Lett. 2017;13:770–776. doi: 10.3892/ol.2016.5476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang R. Two’s company, three’s a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;16:1792–1798. doi: 10.1096/fj.02-0211hyp. [DOI] [PubMed] [Google Scholar]
- 15.Cai WJ, Wang MJ, Ju LH, Wang C, Zhu YC. Hydrogen sulfide induces human colon cancer cell proliferation: role of Akt, ERK and p21. Cell Biol Int. 2010;34:565–572. doi: 10.1042/CBI20090368. [DOI] [PubMed] [Google Scholar]
- 16.Cao Q, Zhang L, Yang G, Xu C, Wang R. Butyrate-stimulated H2S production in colon cancer cells. Antioxid Redox Signal. 2010;12:1101–1109. doi: 10.1089/ars.2009.2915. [DOI] [PubMed] [Google Scholar]
- 17.Pupo E, Pla AF, Avanzato D, Moccia F, Cruz JE, Tanzi F, Merlino A, Mancardi D, Munaron L. Hydrogen sulfide promotes calcium signals and migration in tumor-derived endothelial cells. Free Radic Biol Med. 2011;51:1765–1773. doi: 10.1016/j.freeradbiomed.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 18.Rose P, Moore PK, Ming SH, Nam OC, Armstrong JS, Whiteman M. Hydrogen sulfide protects colon cancer cells from chemopreventative agent beta-phenylethyl isothiocyanate induced apoptosis. World J Gastroenterol. 2005;11:3990–3997. doi: 10.3748/wjg.v11.i26.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Szabo C, Coletta C, Chao C, Modis K, Szczesny B, Papapetropoulos A, Hellmich MR. Tumor-derived hydrogen sulfide, produced by cystathionine-beta-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc Natl Acad Sci U S A. 2013;110:12474–12479. doi: 10.1073/pnas.1306241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhen Y, Pan W, Hu F, Wu H, Feng J, Zhang Y, Chen J. Exogenous hydrogen sulfide exerts proliferation/anti-apoptosis/angiogenesis/migration effects via amplifying the activation of NF-kappaB pathway in PLC/PRF/5 hepatoma cells. Int J Oncol. 2015;46:2194–2204. doi: 10.3892/ijo.2015.2914. [DOI] [PubMed] [Google Scholar]
- 21.Zhen Y, Zhang W, Liu C, He J, Lu Y, Guo R, Feng J, Zhang Y, Chen J. Exogenous hydrogen sulfide promotes C6 glioma cell growth through activation of the p38 MAPK/ERK1/2-COX-2 pathways. Oncol Rep. 2015;34:2413–2422. doi: 10.3892/or.2015.4248. [DOI] [PubMed] [Google Scholar]
- 22.Lei Y, Zhen Y, Zhang W, Sun X, Lin X, Feng J, Luo H, Chen Z, Su C, Zeng B, Chen J. Exogenous hydrogen sulfide exerts proliferation, anti-apoptosis, angiopoiesis and migration effects via activating HSP90 pathway in EC109 cells. Oncol Rep. 2016;35:3714–3720. doi: 10.3892/or.2016.4734. [DOI] [PubMed] [Google Scholar]
- 23.Sakamoto K, Suzuki Y, Kurauchi Y, Mori A, Nakahara T, Ishii K. Hydrogen sulfide attenuates NMDA-induced neuronal injury via its anti-oxidative activity in the rat retina. Exp Eye Res. 2014;120:90–96. doi: 10.1016/j.exer.2014.01.008. [DOI] [PubMed] [Google Scholar]
- 24.Yin WL, He JQ, Hu B, Jiang ZS, Tang XQ. Hydrogen sulfide inhibits MPP(+)-induced apoptosis in PC12 cells. Life Sci. 2009;85:269–275. doi: 10.1016/j.lfs.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 25.Jia J, Xiao Y, Wang W, Qing L, Xu Y, Song H, Zhen X, Ao G, Alkayed NJ, Cheng J. Differential mechanisms underlying neuroprotection of hydrogen sulfide donors against oxidative stress. Neurochem Int. 2013;62:1072–1078. doi: 10.1016/j.neuint.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurosaka M, Machida S. Interleukin-6-induced satellite cell proliferation is regulated by induction of the JAK2/STAT3 signalling pathway through cyclin D1 targeting. Cell Prolif. 2013;46:365–373. doi: 10.1111/cpr.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Cruijsen H, Oosterhoff D, Lindenberg JJ, Lougheed SM, Fehres C, Weijers K, van Boerdonk R, Giaccone G, Scheper RJ, Hoekman K, de Gruijl TD. Glioblastoma-induced inhibition of Langerhans cell differentiation from CD34(+) precursors is mediated by IL-6 but unaffected by JAK2/STAT3 inhibition. Immunotherapy. 2011;3:1051–1061. doi: 10.2217/imt.11.107. [DOI] [PubMed] [Google Scholar]
- 28.Zhao J, Li G, Zhang Y, Su X, Hang C. The potential role of JAK2/STAT3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhEPO) after experimental traumatic brain injury of rats. Cytokine. 2011;56:343–350. doi: 10.1016/j.cyto.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 29.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou W, Bi X, Gao G, Sun L. miRNA-133b and miRNA-135a induce apoptosis via the JAK2/STAT3 signaling pathway in human renal carcinoma cells. Biomed Pharmacother. 2016;84:722–729. doi: 10.1016/j.biopha.2016.09.074. [DOI] [PubMed] [Google Scholar]
- 31.Demers M, Couillard J, Belanger S, St-Pierre Y. New roles for matrix metalloproteinases in metastasis. Crit Rev Immunol. 2005;25:493–523. doi: 10.1615/critrevimmunol.v25.i6.30. [DOI] [PubMed] [Google Scholar]
- 32.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–174. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 33.Jones JL, Shaw JA, Pringle JH, Walker RA. Primary breast myoepithelial cells exert an invasion-suppressor effect on breast cancer cells via paracrine down-regulation of MMP expression in fibroblasts and tumour cells. J Pathol. 2003;201:562–572. doi: 10.1002/path.1483. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Li F, Zhao K, Yao J, Cheng Y, Zhao L, Li Z, Lu N, Guo Q. LFG-500 inhibits the invasion of cancer cells via down-regulation of PI3K/AKT/NF-kappaB signaling pathway. PLoS One. 2014;9:e91332. doi: 10.1371/journal.pone.0091332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Puzovic V, Brcic I, Ranogajec I, Jakic-Razumovic J. Prognostic values of ETS-1, MMP-2 and MMP-9 expression and co-expression in breast cancer patients. Neoplasma. 2014;61:439–446. doi: 10.4149/neo_2014_054. [DOI] [PubMed] [Google Scholar]
- 36.Ruan M, Zhang Z, Li S, Yan M, Liu S, Yang W, Wang L, Zhang C. Activation of Toll-like receptor-9 promotes cellular migration via up-regulating MMP-2 expression in oral squamous cell carcinoma. PLoS One. 2014;9:e92748. doi: 10.1371/journal.pone.0092748. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37.Kohn C, Dubrovska G, Huang Y, Gollasch M. Hydrogen sulfide: potent regulator of vascular tone and stimulator of angiogenesis. Int J Biomed Sci. 2012;8:81–86. [PMC free article] [PubMed] [Google Scholar]