

Abstract
This study aimed to investigate the effect of miR-26b expression on neurites outgrowth and cells apoptosis in PC12 cellular model of Alzheimer’s disease (AD). PC12 cells were stimulated by nerve growth factor and insulted by Aβ1-42 to establish PC12 cellular AD model. Methyl thiazolyl tetrazolium (MTT) assay was then used to detect cells viability. Blank mimic, miR-26b mimic, blank inhibitor and miR-26b inhibitor plasmids were transferred into PC12 cellular AD models as NC1-mimic, miR-26b mimic, NC2-inhibitor and miR-26b inhibitor groups respectively. mRNA level, protein level, total neurite outgrowth and cells apoptosis were determined by qPCR, western blot, microscope and Hoechst/PI, respectively. MTT reduction rate was decreased in Aβ1-42 insult group compared to control group (P<0.001). After plasmids transfection, the total neuritis growth was found to be reduced in miR-26b mimic group compared with NC1-mimic group (P<0.05) while was elevated in miR-26b inhibitor group compared with NC2-inhibitor group (P<0.01). As to cells apoptosis, the percentage of apoptosis cells was increased in miR-26b mimic group than NC1-mimic group (P<0.05), and was decreased in miR-26b inhibitor group than NC2-inhibitor group (P<0.05). In addition, neprilysin (NEP) protein and mRNA expressions were decreased in miR-26b mimic group than NC1-mimic group and was increased in miR-26b inhibitor group than NC2-inhibitor group. However, protein or mRNA expression of EIF2S1 and αTTP was not affected by miR-26b. In conclusion, miR-26b inhibits neurite outgrowth, induces cells apoptosis and downregulates NEP expression in PC12 cellular AD model.
Keywords: miR-26b, PC12, Alzheimer’s disease, neurite outgrowth, apoptosis
Introduction
Alzheimer’s disease (AD) is a progressive and irreversible neurological disorder with marked atrophy of cerebral cortex and loss of cortical or subcortical neurons, which is characterized pathologically by deposition of amyloid beta-protein (Aβ) in brain to form senile plaques (SP), and numerous neurofibrillary tangles (NFT) derived from filaments of microtubule associated highly phosphorylated tau proteins. In addition, neuronal synaptic dysfunction and vertebral nerve cell loss is also a characteristic of AD [1,2]. Its clinical manifestations include progressive memory impairment, mental decline, altered behavior and personality and even death [3,4]. However, it is not entirely clear how these typical characteristics are formed in AD.
miRNA, a class of small endogenous noncoding RNAs, regulates gene expression at the transcriptional level or post transcriptional level. They are widely distributed in the central nervous system, also play an important role in neurodevelopment, differentiation and maturation [5]. Several studies disclose that some pivotal AD proteins are served as targets of miRNAs, whose dysregulated expressions are correlated with the development and progress of various neurodegenerative diseases [6-9]. While some previous studies have suggested miR-26b upregulation, observed in AD, perturbs signaling pathways associated with neuronal cell cycle, and thereby causes pleiotropic phenotypes associated with the disease [10]. It indicated that miR-26b may be involved in neuronal aging and in the pathogenesis of neurodegenerative disorders, but their precise etiology in AD development and progression are poorly understood. This study aimed to investigate the role of miR-26b on neurite outgrowth and cells apoptosis in PC12 cellular model of AD.
Materials and methods
Cells culture
Rat pheochromocytoma cells (PC12 cells) were obtained from Shanghai Institutes for Biological Science (Shanghai, China). After cells resuscitation, these cells were cultured in Dulbecco Modified Eagle medium (DMEM) (Gibco, USA) with 10% (v/v) horse serum (Gibco, USA), 5% (v/v) fetal bovine serum (FBS) (Gibco, USA), 100 units/ml penicillin, and 100 μg/ml streptomycin (Corning, Lowell, USA). The PC12 cells were incubated in a humidified atmosphere (95% air and 5% CO2) at 37°C. The medium was replaced every 2 days.
Preparation of oligomerized Aβ1-42
Aβ1-42 was purchased from Sigma company (USA) and was dissolved in dimethyl sulfoxide (DMSO) at a concentration of 1 mM. Prior to the treatment, peptides were pre-incubated at 37°C for 7 days to promote aggregation and then diluted in medium to desired concentrations as described previously [11]. Soluble oligomerized Aβ1-42 peptides (equivalent to 1 mM peptides) were added to cells to induce damaging effects.
Neurite differentiation and Aβ1-42 insult
For induction of neuronal differentiation, the PC12 cells were treated with 20 ng/mL nerve growth factor (NGF) (Sigma, UA) for 72 hours at 37°C with 95% air and 5% CO2. The medium was changed every 2 days, and fresh NGF was added each time.
At the day 4, 1 mM of Aβ1-42 soluble oligomerized Aβ1-42 peptides were added to NGF inducted PC12 cells and these cells were incubated for 24 h to establish cell of AD model.
Methyl thiazolyl tetrazolium (MTT) assay
After PC12 cells were induced by NGF and insulted by Aβ1-42, cells viability was assessed by MTT reagent (Sigma, USA). Firstly, 100 mg of MTT was dissolved in 20 mL PBS to make a 5 mg/mL solution. Next, 10 μl of MTT solution was added to each well, which contained 100 μl culture medium, and incubated at 37°C for 4 h. Then the culture medium in the wells was sucked, and 100 μl of DMSO (Dimethyl sulfoxide) was added to each well, the solution was dissolved for 10 min. Finally, the plates were analyzed using a microplate reader (Molecular Devices, USA) at 490 nm. All processing groups were triplicated.
miR-26b transfection and subsequent assays
The PC12 cellular PC12 cellular AD model were divided into four groups and then transfected with blank mimic, miR-26b mimic, blank inhibitor, miR-26b inhibitor plasmids as NC1-mimic group, miR-26b-mimic group, NC2-inhibitor group and miR-26b-inhibitor group. All processing groups were triplicated.
After 24 h plasmids transfection, the expression of miR-26b in each group was detected by qRT-PCR. Then, cells morphology was observed and total neurite outgrowth was quantified by inversion fluorescence microscope. Cells apoptosis was detected by Hoechst/propidium (PI) method, and the expressions of apoptosis-related proteins were determined by western blot. In addition, mRNA and protein expressions of miR-26b predicted target genes were detected by qRT-PCR and western blot respectively.
qRT-PCR assay
miRNAs and mRNA expression were evaluated by qRT-PCR. The total RNA from cells was extracted using TRIZol Reagents (Invitrogen, USA), according to the manufacturer’s instructions. The quantitation of RNA samples was performed by OD 260. 1 μg of total RNA from each sample were used for cDNA synthesis with transcription kit (TOYOBO, Japan). And then cDNA products were subjected to qPCR with SYBR Green kit (KAPA, USA). The PCR amplification was performed as follows: 95°C for 5 min, followed by 40 cycles of 95°C for 5 s, 61°C for 30 s. U6 or GAPDH was used as reference gene. All experiments were triplicated. The qRT-PCR results were calculated by the 2-ΔΔCt method. The primers information was presented in Table 1.
Table 1.
Primers information
| Gene name | Sequence (5’→3’) |
|---|---|
| MiR-26b | F: CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACCTATCC |
| R: ACACTCCAGCTGGGTTCAAGTAATTCAGGATA | |
| U6 | F: CTCGCTTCGGCAGCACA |
| R: AACGCTTCACGAATTTGCGT | |
| GAPDH | F: CCTTCTCTTGTGACAAAGTGGACA |
| R: CATTTGATGTTAGCGGGATCTCG | |
| NEP | F: GGAAGAAGTGGTTGTTTATGCTCC |
| R: AGCATTTCTGGACTCCTTGTAGTT | |
| αTTP | F: AGAATGTCCAGAACTAAGTGCAGA |
| R: CTCCTGTACAATGAGCTCTGATGT | |
| EIF2S1 | F: GCTATGGTTACGAAGGCATTGATG |
| R: GCTCCATCTGAACATTGAACACTC |
Total neurite outgrowth measurement
After cells morphology was observed by inverted microscope (Nikon, Japan), Imaging Software Presage (Advanced imaging Concepts, lnc., USA) was used to quantify the total neurite outgrowth per cell which was defined as total length of neurite outgrowth of cells divided by number of cells included.
Hoechst/PI assay
Hoechst 33342 (λex350 nm, λem461 nm) and PI (λex535 nm, λem617 nm) were added to the culture medium at final concentrations of 8 and 1.5 M, respectively, and cultured at 37°C for 30 min. Images were collected by inversion fluorescence microscope (Nikon, Japan), the damaged cells and total cells were calculated under ×200 magnification, and the percentage of damaged cell was calculated. All experiments were triplicated.
Western blot assay
Total proteins were extracted from cells of each group with 1 ml ice-cold RIPA buffer (Thermo Fisher Scientific, USA), then total protein concentration in each sample was measured using the bicinchoninic acid (BCA) kit (Pierce Biotechnology, Rockford, IL, USA) and compared with the standard curve. Next, 20 μg total protein samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed, and separated proteins were electrophoretically transferred to polyvinylidene fluoride membranes (PVDF) membrane (Millipore, Bedford, USA). After blocked with 5% skim milk for 2 h, membranes were incubated with the corresponding primary antibody overnight at 4°C. Then, membranes were incubated with the appropriate HRP-conjugated secondary antibody for 1 h at room temperature. The bands were visualized using an enhanced chemiluminescence (ECL) kit (Millipore, Bedford, USA) followed by exposure to X-ray film. All experiments were triplicated. Specific antibody was used to detect the corresponding protein. Antibodies used for Western blot are listed in Table 2.
Table 2.
Antibodies information
| Antibody name | Company | Country | Dilution ratio |
|---|---|---|---|
| Primary antibody | |||
| Rabbit polyclonal Anti-Caspase-3 antibody | Abcam | USA | 1:2000 |
| Rabbit monoclonal Anti-cleaved Caspase-3 antibody | Abcam | USA | 1:2000 |
| Rabbit monoclonal Anti-P38 antibody | Abcam | USA | 1:2000 |
| Rabbit polyclonal Anti-P38 (phosphoT180+Y182) antibody | Abcam | USA | 1:2000 |
| Rabbit monoclonal Anti-GAPDH antibody | Abcam | USA | 1:2000 |
| Rabbit polyclonal Anti-Nephrin antibody | Abcam | USA | 1:2000 |
| Rabbit monoclonal Anti-EIF2S1 antibody | Abcam | USA | 1:2000 |
| Rabbit polyclonal Anti-TTPA antibody | Abcam | USA | 1:2000 |
| Second antibody | |||
| Goat Anti-Rabbit IgG H&L | Abcam | USA | 1:2000 |
miR-26b target genes assay
The miR-26b target gene was predicted by these following analysis: (1) Target mRNAs of miR-26b were firstly predicted by miRwalk 2.0 with 6 or above positive results by 12 methods (miRwalk, Microt4, miRanda, mirbridge, miRDB, miRMap, miRNAMap, pictar2, PITA, RNA22, RNAhybrid, Targetsacn) (zmf.umm.uni-Heidelberg.de/apps/zmf/mirwalk2) [12]. (2) Genes correlated with AD pathology or risk were analyzed by DisGene (www.disgenet.org) [13]. (3) Potential targeted genes were then analyzed by combining of miR-26b target genes and AD related genes. Thus, top three miR-26b gene (neprilysin (NEP), α-tocopherol transfer protein (αTTP) and in eukaryotic translation initiation factor 2 subunit 1 (EIF2S1) were selected to be determined by qRT-PCR and western blot.
Statistics
Statistical analysis was performed using SPSS software 22.0 (IBM, USA) and GrapgPad prism V5.01 (IBM, USA). Data was presented as mean ± standard. Comparison between two groups was determined by t test. P<0.05 was considered significant.
Results
Cells viability after Aβ1-42 insult
In order to detect the establishment of AD model, we used MTT assay to determine the reduction rate of NGF inducted PC12 cells after Aβ1-42 insult. We found that MTT reduction rate was decreased in Aβ1-42 insult group compared to control group (P<0.001, Figure 1).
Figure 1.
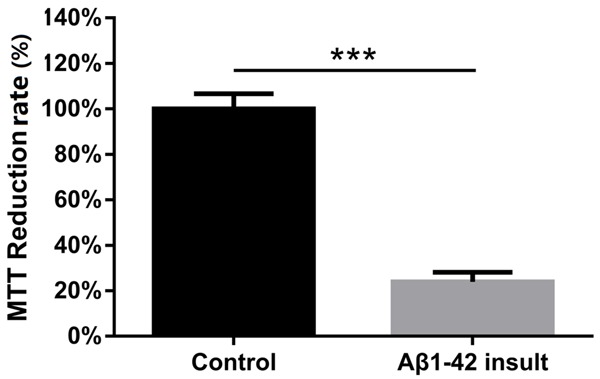
Effect of Aβ1-42 insult on PC12 cellular AD model. Comparison between two groups was determined by t test. P<0.05 was considered significant. *P<0.05, **P<0.01, ***P<0.001.
miR-26b expression after plasmids transfection
At 24 h following plasmids transfection, the result of qRT-PCR showed that, compared to NC1-mimic group, miR-26b-mimics induced an upregulation of miR-26b level in miR-26b-mimic group (P<0.001, Figure 2), and compared with NC2-inhibitor group, the miR-26b expression in miR-26b-inhibitor group was markedly decreased (P<0.01, Figure 2).
Figure 2.
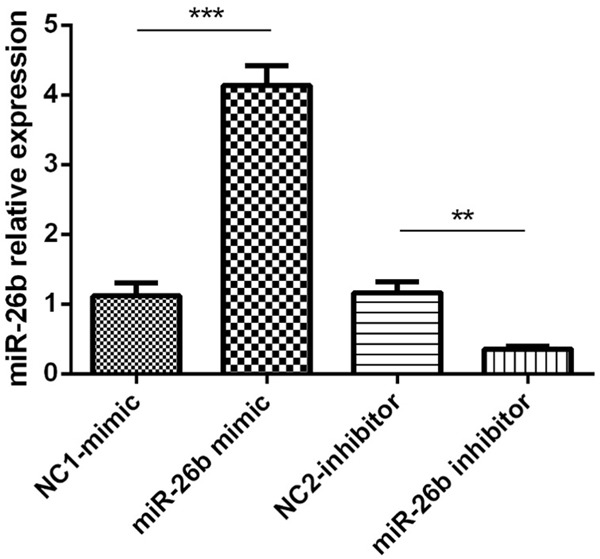
Effect of plasmids transfection on miR-26b expression in PC12 cellular AD model. Comparison between two groups was determined by t test. P<0.05 was considered significant. *P<0.05, **P<0.01, ***P<0.001.
Effect of miR-26b on total neurite outgrowth
The effect of miR-26b on total neurite outgrowth was presented in Figure 3A, after transfection for 24 h, The mean length of total neurite outgrowth was markedly shorter in miR-26b-mimic group compared with the NC1-mimic group (P<0.05, Figure 3B) and was longer in miR-26b-inhibitor group compared with NC2-inhibitor group (P<0.01, Figure 3B).
Figure 3.
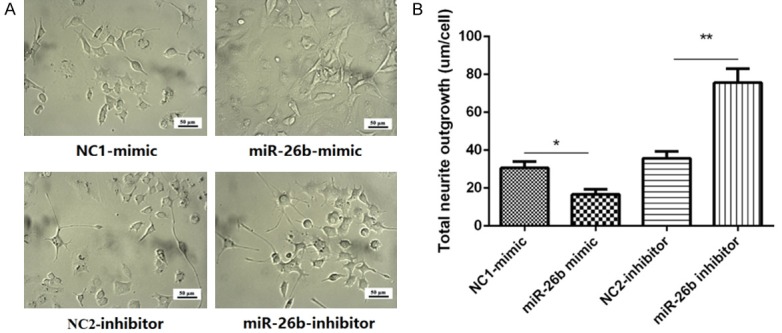
Effect of plasmids transfection on total neurite outgrowth in PC12 cellular AD model. A. The morphology of PC12 cellular AD model after transfected with miR-26b mimic or inhibitor. B. The mean length of neurite outgrowth in PC12 cellular AD model. Comparison between two groups was determined by t test. P<0.05 was considered significant. *P<0.05, **P<0.01, ***P<0.001.
miR-26b promoted cells apoptosis
Cells apoptosis were presented in Figure 4A, 24 h following plasmids transfection, cells apoptosis percentage in miR-26b-mimic group was obviously elevated than NC1-mimic group (P<0.05, Figure 4B), besides, cells apoptosis percentage was decreased in miR-26b-inhibitor group compared with NC2-inhibitor group (P<0.05, Figure 4B).
Figure 4.
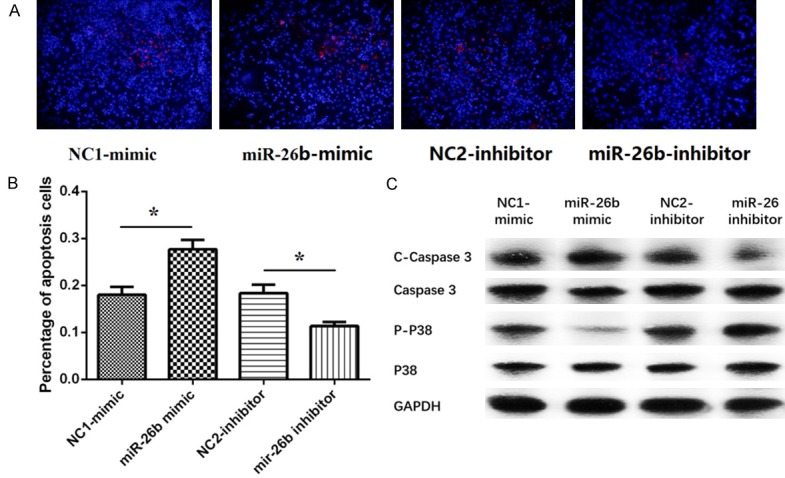
Effect of plasmids transfection on apoptosis in PC12 cellular AD model. A. The cells apoptosis of PC12 cellular AD model under fluorescence microscope. B. The cells apoptosis percentage of PC12 cellular AD model in miR-26b-mimic group and miR-26b-inhibitor group. C. The expression of C-Caspases 3, Caspases 3, P-P38 and P38 activity in miR-26b-mimic group and miR-26b-inhibitor group. Comparison between two groups was determined by t test. P<0.05 was considered significant. *P<0.05, **P<0.01, ***P<0.001.
Meanwhile, compared with NC1-mimic group, the cleaved caspase-3 (C-Caspase 3) levels was elevated and the Phosphorylated P38 (P-P38) was reduced in miR-26b-mimic group (P>0.05, Figure 4C). While, the C-Caspase 3 levels was diminished and the P-P38 was enhanced in miR-26b-mimic group compared with NC2-inhibitor group (P>0.05, Figure 4C). Furthermore, the levels of total caspase 3 and P38 protein in miR-26b-mimic group and miR-26b-inhibitor group were not different from those in the control groups (All P>0.05, Figure 4C). This indicates that miR-26b promoted cells apoptosis in PC12 cellular AD model.
Expression of candidate target genes
Compared with NC1-mimic group, the mRNA and protein expression of NEP gene was reduced in miR-26b-mimic group (P<0.05, Figure 5A, 5D), and their expressions were increased in miR-26b-inhibitor group (P<0.01, Figure 5A, 5D). However, no difference of the mRNA and protein expression of EIF2S1 and αTTP was found (all P>0.05, Figure 5B-D).
Figure 5.
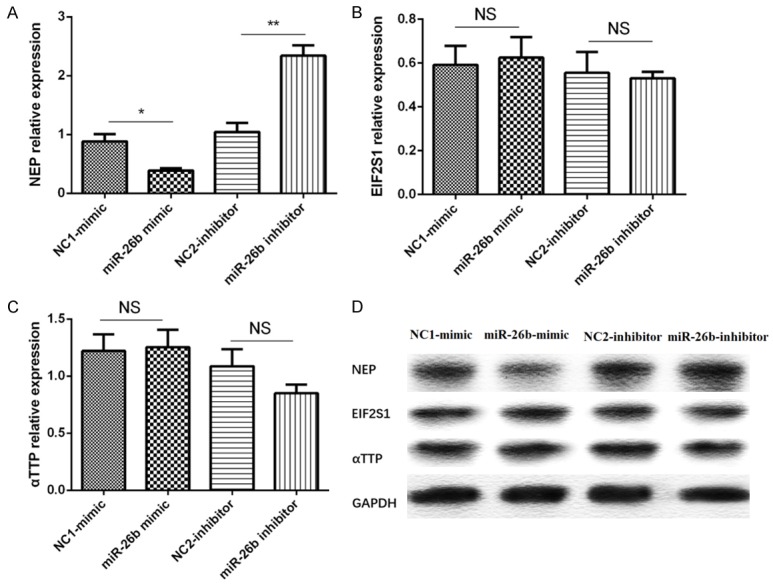
Effect of miR-26b levels on NEP, EIF2S1, αTTP genes. A. The expression of NEP mRNA in different groups. B. The expression of EIF2S1 mRNA in different groups. C. The expression of αTTP mRNA in different groups. D. The expression of NEP, EIF2S1, αTTP protein in different groups. Comparison between two groups was determined by t test. P<0.05 was considered significant. *P<0.05, **P<0.01, ***P<0.001.
Discussion
In this study, we found that (1) miR-26b inhibited the total neurite outgrowth and promoted cells apoptosis in PC12 cellular AD model. (2) miR-26b reduced the NEP expression but not αTTP or EIF2S1 expression in PC12 cellular AD model.
miRNAs are involved in post-transcriptional regulation of gene expression in multicellular organisms by affecting both the stability and translation of mRNAs [14]. A deal of miRNAs has been proposed to be involved in pathogenesis of AD and other neurological disorders [10,15-17]. A research shows that overexpression of miR-125b in primary neurons is associated with tau hyperphosphorylation and cognitive deficits [18]. In AD mouse models, the miR-15 family regulates the level of tau phosphorylation by targeting Erk1 kinase [19]. Moreover, miR-98 increases Aβ formation and tau phosphorylation by inhibiting the translation of IGF-1 in HEK293 cells, which leads to the occurrence of AD [20]. These indicate miRNAs play crucial functions in AD development and progression.
miR-26b, a member of the miR-26 family, is located on human chromosome 2q35, which is abundantly expressed in nerve [21-23]. Several studies illustrate that miR-26b expression is dysregulated in AD samples. A research exhibits that miR-26b expression is upregulated in AD human postmortem brains compared with healthy brains [18]. Another investigation indicates that miR-26b is increased in blood of 48 AD patients compared with 22 normal control [17]. Furthermore, an animal experiment shows that miR-26b level is elevated in cholesterol fed rabbit model of AD compared to the control samples [24]. These suggest that miR-26b expression correlates with the risk of AD. An in vitro experiment also confirms that miR-26b activates cell cycle entry (CCE), tau-phosphorylation, and promotes cells apoptosis in rat primary post mitotic neurons, which further indicates miR-26b might be involved in AD pathogenesis [10]. Consistent with prior studies, we observed that miR-26b mimic reduced total neurite outgrowth and promoted cells apoptosis in PC12 cellular AD model, while the result of miR-26b inhibitor group was opposite. This indicated miR-26b was involved in the pathology of AD by regulating neurite outgrowth and cells apoptosis. And the possible explanations might be as follows: (1) miR-26b induces CCE and promotes cells apoptosis through activating the expression of cyclin E1 (CCNE1), DNA polymerase excitability factor (PCNA) and ki67 protein [10]. (2) miR-26b stimulates NFT formation by increasing Rb1 mRNA to increase cyclin-dependent kinase 5 (Cdk5) activity level and to induce tau phosphorylation, thus inhibits the neurite outgrowth [10].
NEP, also known as membrane metallo-endopeptidase (MME), is located on human chromosome 3q25.2, whose coding protein is a type II transmembrane glycoprotein [25]. NEP is found in a variety of normal tissues, particularly on presynaptic membranes of neuron [26]. A research observes that in brain tissue of AD patients, NEP mRNA expression is remarkably decreased compared with normal control [27]. Recently, an animal experiment exhibits that upregulation of NEP expression reduces the concentration of Aβ in the mouse brain [26]. In addition, another in vivo and in vitro experiment reveals that a decreased NEP level in AD mice model and primary neurons results in an increased Aβ level and vice versa [28-30]. These imply that NEP plays an important role in delaying disease progression in AD. However, the interaction between miR-26b and NEP has not been studied so far. In our study, we observed that miR-26b inversely regulated expression of NEP but not EIF2S1 or αTTP in cellular AD model. These indicate that miR-26b might be involved in delaying AD progress by regulating NEP but not EIF2S1 or αTTP.
In conclusion, miR-26b inhibits neurite outgrowth, induces cells apoptosis and downregulates NEP expression in PC12 cellular AD model.
Disclosure of conflict of interest
None.
References
- 1.Scheltens P, Blennow K, Breteler MM, de Strooper B, Frisoni GB, Salloway S, Van der Flier WM. Alzheimer’s disease. Lancet. 2016;388:505–517. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- 2.Nasica-Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete NV, Cote S, De Simone A, Doig AJ, Faller P, Garcia A, Laio A, Li MS, Melchionna S, Mousseau N, Mu Y, Paravastu A, Pasquali S, Rosenman DJ, Strodel B, Tarus B, Viles JH, Zhang T, Wang C, Derreumaux P. Amyloid beta protein and Alzheimer’s disease: when computer simulations complement experimental studies. Chem Rev. 2015;115:3518–3563. doi: 10.1021/cr500638n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spiegel J, Pirraglia E, Osorio RS, Glodzik L, Li Y, Tsui W, Saint Louis LA, Randall C, Butler T, Xu J, Zinkowski RP, Zetterberg H, Fortea J, Fossati S, Wisniewski T, Davies P, Blennow K, de Leon MJ. Greater specificity for cerebrospinal fluid P-tau231 over P-tau181 in the differentiation of healthy controls from Alzheimer’s disease. J Alzheimers Dis. 2016;49:93–100. doi: 10.3233/JAD-150167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sheng M, Sabatini BL, Sudhof TC. Synapses and Alzheimer’s disease. Cold Spring Harb Perspect Biol. 2012:4. doi: 10.1101/cshperspect.a005777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill JM, Lukiw WJ. MicroRNA (miRNA)-mediated pathogenetic signaling in Alzheimer’s disease (AD) Neurochem Res. 2016;41:96–100. doi: 10.1007/s11064-015-1734-7. [DOI] [PubMed] [Google Scholar]
- 6.Zovoilis A, Agbemenyah HY, Agis-Balboa RC, Stilling RM, Edbauer D, Rao P, Farinelli L, Delalle I, Schmitt A, Falkai P, Bahari-Javan S, Burkhardt S, Sananbenesi F, Fischer A. microRNA-34c is a novel target to treat dementias. EMBO J. 2011;30:4299–4308. doi: 10.1038/emboj.2011.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao Y, Bhattacharjee S, Jones BM, Hill J, Dua P, Lukiw WJ. Regulation of neurotropic signaling by the inducible, NF-kB-sensitive miRNA-125b in Alzheimer’s disease (AD) and in primary human neuronal-glial (HNG) cells. Mol Neurobiol. 2014;50:97–106. doi: 10.1007/s12035-013-8595-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu HC, Wang LM, Wang M, Song B, Tan S, Teng JF, Duan DX. MicroRNA-195 downregulates Alzheimer’s disease amyloid-beta production by targeting BACE1. Brain Res Bull. 2012;88:596–601. doi: 10.1016/j.brainresbull.2012.05.018. [DOI] [PubMed] [Google Scholar]
- 9.Gupta P, Bhattacharjee S, Sharma AR, Sharma G, Lee SS, Chakraborty C. miRNAs in Alzheimer disease-a therapeutic perspective. Curr Alzheimer Res. 2017;14:1198–1206. doi: 10.2174/1567205014666170829101016. [DOI] [PubMed] [Google Scholar]
- 10.Absalon S, Kochanek DM, Raghavan V, Krichevsky AM. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J Neurosci. 2013;33:14645–14659. doi: 10.1523/JNEUROSCI.1327-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, Liu C, Zhu J, Shu P, Yin B, Gong Y, Qiang B, Yuan J, Peng X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol Aging. 2012;33:522–534. doi: 10.1016/j.neurobiolaging.2010.04.034. [DOI] [PubMed] [Google Scholar]
- 12.Dweep H, Gretz N, Sticht C. miRWalk database for miRNA-target interactions. Methods Mol Biol. 2014;1182:289–305. doi: 10.1007/978-1-4939-1062-5_25. [DOI] [PubMed] [Google Scholar]
- 13.Pinero J, Bravo A, Queralt-Rosinach N, Gutierrez-Sacristan A, Deu-Pons J, Centeno E, Garcia-Garcia J, Sanz F, Furlong LI. DisGeNET: a comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017;45:D833–D839. doi: 10.1093/nar/gkw943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–524. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Veremeyko T, Wong AH, El Fatimy R, Wei Z, Cai W, Krichevsky AM. Downregulation of miR-132/212 impairs S-nitrosylation balance and induces tau phosphorylation in Alzheimer’s disease. Neurobiol Aging. 2017;51:156–166. doi: 10.1016/j.neurobiolaging.2016.12.015. [DOI] [PubMed] [Google Scholar]
- 16.Dill H, Linder B, Fehr A, Fischer U. Intronic miR-26b controls neuronal differentiation by repressing its host transcript, ctdsp2. Genes Dev. 2012;26:25–30. doi: 10.1101/gad.177774.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satoh J, Kino Y, Niida S. MicroRNA-Seq data analysis pipeline to identify blood biomarkers for Alzheimer’s disease from public data. Biomark Insights. 2015;10:21–31. doi: 10.4137/BMI.S25132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banzhaf-Strathmann J, Benito E, May S, Arzberger T, Tahirovic S, Kretzschmar H, Fischer A, Edbauer D. MicroRNA-125b induces tau hyperphosphorylation and cognitive deficits in Alzheimer’s disease. EMBO J. 2014;33:1667–1680. doi: 10.15252/embj.201387576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delay C, Hebert SS. MicroRNAs and Alzheimer’s disease mouse models: current insights and future research avenues. Int J Alzheimers Dis. 2011;2011:894938. doi: 10.4061/2011/894938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu YK, Wang X, Li L, Du YH, Ye HT, Li CY. MicroRNA-98 induces an Alzheimer’s disease-like disturbance by targeting insulin-like growth factor 1. Neurosci Bull. 2013;29:745–751. doi: 10.1007/s12264-013-1348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhuang H, Zhang R, Zhang S, Shu Q, Zhang D, Xu G. Altered expression of microRNAs in the neuronal differentiation of human Wharton’s Jelly mesenchymal stem cells. Neurosci Lett. 2015;600:69–74. doi: 10.1016/j.neulet.2015.05.061. [DOI] [PubMed] [Google Scholar]
- 22.Chang WS, Wang YH, Zhu XT, Wu CJ. Genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease. Med Sci Monit. 2017;23:2721–2731. doi: 10.12659/MSM.905064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MIR26B microRNA 26b [Homo sapiens (human)] Gene ID: 407017, updated on 15-Oct-2017 [Google Scholar]
- 24.Liu QY, Chang MN, Lei JX, Koukiekolo R, Smith B, Zhang D, Ghribi O. Identification of microRNAs involved in Alzheimer’s progression using a rabbit model of the disease. Am J Neurodegener Dis. 2014;3:33–44. [PMC free article] [PubMed] [Google Scholar]
- 25.MME membrane metalloendopeptidase [Homo sapiens (human)] Gene ID: 4311, updated on 8-Oct-2017 [Google Scholar]
- 26.Lin CY, Perche F, Ikegami M, Uchida S, Kataoka K, Itaka K. Messenger RNA-based therapeutics for brain diseases: an animal study for augmenting clearance of beta-amyloid by intracerebral administration of neprilysin mRNA loaded in polyplex nanomicelles. J Control Release. 2016;235:268–275. doi: 10.1016/j.jconrel.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Yasojima K, McGeer EG, McGeer PL. Relationship between beta amyloid peptide generating molecules and neprilysin in Alzheimer disease and normal brain. Brain Res. 2001;919:115–121. doi: 10.1016/s0006-8993(01)03008-6. [DOI] [PubMed] [Google Scholar]
- 28.Park MH, Lee JK, Choi S, Ahn J, Jin HK, Park JS, Bae JS. Recombinant soluble neprilysin reduces amyloid-beta accumulation and improves memory impairment in Alzheimer’s disease mice. Brain Res. 2013;1529:113–124. doi: 10.1016/j.brainres.2013.05.045. [DOI] [PubMed] [Google Scholar]
- 29.Iwata N, Sekiguchi M, Hattori Y, Takahashi A, Asai M, Ji B, Higuchi M, Staufenbiel M, Muramatsu S, Saido TC. Global brain delivery of neprilysin gene by intravascular administration of AAV vector in mice. Sci Rep. 2013;3:1472. doi: 10.1038/srep01472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hama E, Shirotani K, Masumoto H, Sekine-Aizawa Y, Aizawa H, Saido TC. Clearance of extracellular and cell-associated amyloid beta peptide through viral expression of neprilysin in primary neurons. J Biochem. 2001;130:721–726. doi: 10.1093/oxfordjournals.jbchem.a003040. [DOI] [PubMed] [Google Scholar]