

Abstract
Whole-exome sequencing (WES) was used to explore pathogenic variants present in a Zhuang family with coronary artery disease (CAD). The 11 Zhuang family members enrolled in this study include three generations of direct relatives with a history of CAD. All members currently live in Guangxi Zhuang Autonomous Region (China), in which all members were also born. Clinical and biochemical tests were performed on the members who were divided into the CAD group (four members), the high-risk CAD group (four members), or normal control group (three members). Whole-exome sequencing was performed on two randomly selected CAD members and one control member in order to screen for candidate genes. All members of the family were genetically verified using Sanger sequencing. Bioinformatics analysis performed on WES data led to the discovery of five candidate genes. Having been confirmed using Sanger sequencing, two pathogenic mutations, PPP2R3A (c.721G; p.Glu241Lys>A) and TMX3 (c.1035 + 38delA), were found in four members of the CAD group and two members of the high-risk group. These variants were not detected in the three control group members. PPP2R3A and TMX3 may be new pathogenic mutations associated with CAD in the Zhuang population of Guangxi province.
Keywords: Coronary artery disease, pedigree, PPP2R3A, TMX3, whole-exome sequencing, Zhuang ethnic group
Introduction
Coronary artery disease (CAD) is a complex, multi-gene disease, caused by genetic as well as environmental factors, and commonly leads to death [1,2]. Traditional environmental risk factors for CAD include smoking, obesity, diabetes, hypertension, and hyperlipidemia. With in-depth study of the pathogenesis of CAD, genetic factors have in recent years been found to play an important role in development of this condition. Studies have shown that genetic factors account for the onset of approximately 40%-60% of CAD cases [3,4]. At present, CAD-related genes are mainly identified through genome-wide association analysis. More than 60 susceptibility loci have been found [5], with most being discovered by studying population distribution and through big data analysis. However, this approach lacks supportive research from pedigrees of families with a history of CAD. Since the occurrence of CAD shows significant familial aggregation, family history is of high predictive value for genetic susceptibility studies [6]. Some advantages to performing WES include low cost, high efficiency, and high sensitivity to common and rare mutations. Furthermore, this method has become an important guideline for detecting disease-causing mutations [7].
The prevalence of CAD varies in different ethnic groups [8], and is related to gene polymorphisms [9]. Compared to Caucasians, Blacks have a higher prevalence of CAD and unstable angina [10], and their risk of developing cardiovascular and cerebrovascular stroke is two to four times higher than that of Caucasians. The interleukin-6 (IL-6) C-572G gene is associated with the occurrence and development of CAD. This variant is differentially distributed across race groups and regions [11]. It has been reported that the 572C/G locus of the IL-6 gene is similar in Chinese and Japanese populations, but different from populations based in Europe and the United States [12]. China is a multi-ethnic country composed of Han Chinese and 55 ethnic minority populations. Living mostly in the Guangxi Zhuang Autonomous Region, the Zhuang are the most populous ethnic minority in China with more than 16 million people belonging to this group. In the sixth National demographic census, the number of Zhuang people living in Guangxi totaled 14.4 million. In this study, the clinical and biochemical tests of members of a Zhuang family with CAD history were analyzed. It is the first, to our knowledge, to describe the presence of CAD-related genes in members of the Guangxi Zhuang population. WES and Sanger sequencing were used to explore the pathogenic variants in this Zhuang family, which provides a basis for the further study of CAD pathogenesis.
Materials and methods
Pedigree information
The clinical data used to derive the Zhuang family’s pedigree was obtained from the First Affiliated Hospital of the Guangxi Medical University. The members enrolled in the study live in a remote, mountainous area in Hechi, Guangxi province. The pedigree spanning three generations was constructed from immediate Zhuang family members. No consanguineous marriages were observed. Of the eight members of the family to suffer from CAD, four are deceased. We surveyed members of the pedigree and collected clinical data. This included physical examinations of the 11 living members and extraction of 10 mL of peripheral venous blood for genomic DNA isolation and biochemical examination. The study was approved by the Guangxi Medical University First Affiliated Hospital Ethics Committee.
DNA extraction and whole-exome sequencing
Genomic DNA was extracted from whole blood using a genomic DNA isolation kit (Tiangen Biochemical Technology, Beijing, China.). The concentration and quality of DNA was determined by a nucleic acid and protein detector, as well as agarose gel electrophoresis. WES was performed by the Beijing Genomics Institute on > 2 μg of DNA from two CAD patients (II-6, II-15) and one control member (II-1). The whole-exome test work flow included randomly dividing DNA into small fragments for genomic DNA library construction and target area capture, which was followed by sequencing with a Hiseq2000 sequencer.
Sequencing data analysis
Raw WES data was filtered to remove redundant and incorrect data. The filtered data was then aligned with the GRCh37/HG19 reference genome using the Burrows-Wheeler Alignment tool, which was followed by re-alignment and calibration using Picard and the Genome Analysis Tool kit (GATK) to ensure the accuracy of mutation detection. The sequencing Raw reads, average depth and coverage of sequencing were calculated. GATK (v3.3.0) HaplotypeCaller was used to detect genomic variations, which included SNPs and indels. Variation results were annotated using SnpEff, with data analyzed and screened using dbSNP and case-control data from the 1000 Genomes Project. Finally, SIFT and Polyphen were used to predict and screen for harmful mutations.
Validation of candidate genes
Reflective of the pedigree, disease-associated candidate genes were identified by WES in family members shown to suffer from CAD. Candidate gene primers were designed using Primer 5.0 (Table 1), were synthesized by Beijing Genomics Institute, and verified by Sanger sequencing. Sanger sequencing was performed using an ABI 3730XL DNA Sequencer (Applied Biosystems). The sequencing reaction solution had a total volume of 5 μL, and the reaction program included denaturing at 95°C for 15 s, followed by annealing at 50°C for 5 s and extension at 60°C for 90 s. The sequencing reaction was repeated for 35 cycles.
Table 1.
PCR primers for five candidate genes
| Gene | Positive amplification primer sequence | Reverse amplification primer sequence |
|---|---|---|
| RNF25 | TGCCGTTCCTGTTCCTGC | GAGCGTCACCAAGCCAGA |
| TMX3 | TCAACCTTAGCCAACAGC | AGATGAATTGCGTATCCT |
| CCR5 | GGTGGAACAAGATGGATT | GACACCGAAGCAGAGTTT |
| PPP2R3A | CCCATCCTTTGGTTTACT | CATTATTCCCAGACTGCA |
| MRPL42 | CCCCTCAACTGGAACAT | CCTGAGAATGAGAACCCT |
Results
Clinical data and the derivation of the pedigree
The Zhuang CAD family under investigation had a total of 44 members spanning three generations (Figure 1). Of these, 11 members agreed to participate in this study since the age of the family members varied greatly and some members lived outside of the Hechi area. The detailed clinical data and biochemical test results are shown in Table 2. The 11 enrolled members were divided into three groups: the CAD group, the high-risk CAD group, and the healthy control group. For this study, CAD diagnosis was based on WHO diagnostic criteria for CAD [13], which include: a stenosis rate of vascular diameter of coronary angiography left main artery, left anterior descending artery, left circumflex artery, right coronary artery or its main branch ≥50%. Criteria for the high-risk CAD group included no clinical symptoms of CAD, but the presentation of more than one CAD risk factor. The healthy control group had no clinical symptoms or CAD risk factors, and also showed normal biochemical test results. A total of four members (36.4%) had already been diagnosed with CAD. Three of these cases (II-6, II-11, and II-15) were confirmed by coronary angiography and the insertion of two or more stents. One member (II-13) had angina, presented with CAD risk factors (hypertension, obesity, and smoking habits), and was thus clinical diagnosed with CAD. Four members (II-18, III-4, III-5, III-13) presented with different risk factors for CAD, including hypertension, hyperlipemia, obesity, while the remaining three members (II-2, III-3, III-16) lacked a diagnosis of CAD, did not have any related risk factors, and were put in the normal control group.
Figure 1.
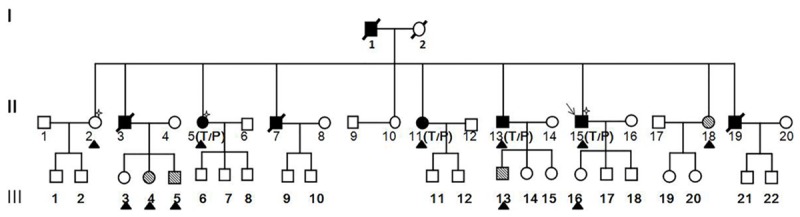
Pedigree structure of a family. ■ and ●, CAD affected; □ and ○, not CAD affected; MARK 1 and MARK 2, Individuals reported to have High coronary heart disease factors; MARK 3, included in exome sequencing analysis; ▲, Individuals reported Members of the pedigree agreeing to participate in this study; P, mutated PPP2R3A allele; T, mutated TMX3A allele.
 |
 |
Table 2.
Clinical characteristics of family members
| ID | Age (years) | Gender | BP | Smoker | BMI (kg/m2) | CHOL (mmol/L) | TG (mmol/L) | HDL (mmol/L) | LDL (mmol/L) | FBG (mmol/L) |
|---|---|---|---|---|---|---|---|---|---|---|
| II-2 | 68 | F | No | No | 28.9 | 4.6 | 0.83 | 1.87 | 2.42 | 1.37 |
| II-5 | 64 | F | No | No | 22.22 | 5.35 | 1.5 | 1.21 | 3.15 | 3.27 |
| II-11 | 56 | F | No | No | 31.24 | 4.81 | 2.32 | 0.90 | 2.63 | 4.02 |
| II-13 | 51 | M | Yes | Yes | 27.34 | 5.05 | 0.66 | 1.83 | 2.60 | 2.55 |
| II-15 | 49 | M | Yes | Yes | 27.74 | 2.84 | 1.09 | 0.87 | 1.29 | 4.26 |
| II-18 | 46 | F | No | No | 20.5 | 7.23 | 0.82 | 1.50 | 5.05 | 1.07 |
| III-3 | 44 | F | No | No | 19.11 | 4.88 | 1.61 | 1.10 | 2.83 | 3.23 |
| III-4 | 41 | F | Yes | No | 32.05 | 8.04 | 1.18 | 1.15 | 5.91 | 3.63 |
| III-5 | 37 | M | No | No | 25.95 | 5.27 | 0.86 | 1.76 | 2.88 | 3.67 |
| III-13 | 28 | M | No | No | 23.46 | 7.13 | 3.47 | 0.92 | 4.30 | 3.04 |
| III-16 | 22 | F | No | No | 18.73 | 3.89 | 0.71 | 0.91 | 2.28 | 4.5 |
BMI: 18.5-23.9, CHOL 3.1-5.2, TG 0.56-1.70, HDL 1.16-1.42, LDL 2.7-3.13, FBG 3.9-6.1. M: male, F: female, BP: blood pressure, BMI: body mass index, CHOL: cholesterol, TG: triglycerides, HDL: high-density lipoprotein, LDL: low-density lipoprotein, FBG: fasting blood sugar.
Whole-exome sequencing data analysis
WES yielded an average of over 8 G of data per sample. The average target depth of sequencing was approximately 70.40×. The average 10× target area coverage was 95.92%. Candidate genes for CAD were filtered by 1) selecting the genes discovered in the CAD group that were not present in the control group; 2) excluding synonymous mutations and selecting those with a minor allele frequency < 0.01% from the 1000 Genomes Project database; and 3) testing amino acid function of the identified mutations using SIFT and PolyPhen-2. This strategy identified a total of 588 preliminary SNP and 428 indel mutations. Mutations that in both programs were predicted to have harmful consequences were selected as SNPs of interest. A total of four SNP and one indel mutations were found (Table 3).
Table 3.
Five candidate genes
| Gene | Chromosome | Mutation site | Function mutation | SIFT | Polyphen-2 |
|---|---|---|---|---|---|
| RNF25 | Chr2 | c.419A>G | Missense mutation | 0.01 (D) | 0.998 (D) |
| CCR5 | Chr3 | c.316G>A | Missense mutation | 0 (D) | 0.971 (D) |
| PPP2R3A | Chr3 | c.721G>A | Missense mutation | 0 (D) | 0.994 (D) |
| MRPL42 | Chr12 | c.364C>T | Missense mutation | 0 (D) | 0.995 (D) |
| TMX3 | Chr18 | c.1035 + 38delA | Intron mutations |
Validation of candidate genes
The candidate genes were re-validated by PCR and confirmed via Sanger sequencing. Two pathogenic genes PPP2R3A (c.721G; p.Glu241Lys>A) and TMX3 (c.1035 + 38delA), were found (Figure 2). The c.721G>A (p.Glu241Lys) variant is located in the PPP2R3A gene on chromosome 3, and results in a missense mutation from a glutamic acid codon (CAG) to a lysine codon (AAG). This gene variant was found in four CAD patients and one high risk CAD member (II-18). The c.1035 + 38delA mutation is located in the TMX3 gene on chromosome 18.A cytidine deletion at loci 454 results in the occurrence of this mutation. This mutation was not found in the dbSNP database and is therefore considered to be a new SNP site. The TMX3 gene variant was found in four CAD patients and two high risk CAD members. Neither of the two pathogenic gene variants was found in the three control members.
Figure 2.
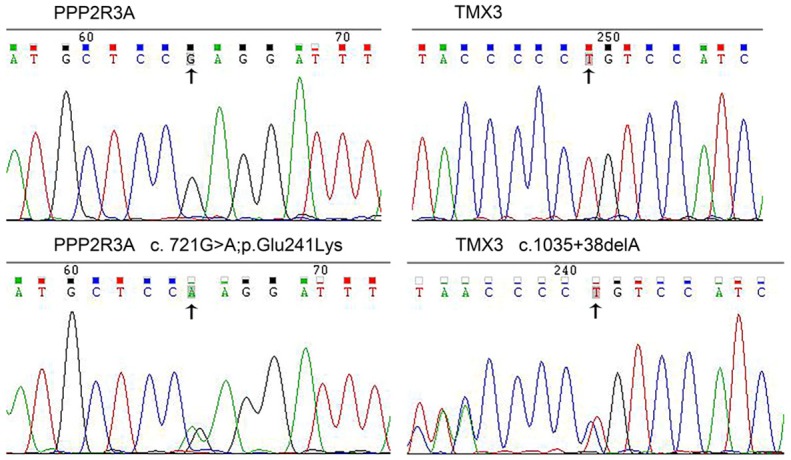
Sequence chromatograms of PPP2R3A and TMX3. The top and bottom panels, respectively, show chromatograms of wild-type and mutated sequences.
Discussion
By using whole-exome and Sanger sequencing, this study identified two putative pathogenic genes, PPP2R3A and TMX3, in a Zhuang family living in the Guangxi Zhuang Autonomous Region with a family history of CAD. These two variants might be pathogenic mutations specific to the Zhuang population in the Guangxi province. The PPP2R3A and TMX3 mutations were collectively found in six members enrolled in this study. Four members were confirmed CAD patients by coronary angiography or clinical diagnosis, and the remaining two possessed multiple high-risk factors for CAD, which included dyslipidemia and high blood pressure. No variant was found in control group family members.
The genetic mechanism of CAD can be explained by the superposition effects of multiple minor alleles [14]. It is, however, a complex multi-gene disease. Studies have reported that the prevalence of CAD varies in different ethnic groups, while genome-wide association analysis is the primary tool through which studies on genes related to CAD are conducted. However, only 33% of the genes identified using this method have definitively been shown to relate to CAD [15,16]. In addition, this method is mainly used for independent CAD patients instead of pedigree with CAD history. The use of WES technology is not restricted by the family history of the individual(s) under investigation and is widely applied to mono and polygenic disease research. Recent studies have therefore started to focus on the exploration of CAD-related gene mutation loci through pedigree research using WES. For example, Erdmann et al. [6] found two heterozygous mutations, GUCY1A3 and CCT7, in a pedigree that included 32 CAD patients; and Inanloo Rahatloo K et al. [17] found a pathogenic ST6GALNAC5 gene variant in a large Iranian family.
In this study, we found new pathogenic alleles within the PPP2R3A and TMX3 genes in a Zhuang family living in the Guangxi Zhuang Autonomous Region with a family history of CAD. PPP2R3A is part of the protein phosphatase 2A regulatory subunit B, which is a serine/threonine phosphatase that plays an important role in regulating growth and division. PP2A is one of the major phosphatases in the human heart and regulates membrane excitability and cardiac contractions by dephosphorylating ion channel arrays and cardiac proteins [18]. Phosphorylation disorders are associated with many cardiovascular diseases, including heart failure, myocardial infarction, atrial fibrillation, and sinus node disease [19]. PPP2R3A has two subunits: PR72 and PR130. PR130 is the largest transcription factor of PPP2R3A and is highly expressed in heart [20]. It has been reported that calcium released by PR130 and the sarcoplasmic reticulum act as targets for the ryanodine receptor 2. This process is associated with cardiac excitatory/contractile coupling [18]. These studies have shown that PPP2R3A gene mutations have some impact on the contraction of the heart, which may be related to the presentation of CAD. The TMX3 gene variant is a new SNP site. TXM3 is a member of the endoplasmic reticulum disulfide isomerase (PDI) family. Previous studies have shown that PDI is present on the platelet surface, is secreted by platelet activity, and is related to the formation of thrombi [21]. Although studies have confirmed that TMX3 is also present on the platelet surface [22], its specific biological function is not yet clear. It is well-known that platelet aggregation and thrombosis are the main causes of acute coronary syndrome. Therefore, we believe that there is a relationship between TMX3 and CAD, which warrants further investigation.
This study also found that among the four enrolled members with risk factors for CAD, one carried both the PPP2R3A and TMX3 variants, one only carried the TMX3 mutation, and the remaining two carried neither of the two variants. Although this result is not consistent with a co-segregation model, it could be as a result of environment, age and gender. For example, young patients may not show clinical symptoms of CAD, while women show clinical symptoms relatively late due to the protective effects of estrogen [23]. Nevertheless, we will conduct long-term follow-ups on these four members in order to track the relationship between the two variants and CAD.
The limitations of this study are due to the fact that 1) the sample size is too small; and 2) verification was performed within one family only. More cases occurring in families of the same ethnic group will be included in future studies. We will also include a larger group of independent CAD patients from the Guangxi Zhuang population to further explore the relationship between PPP2R3A, TMX3 and CAD in the Zhuang Chinese population.
Acknowledgements
This study was partly supported by Guangxi Science and Technology Plan Project (No: 1598011-2) and Guangxi Key Laboratory Base of Precision Medicine in Cardio-cerebrovascular Diseases Control and Prevention.
Disclosure of conflict of interest
None.
References
- 1.Graham G. Population-based approaches to understanding disparities in cardiovascular disease risk in the United States. Int J Gen Med. 2014;7:393–400. doi: 10.2147/IJGM.S65528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellison SR. Sudden cardiac death in adolescents. Prim Care. 2015;42:57–76. doi: 10.1016/j.pop.2014.09.012. [DOI] [PubMed] [Google Scholar]
- 3.Rosenzweig A. Scanning the genome for coronary risk. N Engl J Med. 2007;357:497–499. doi: 10.1056/NEJMe078121. [DOI] [PubMed] [Google Scholar]
- 4.Vinkhuyzen AA, Wray NR, Yang J, Goddard ME, Visscher PM. Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu Rev Genet. 2013;47:75–95. doi: 10.1146/annurev-genet-111212-133258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nurnberg ST, Zhang H, Hand NJ, Bauer RC, Saleheen D, Reilly MP, Rader DJ. From loci to biology: functional genomics of genome-wide association for coronary disease. Circ Res. 2016;118:586–606. doi: 10.1161/CIRCRESAHA.115.306464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Erdmann J, Stark K, Esslinger UB, Rumpf PM, Koesling D, de Wit C, Kaiser FJ, Braunholz D, Medack A, Fischer M, Zimmermann ME, Tennstedt S, Graf E, Eck S, Aherrahrou Z, Nahrstaedt J, Willenborg C, Bruse P, Braenne I, Nothen MM, Hofmann P, Braund PS, Mergia E, Reinhard W, Burgdorf C, Schreiber S, Balmforth AJ, Hall AS, Bertram L, Steinhagen-Thiessen E, Li SC, Marz W, Reilly M, Kathiresan S, McPherson R, Walter U, Ott J, Samani NJ, Strom TM, Meitinger T, Hengstenberg C, Schunkert H. Dysfunctional nitric oxide signalling increases risk of myocardial infarction. Nature. 2013;504:432–436. doi: 10.1038/nature12722. [DOI] [PubMed] [Google Scholar]
- 7.Isakov O, Perrone M, Shomron N. Exome sequencing analysis: a guide to disease variant detection. Methods Mol Biol. 2013;1038:137–158. doi: 10.1007/978-1-62703-514-9_8. [DOI] [PubMed] [Google Scholar]
- 8.Marrzoq LF, Sharif FA, Abed AA. Relationship between ApoE gene polymorphism and coronary heart disease in Gaza Strip. J Cardiovasc Dis Res. 2011;2:29–35. doi: 10.4103/0975-3583.78584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rubinstein A, Izkhakov E. [Lipoprotein associated phospholipase A2] . Harefuah. 2011;150:136–140. 205. [PubMed] [Google Scholar]
- 10.Saunders E, Ofili E. Epidemiology of atherothrombotic disease and the effectiveness and risks of antiplatelet therapy: race and ethnicity considerations. Cardiol Rev. 2008;16:82–88. doi: 10.1097/CRD.0b013e31815685fa. [DOI] [PubMed] [Google Scholar]
- 11.Li YY, Zhou CW, Xu J, Qian Y, Wang XM. Interleukin-6 C-572G gene polymorphism and coronary artery disease in Asian: a meta-analysis of 2511 subjects. Int J Clin Exp Med. 2015;8:8995–9003. [PMC free article] [PubMed] [Google Scholar]
- 12.Humphries SE, Luong LA, Ogg MS, Hawe E, Miller GJ. The interleukin-6 -174G/C promoter polymorphism is associated with risk of coronary heart disease and systolic blood pressure in healthy men. Eur Heart J. 2001;22:2243–2252. doi: 10.1053/euhj.2001.2678. [DOI] [PubMed] [Google Scholar]
- 13.Nomenclature and criteria for diagnosis of ischemic heart disease. Report of the Joint International Society and Federation of Cardiology/World Health Organization task force on standardization of clinical nomenclature. Circulation. 1979;59:607–609. doi: 10.1161/01.cir.59.3.607. [DOI] [PubMed] [Google Scholar]
- 14.McPherson R, Tybjaerg-Hansen A. Genetics of coronary artery disease. Circ Res. 2016;118:564–578. doi: 10.1161/CIRCRESAHA.115.306566. [DOI] [PubMed] [Google Scholar]
- 15.Roberts R, Stewart AF. 9p21 and the genetic revolution for coronary artery disease. Clin Chem. 2012;58:104–112. doi: 10.1373/clinchem.2011.172759. [DOI] [PubMed] [Google Scholar]
- 16.Lee SE, Kim HS. Unraveling new therapeutic targets of coronary artery disease by genetic approaches. Circ J. 2015;79:8–14. doi: 10.1253/circj.CJ-14-0985. [DOI] [PubMed] [Google Scholar]
- 17.InanlooRahatloo K, Parsa AF, Huse K, Rasooli P, Davaran S, Platzer M, Kramer M, Fan JB, Turk C, Amini S, Steemers F, Gunderson K, Ronaghi M, Elahi E. Mutation in ST6GALNAC5 identified in family with coronary artery disease. Sci Rep. 2014;4:3595. doi: 10.1038/srep03595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Heijman J, Dewenter M, El-Armouche A, Dobrev D. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol. 2013;64:90–98. doi: 10.1016/j.yjmcc.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 19.DeGrande ST, Little SC, Nixon DJ, Wright P, Snyder J, Dun W, Murphy N, Kilic A, Higgins R, Binkley PF, Boyden PA, Carnes CA, Anderson ME, Hund TJ, Mohler PJ. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J Biol Chem. 2013;288:1032–1046. doi: 10.1074/jbc.M112.426957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zwaenepoel K, Louis JV, Goris J, Janssens V. Diversity in genomic organisation, developmental regulation and distribution of the murine PR72/B” subunits of protein phosphatase 2A. BMC Genomics. 2008;9:393. doi: 10.1186/1471-2164-9-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schulman S, Bendapudi P, Sharda A, Chen V, Bellido-Martin L, Jasuja R, Furie BC, Flaumenhaft R, Furie B. Extracellular thiol isomerases and their role in thrombus formation. Antioxid Redox Signal. 2016;24:1–15. doi: 10.1089/ars.2015.6530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holbrook LM, Watkins NA, Simmonds AD, Jones CI, Ouwehand WH, Gibbins JM. Platelets release novel thiol isomerase enzymes which are recruited to the cell surface following activation. Br J Haematol. 2010;148:627–637. doi: 10.1111/j.1365-2141.2009.07994.x. [DOI] [PubMed] [Google Scholar]
- 23.North American Menopause Society. The 2012 hormone therapy position statement of: The North American Menopause Society. Menopause. 2012;19:257–271. doi: 10.1097/gme.0b013e31824b970a. [DOI] [PMC free article] [PubMed] [Google Scholar]