

Abstract
Coronary artery disease (CAD) is one of the leading causes of mortality and morbidity worldwide and the number of individuals at CAD risk is increasing. To better manage cardiovascular disease, improved tools for risk prediction including the identification of novel accurate biomarkers are needed. MicroRNAs (miRNAs) are small non-coding RNAs that modulate the expression of protein-coding genes at the post-transcription level and their dysregulated expression has been implicated in various pathogenic processes including cardiovascular disease. Circulating miRNAs have been widely recommended as potential biomarkers for many diseases including coronary artery disease. In the present study, we found that miR-3646 was significantly upregulated in the serum samples of CAD patients and in the mice with acute myocardial infarction (AMI) compared with the healthy control group via using quantitative reverse transcription polymerase chain reaction (qRT-PCR). Moreover, the serum levels of miR-3646 were significantly positively correlated with the expression of IL-6 both in CAD patient samples and AMI mice samples. In human THP-1 macrophages, transfection with miR-3646 mimic elevated the expression of IL-6 while silence of miR-3646 suppressed the IL-6 level. Further exploration of the downstream targets of miR-3646 identified that blocking RHOH expression also could upregulate IL-6 expression. In addition, our findings also showed that miR-3646 promoted vascular smooth muscle cell (VSMC) proliferation and migration by targeting RHOH. These results demonstrate that the miR-3646-RHOH axis may serve as a key regulator in the progression of CAD by modulating vascular inflammation and regulating the biologic behaviors of VSMCs.
Keywords: Coronary artery disease, miR-3646, RHOH, vascular inflammation, vascular smooth muscle cells
Introduction
Coronary artery disease (CAD), one of the leading causes of mortality and disability worldwide, is well-recognized as a complex disease caused by the combined impact of genetic factors and environments [1-3]. CAD is caused by atherosclerotic plaque, which is an inflammatory disease that involves multiple cell types, including immune system cells and cells in the vessel wall [4,5]. Immune cells and cytokines play pivotal roles in the pathogenic inflammation of atherosclerosis. Experimental data support a critical role of macrophages in the initiation, propagation, and progression of atherosclerosis [6-8]. Tumor necrosis factor-alpha (TNF-α) is regarded as a pro-inflammatory cytokine that is involved in the pathophysiology of CAD and is often expressed in atherosclerotic plaques [9-12].
Dysregulated proliferation of vascular smooth muscle cells (VSMCs) also contributes to atherosclerotic plaque formation [13,14]. In normal vessels, the majority of VSMCs is quiescent and contractile. In disease states such as atherosclerosis, VSMCs can switch from the adult, quiescent, contractile phenotype to the synthetic, proliferative and migratory phenotype [13-16]. Therefore, finding therapies to effectively inhibit VSMC proliferation and migration is a major focus for prevention and treatment of CAD. However, the detail mechanisms underlying the VSMC proliferation and migration have not been fully elucidated.
MicroRNAs (miRNAs) represent a subset of endogenous, single-strand, small non-coding RNA with 19-25 nucleotides in length, which post-transcriptionally suppress target gene expression through binding to complementary sequences in the 3’-untranslated region (3’UTR) [17-19]. miRNAs have been widely reported to be involved in multiple biologic processes, including cell proliferation, apoptosis, and differentiation [20,21]. Increasing evidence has suggested that miRNAs impact the initiation and progression of CAD [22-27]. For instance, Lai et al. have indicated that miR-574-5p serves as a CAD-related factor and promotes VSMC proliferation and inhibits apoptosis by inhibiting ZDHHC14 gene expression [13]. Zhang et al. have also suggested that miR-448 plays an important role in the proliferation and migration of VSMCs by repressing MEF2C expression [28].
miR-3646 is a tumor promoting miRNA located at 20q13.12. Much evidence has shown that miR-3646 is a potential oncogene in breast cancer and it may represent a new biomarker in the diagnosis and prediction of prognosis and chemoresistance [29,30]. A study has shown that miR-3646 play a pivotal role in ischemic disease [31]. Although miR-3646 has been reported to be deregulated in cardiovascular disease [32,33], its role in CAD is understudied, especially in CAD-associated inflammation and the biological behaviors of VSMCs.
In the present study, we found that miR-3646 was significantly upregulated in the serum samples of CAD patients and AMI mice. A positive-correlation between miR-3646 and TNFα was observed in these samples. Importantly, RHOH was a direct target of miR-3646 in THP-1 macrophages and VSMCs. Overexpression of miR-3646 or silence of RHOH in THP-1 cells elevated TNFα expression, while downregulation of miR-3646 suppressed TNFα expression. On the other hand, in VSMCs, overexpression of miR-3646 or silence of RHOH promotes cell proliferation and migration. Our findings not only demonstrate the function of the miR-3646-RHOH axis in CAD, but also provide a putative therapeutic target for related diseases.
Materials and methods
Cell culture
Human THP-1, VSMCs and 293T cells were purchased from American Type Culture Collection (ATCC, Rockville, MD, USA). THP-1 cells were cultured in RPMI 1640 medium (Gibco, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS, Gibco). VSMCs were cultured in Medium 199 (M199, Gibco) supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin (Sigma, Shanghai, China). 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% FBS, 100 U/ml penicillin and 100 μg/ml streptomycin. All the cells were incubated in a humidified chamber with 5% CO2 at 37°C.
Patient sample collections
Patients with CAD (n = 24) were collected by the Department of Cardiology, Tangshan Workers’ Hospital between January 2017 and December 2017, which was approved by the Ethics Committee of Tangshan Workers’ Hospital. Nineteen healthy volunteers were enrolled as controls. Venous blood samples (5-10 ml) were collected from all the participants, which were centrifuged at 2000 rpm at 4°C for 30 min. The serum samples were isolated and stored at -80°C until analysis.
AMI mouse model constructions
All animal procedures were fully reviewed and approved by the Institutional Animal Care and Use Committee of Tangshan Workers’ Hospital. Six-eight-week-old male C57BL/6J mice were obtained from the Institute of Laboratory Animal Science, Chinese Academy of Medical Sciences (Beijing, China). Mice were anaesthetized with sodium pentobarbital (90 mg/kg, Sigma-Aldrich, St Louis, MO, USA) by intraperitoneal injection and were then intubated and mechanically ventilated. The pericardium of each mouse was opened after thoracotomy, and the left coronary artery was identified and ligated with a 7/0 no-damage silk suture to induce AMI. After full recovery, the animals were returned to independently ventilated cages. Blood samples were collected 24 hours after the surgery was performed.
miRNA mimic, miRNA inhibitor and siRNA transfection
Cells were grown in the indicated medium 24 hours before transfection. The cells were transfected with 50 nM of miR-3646 mimics, inhibitor, RHOH siRNA or the scramble mimics/inhibitor/siRNA using lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. The miRNA mimics, inhibitors, and siRNA were synthesized by Guangzhou RiboBio Co., Ltd. The siRNA sequences were as follows: GGUGCUGAUGUGCUACUCUGU (RHOH siRNA), GGCAAGCUGACCCUGAAGUU (Control siRNA).
Semi-quantitative RT-PCR and quantitative RT-PCR
Total RNA was isolated from the serum samples and harvested cells by using TRIzol LS (Invitrogen) or TRIzol (Invitrogen) respectively following the manufacturer’s manual. For mRNA detection of RHOH, Quantitative RT-PCR was performed using the One Step SYBR® PrimeScript™ RT-PCR Kit II (Takara, Tokyo, Japan) on the LightCycler® 96 Real-Time PCR System (BioRad, Hercules, CA, USA). β-ACTIN was used as an internal control. Primers were as follows: 5’-CAAATGAAACCCCCTCTGCG-3’ (RHOH, forward), 5’-TTGGGTTAGTGTGTGTGGGG-3’ (RHOH, reverse), 5’-AAAGACCTGTACGCCAACAC-3’ (β-ACTIN, forward), and 5’-GTCATACTCCTGCTTGCTGAT-3’ (β-ACTIN, reverse). For miR-3646 detection. 5 ng of total RNA were reverse transcribed to cDNA with stem-loop primers by using the QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). Quantitative real-time PCR (qRT-PCR) was done by using the SYBR® Green detection reagent (Takara). U6 RNA was used as internal control. Primers were as follows: 5’-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTGGGCT-3’ (specific miR-3646 RT primer), 5’-GTGCAGGGTCCGAGGT-3’ (miR-3646, forward), 5’-GCAAAATGAAATGAGCCCAG-3’ (miR-3646, reverse). 5’-CTCGCTTCGGCAGCACA-3’ (U6, forward), 5’-AACGCTTCACGAATTTGCGT-3’ (U6, reverse).
Luciferase reporter assay
Reporter plasmid containing wild type RHOH or mutant plasmid was transfected into 293T cells along with miR-3646 mimic or control mimic, using lipofectamine 2000 reagent according to the manufacturer’s manual. Forty-eight hours later, cells were lysed with 100 µl of Passive Lysis Buffer (Promega, Fitchburg, WI, USA), and their Renilla luciferase levels were analyzed using the Dual Glo Luciferase Assay System (Promega). The firefly luciferase activity was used as internal control. The primers used for cloning and site directed mutagenesis are as follows: 5’-GACCGCGATCGCACCCCAAGAGACTTCACA-3’ (RHOH-WT-forward), 5’-CTTAGTTTAAACGGAGCAGTCTGATTCTCTAC-3’ (RHOH-WT-reverse); 5’-TTCTGAAATGATAAAACATCAAAGTAAAATTTACTATAT-3’ (RHOH-Mut-forward), 5’-ATATAGTAAATTTTACTTTGATGTTTTATCATTTCAGAA-3’ (RHOH-Mut-reverse).
Measurement of TNFα by ELISA
Commercial ELISA kits (Invitrogen, USA) were used to measure the TNFα in the serum samples and cell culture supernatant, following the manufacturer’s instructions.
Cell proliferation assay
One thousand cells were seeded in a 96-well plate and transfection was performed when cell confluence reached 50-70%. Proliferation rates were determined at 0, 24, 48 and 72 hours after transfection. At each timepoint, cell culture medium was changed with fresh culture medium pre-mixed with 10% CCK-8 reagent (Dojindo Laboratories, Kumamoto, Japan). Then the cells were incubated at 37°C for about 1 hour and analyzed for absorbance by an auto microplate reader (Thermo Scientific, Waltham, Massachusetts, USA) at 450 nm.
Western blot
Western blot was performed as previously described [34]. Rabbit monoclonal RHOH (Cat# 118507), CDK2 (Cat# 32147), Cyclin D1 (Cat# 134175), p21 (Cat# 109520) and p27 (Cat# 32034) antibody was purchased from Abcam (Cambridge, MA, USA). Mouse monoclonal β-actin antibody was from Beyotime (Cat# AA128, Shanghai, China).
Wound healing assay
Six-well plates were seeded with 5 × 105 cells transfected with either control or miR-3646 mimic and cultured in an incubator until 100% confluence was reached. The layer of cells was scratched with a 10 μl pipette tip and washed with fresh medium three times. The incubation was continued in culture medium with 1% FBS for 12 hours. The distance of cell migration was determined by microscopy.
Migration assay
One million VSMCs were suspended into 100 μl of basic culture medium and seeded in the top chamber of an insert (8 μm pores), which was inserted into a 24-well plate. Five hundred μl of culture medium with 10% FBS was added to the bottom chambers. Six hours later, the cells passing through the membrane onto the lower side of the chamber were fixed in 4% paraformaldehyde (Beyotime), stained with 0.1% crystal violet (Beyotime) and detected under a Nikon inverted microscope (Nikon Instruments, Melville, NY, USA). Stained crystal violet was then dissolved with 33% acetic acid and quantified at a microplate reader at 570 nm.
Statistical analysis
The statistical significance between two groups was determined by unpaired Student’s t test. The correlation between miR-3646 and RHOH was analyzed by Pearson method. A P-value smaller than 0.05 was considered significant.
Results
miR-3646 expression is upregulated and positively correlated with TNFα in serum samples of CAD patients and AMI mice
Initially, to evaluate the role of miR-3646 in CAD, we detected the expression levels of circulating miR-3646 in patients with CAD (n = 24) and healthy controls (n = 19) by using quantitative RT-PCR. We found that circulating miR-3646 was significantly upregulated in patients with CAD compared with healthy controls (P = 0.0172, Figure 1A). Then, we established an AMI model in mice and determined the expression levels of circulating miR-3646 in these samples. Compared with the sham group, the expression of miR-3646 was also significantly higher in the AMI mice (P = 0.0010, Figure 1D). Next, we also examined the inflammatory factor TNFα in these samples. The results demonstrated that TNFα was also upregulated in CAD patients and AMI mice compared with healthy control (Figure 1B and 1E). Importantly, we found that the expression levels of miR-3646 were positively correlated with TNFα both in CAD patients and AMI mice (Figure 1C and 1F). These findings suggest that upregulation of miR-3646 may contribute to CAD by elevating TNFα.
Figure 1.
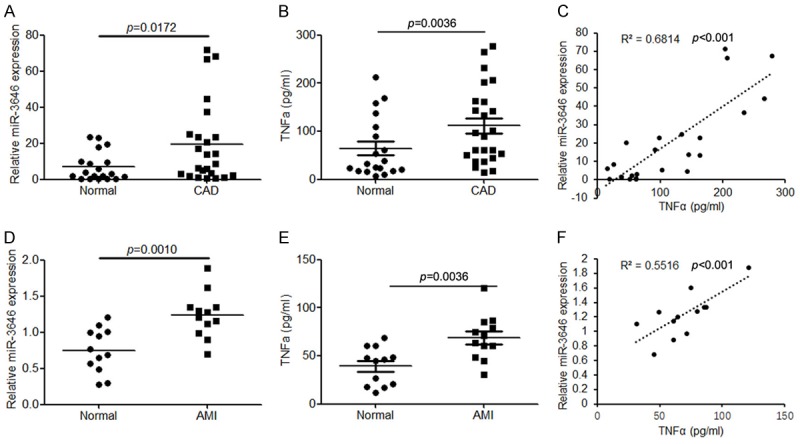
miR-3646 expression is significantly upregulated in the serum samples derived from patients with CAD and from mice with AMI, and its expression is positively correlated with TNFα. A. The expression of circulating miR-3646 in patients with CAD (n = 24) compared with healthy control (n = 19). B. ELSIA analysis to determine the protein level of TNFα in the serum samples derived from patients with CAD compared with healthy controls. C. Correlation analysis of miR-3646 and TNFα expression in the serum samples of CAD patients. D. The expression of miR-3646 in the serum samples of AMI mice (n = 12) compared with normal controls (n = 12). E. The TNFα expression in the serum samples of AMI mice compared with normal control. F. Correlation analysis of miR-3646 and TNFα expression in the serum samples of AMI mice.
miR-3646 upregulates the levels of TNFα in THP-1 macrophages
To elucidate whether miR-3646 regulate the expression of TNFα. We transfected miR-3646 mimic or inhibitor into THP-1 macrophages. The results indicated that transfection of miR-3646 mimic (Figure 2A) elevated the levels of TNFα (Figure 2C), while silence of miR-3646 (Figure 2B) suppressed TNFα levels (Figure 2C). Bioinformatics analysis predicted that RHOH was a direct target of miR-3646 (Figure 2D). To elucidate the relationship between miR-3646 and RHOH, a luciferase reporter assay was conducted. Co-transfection of miR-3646 mimic into 293T cells with the reporter plasmid containing the conserved miR-3646 predicted binding site for RHOH suppressed the luciferase activity of the reporter gene, compared with the control miRNA mimic (Figure 2E). Disruption of the predicted binding sites reduced the inhibitory activity of miR-3646 mimic on the luciferase activity (Figure 2E). Moreover, overexpression of miR-3646 in THP-1 cells (Figure 2A) significantly suppressed RHOH protein levels (Figure 2F). These results indicate that RHOH is a direct target of miR-3646 in macrophages. Importantly, silence of RHOH in THP-1 cells (Figure 2G) also significantly inhibited the level of TNFα, which was determined by the ELISA assay (Figure 2H). These data demonstrate that miR-3646 upregulated TNFα expression in macrophages may via targeting to RHOH.
Figure 2.
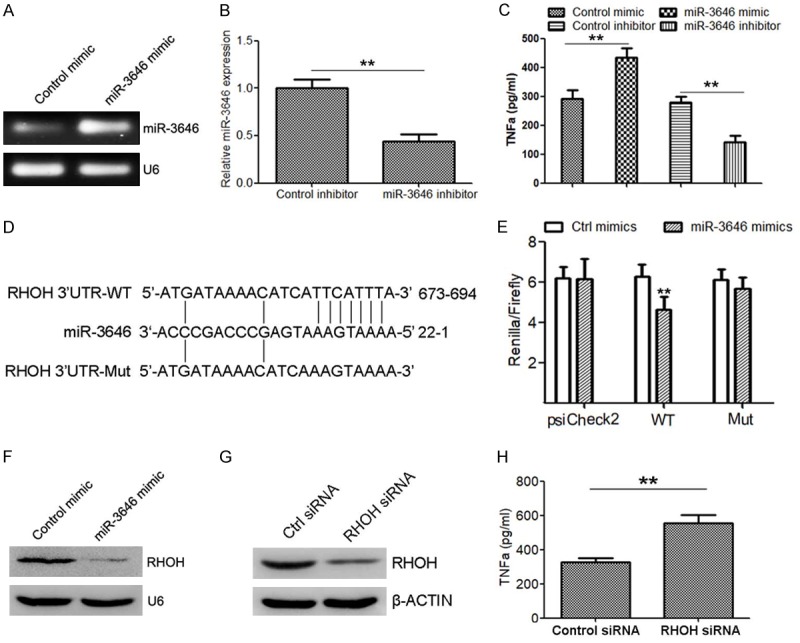
miR-3646 promotes TNFα secretion in THP-1 cells potentially by suppressing RHOH. A. Semi-quantitative RT-PCR to determine the expression of miR-3646 in THP-1 cells transfected with miR-3646 mimic compare with control. B. Quantitative RT-PCR to detect the expression of miR-3646 in THP-1 cells transfected with miR-3646 inhibitor compared with control. C. The protein levels of TNFα in supernatant of cells with indicated treatment, determined by western blot analysis. D. Schema of the miR-3646 binding site within the RHOH 3’UTR. The mutated site is shown below. E. The psiCHECK-2 sensor plasmids containing the RHOH 3’UTR or mutated 3’UTR were co-transfected in 293T cells with miR-3646 mimic, or control mimic. Overexpression of miR-3646 decreased the luciferase expression compared to controls. No downregulation of luciferase was observed for the negative control or mutated plasmid with miR-3646 overexpression. F. Western blot analysis to determine RHOH protein expression in THP-1 cells transfected with miR-3646 mimic or control mimic. G. Western blot to determine the protein level of RHOH in THP-1 cells transfected with RHOH siRNA or control siRNA. H. Determination of TNFα expression in THP-1 cells transfected with RHOH siRNA or control siRNA. **P < 0.01 versus control.
miR-3646 promotes cell proliferation, migration in VSMCs
It is well recognized that the proliferation and migration of VSMCs play a critical role in the development of CAD. We next determined the effect of miR-3646 and its target RHOH in the proliferation and migration of VSMCs. QRT-PCR analysis illustrated that PDGF-bb (20 ng/ml) can induce miR-3646 expression (Figure 3A). Moreover, CKK8 analysis demonstrated that ectopic expression of miR-3646 in VSMCs (Figure 3B) significantly elevated the cell proliferate rate (Figure 3C). Meanwhile, we also determined the expression of cell cycle-related proteins in miR-3646 mimic transfected VSMCs. The results showed that the cell cycle promoting genes CDK2 and Cyclin D1 was upregulated by miR-3646 in VSMCs, while the expression of cell cycle inhibiting genes p21 and p27 was suppressed (Figure 3D). Moreover, we also evaluated the effect of miR-3646 on VSMC migration. Both wound healing assay (Figure 3E and 3G) and Boyden chamber migration assay (Figure 3F and 3H) demonstrated that ectopic expression of miR-3646 drastically elevated the migrative ability of VSMCs.
Figure 3.
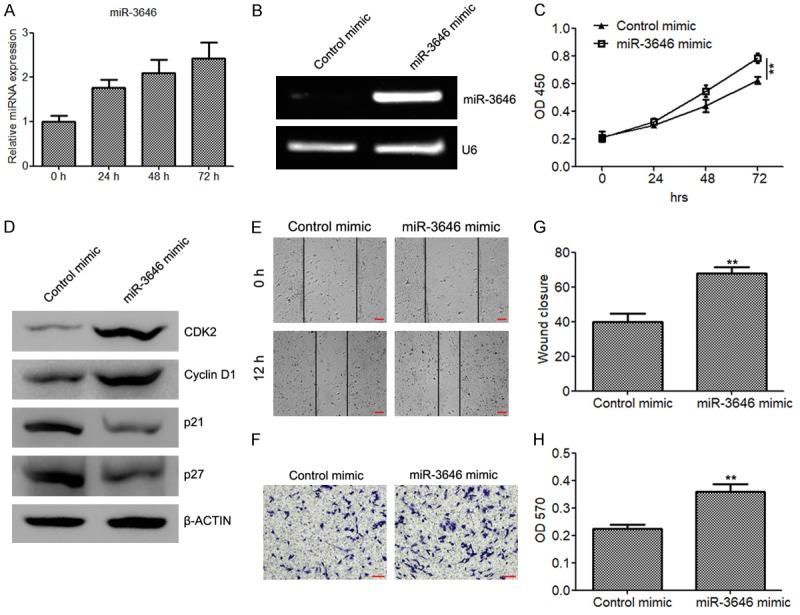
miR-3646 promotes cell proliferation and migration of VSMCs. (A) Quantitative RT-PCR to determine the expression of miR-3646 in VSMCs treated with 20 ng/ml of PDGF-bb for 0, 24, 48 and 72 h. (B) Semi-quantitative RT-PCR to determine the expression of miR-3646 in VSMCs transfected with miR-3646 mimic or control mimic. (C) CKK8 assay to evaluate the proliferate rate of VSMCs transfected with miR-3646 mimic or control mimic. (D) Determination of cell cycle proteins CDK2, Cyclin D1, p21, and p27 in VSMCs transfected with miR-3646 mimic or control mimic. (E and H) Wound healing assay (E and G) and migration assay (F and H) to determine the and migration abilities of VSMCs transfected with miR-3646 mimic or control mimic. Cells invading or migrating to the bottom side of the upper chamber were stained with crystal violet, dissolved with acetic acid and quantified at OD 570 (H). **P < 0.01 versus control.
Silence of RHOH also promotes cell proliferation, migration in VSMCs
To evaluate the potential mechanism of miR-3646 in regulating cell proliferation and apoptosis in VSMCs, we also detected the expression of RHOH in VSMCs transfected with miR-3646. The results demonstrated that miR-3646 also suppressed RHOH translation in VSMCs (Figure 4A). In addition, PDGF-bb also inhibited RHOH expression, determined by quantitative RT-PCR and western blot analysis (Figure 4B and 4C). After a series of cellular function experiments, we found that silencing of RHOH (Figure 4D) also increased cell proliferation (Figure 4E) and promoted cell migration (Figure 4F and 4G).
Figure 4.
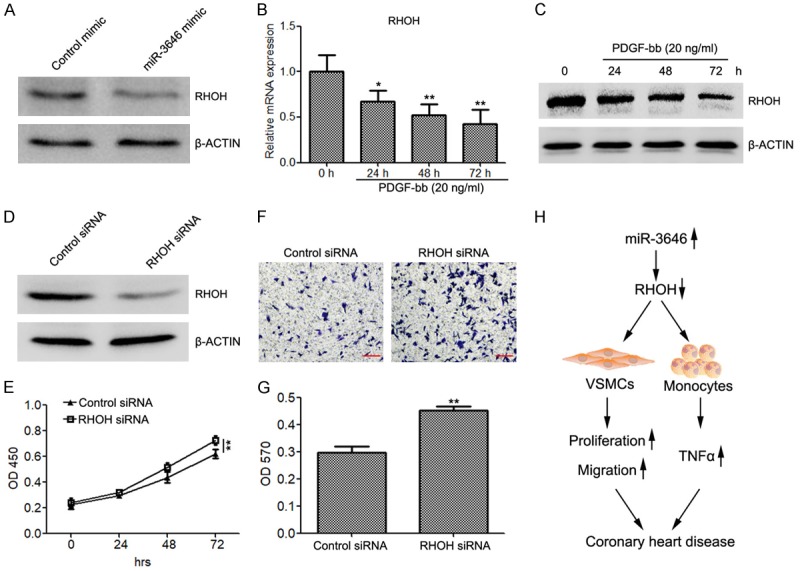
Silencing of RHOH restores cell proliferation and migration of VSMCs. (A) Western blot analysis to evaluate the protein levels of RHOH in VSMCs transfected with miR-3646 mimic or control mimic. (B and C) Quantitative RT-PCR (B) and western blot (C) to determine the expression of RHOH in VSMCs treated with 20 ng/ml of PDGF-bb for 0, 24, 48 and 72 h. (D) The expression of RHOH in VSMCs transfected with RHOH siRNA or control siRNA. (E) CKK8 assay to evaluate the proliferation rate of VSMCs transfected with RHOH siRNA and control siRNA. (F and G) Migration assay to determine the migration abilities of VSMCs transfected with RHOH siRNA and control siRNA. (H) Schematic view to present the regulatory role of the miR-3646-RHOH axis in CAD. *P < 0.05, **P < 0.01 versus control.
Taken together, these findings indicate that miR-3646 promotes vascular inflammation and increased cell proliferation and migration of VSMCs by directly targeting RHOH (Figure 4H).
Discussion
Dysregulation of miRNAs contributes to various pathologic diseases, including CAD [22-26,35]. Recent studies have shown that a variety of circulating miRNAs are aberrantly expressed in the sera of CAD patients and can be potential diagnostic biomarkers and therapeutic options for CAD [22,36]. In the present study, we found that the expression of miR-36466 was drastically upregulated in patients with CAD and mice with AMI. Moreover, we demonstrated that miR-3646 promoted vascular inflammation and biological behaviors of VSMCs by targeting to RHOH. These results suggest that the miR-3646-RHOH axis is of great importance in the pathogenesis of CAD. Thus, clinically modulating miR-3646 and RHOH expressions could be promising means of reducing inflammation and decreasing VSMC proliferation and migration, improving endothelial regeneration after vascular injury.
In this study, we focused on miR-3646, as this miRNA has been previously reported as a tumor promoting miRNA in carcinogenesis and associated with ischemic diseases. Several studies also demonstrate that this miRNA is aberrantly expressed in CAD patients based on microarray analysis. Our unpublished high throughput sequencing data also showed that miR-3646 was upregulated in the serum samples of CAD patients. In the present study, we systematically identified the expression, function, and underlying mechanisms of miR-3646 in CAD.
Small G proteins of the Rho family are defined as major regulators of cell functions including migration, proliferation, differentiation and gene transcription [37]. Recent studies have demonstrated that abnormal activation of the RHOA/ROCK pathway has been observed in major cardiovascular disorders such as restenosis, hypertension, pulmonary hypertension, and cardiac hypertrophy. Functional analyses have further revealed that RHOA-dependent pathways are involved in excessive contraction, migration, and proliferation associated with arterial diseases [37-39]. However, the exact role of RHOH in vascular inflammation and the biological behaviors of VSMCs have not been fully elucidated. In the present study, we found that RHOH not only suppressed inflammatory factor release of macrophages, but also inhibited cell proliferation and migration of VSMCs. Previous studies have shown that RHOH is expressed in hematopoietic cells and functions as a negative regulator of cell growth and survival. Consistently, our findings also demonstrate that RHOH negatively regulates VSMC proliferation and migration [40]. Li et al. have found that RhoH functions differently from other Rho GTPases and is a potent inhibitor of the activation of NFkappaB and p38 by other Rho GTPases [40]. Furthermore, reduction of RhoH levels in T cells augments the response to Rac activation [40]. The inhibitory function of RhoH in Rho GRPases supports our and other researchers’ results in which RHOH is inactivated and the RHOA/ROCK pathway is activated in CAD.
Acknowledgements
This research was self-financed and was received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. The first author greatly thanks her family for their kindly helps.
Disclosure of conflict of interest
None.
References
- 1.Duan L, Liu C, Hu J, Liu Y, Wang J, Chen G, Li Z, Chen H. Epigenetic mechanisms in coronary artery disease: the current state and prospects. Trends Cardiovasc Med. 2018;28:311–319. doi: 10.1016/j.tcm.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 2.Duan L, Hu J, Xiong X, Liu Y, Wang J. The role of DNA methylation in coronary artery disease. Gene. 2018;646:91–97. doi: 10.1016/j.gene.2017.12.033. [DOI] [PubMed] [Google Scholar]
- 3.Roberts R. A breakthrough in genetics and its relevance to prevention of coronary artery disease in LMIC. Glob Heart. 2017;12:247–257. doi: 10.1016/j.gheart.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 4.Roberts R. A genetic basis for coronary artery disease. Trends Cardiovasc Med. 2015;25:171–178. doi: 10.1016/j.tcm.2014.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Veerasamy M, Bagnall A, Neely D, Allen J, Sinclair H, Kunadian V. Endothelial dysfunction and coronary artery disease: a state of the art review. Cardiol Rev. 2015;23:119–129. doi: 10.1097/CRD.0000000000000047. [DOI] [PubMed] [Google Scholar]
- 6.Ikonomidis I, Michalakeas CA, Parissis J, Paraskevaidis I, Ntai K, Papadakis I, Anastasiou-Nana M, Lekakis J. Inflammatory markers in coronary artery disease. Biofactors. 2012;38:320–328. doi: 10.1002/biof.1024. [DOI] [PubMed] [Google Scholar]
- 7.Zakynthinos E, Pappa N. Inflammatory biomarkers in coronary artery disease. J Cardiol. 2009;53:317–333. doi: 10.1016/j.jjcc.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 8.Jahn J, Dalhoff K, Katus HA. Coronary artery disease: an inflammatory or infectious process. Basic Res Cardiol. 2000;95(Suppl 1):I59–64. doi: 10.1007/s003950070011. [DOI] [PubMed] [Google Scholar]
- 9.Hassan-Nejhad M, Bagheri M, Khadem-Vatani K, Seyed Mohammad Zad MH, Abdi Rad I, Rahimi B, Rostamzadeh A, Rahimlou A. Tumor necrosis factor-alpha gene expression in PBMCs of Iranian Azeri Turkish patients with premature coronary artery disease (Age. 50 years) Maedica (Buchar) 2018;13:12–16. [PMC free article] [PubMed] [Google Scholar]
- 10.Vrselja Z, Sram M, Andrijevic D, Takac B, Leksan I, Radic R, Curic G. Transcardial gradient of adiponectin, interleukin-6 and tumor necrosis factor-alpha in overweight coronary artery disease patients. Cytokine. 2015;76:321–327. doi: 10.1016/j.cyto.2015.09.009. [DOI] [PubMed] [Google Scholar]
- 11.Cheng Y, An B, Jiang M, Xin Y, Xuan S. Association of tumor necrosis factor-alpha polymorphisms and risk of coronary artery disease in patients with non-alcoholic fatty liver disease. Hepat Mon. 2015;15:e26818. doi: 10.5812/hepatmon.26818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gotsman I, Stabholz A, Planer D, Pugatsch T, Lapidus L, Novikov Y, Masrawa S, Soskolne A, Lotan C. Serum cytokine tumor necrosis factor-alpha and interleukin-6 associated with the severity of coronary artery disease: indicators of an active inflammatory burden? Isr Med Assoc J. 2008;10:494–498. [PubMed] [Google Scholar]
- 13.Lai Z, Lin P, Weng X, Su J, Chen Y, He Y, Wu G, Wang J, Yu Y, Zhang L. MicroRNA-574-5p promotes cell growth of vascular smooth muscle cells in the progression of coronary artery disease. Biomed Pharmacother. 2018;97:162–167. doi: 10.1016/j.biopha.2017.10.062. [DOI] [PubMed] [Google Scholar]
- 14.Motterle A, Pu X, Wood H, Xiao Q, Gor S, Ng FL, Chan K, Cross F, Shohreh B, Poston RN, Tucker AT, Caulfield MJ, Ye S. Functional analyses of coronary artery disease associated variation on chromosome 9p21 in vascular smooth muscle cells. Hum Mol Genet. 2012;21:4021–4029. doi: 10.1093/hmg/dds224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Badin JK, Bruning RS, Sturek M. Effect of metabolic syndrome and aging on Ca(2+) dysfunction in coronary smooth muscle and coronary artery disease severity in Ossabaw miniature swine. Exp Gerontol. 2018;108:247–255. doi: 10.1016/j.exger.2018.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Gonzalo-Calvo D, Cenarro A, Garlaschelli K, Pellegatta F, Vilades D, Nasarre L, Camino-Lopez S, Crespo J, Carreras F, Leta R, Catapano AL, Norata GD, Civeira F, Llorente-Cortes V. Translating the microRNA signature of microvesicles derived from human coronary artery smooth muscle cells in patients with familial hypercholesterolemia and coronary artery disease. J Mol Cell Cardiol. 2017;106:55–67. doi: 10.1016/j.yjmcc.2017.03.005. [DOI] [PubMed] [Google Scholar]
- 17.Vaiman D. Genes, epigenetics and miRNA regulation in the placenta. Placenta. 2017;52:127–133. doi: 10.1016/j.placenta.2016.12.026. [DOI] [PubMed] [Google Scholar]
- 18.van Zonneveld AJ, Rabelink TJ, Bijkerk R. miRNA-coordinated networks as promising therapeutic targets for acute kidney injury. Am J Pathol. 2017;187:20–24. doi: 10.1016/j.ajpath.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 19.Achkar NP, Cambiagno DA, Manavella PA. miRNA biogenesis: a dynamic pathway. Trends Plant Sci. 2016;21:1034–1044. doi: 10.1016/j.tplants.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 20.Gao L, Jiang F. MicroRNA (miRNA) profiling. Methods Mol Biol. 2016;1381:151–161. doi: 10.1007/978-1-4939-3204-7_8. [DOI] [PubMed] [Google Scholar]
- 21.Mizuguchi Y, Takizawa T, Yoshida H, Uchida E. Dysregulated miRNA in progression of hepatocellular carcinoma: a systematic review. Hepatol Res. 2016;46:391–406. doi: 10.1111/hepr.12606. [DOI] [PubMed] [Google Scholar]
- 22.Reddy LL, Shah SA, Ponde CK, Rajani RM, Ashavaid TF. Circulating miRNA-33: a potential biomarker in patients with coronary artery disease (CAD) Biomarkers. 2018:1–27. doi: 10.1080/1354750X.2018.1501760. [DOI] [PubMed] [Google Scholar]
- 23.JF OS, Neylon A, McGorrian C, Blake GJ. miRNA-93-5p and other miRNAs as predictors of coronary artery disease and STEMI. Int J Cardiol. 2016;224:310–316. doi: 10.1016/j.ijcard.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 24.Orenes-Pinero E, Marin F, Lip GY. miRNA-197 and miRNA-223 and cardiovascular death in coronary artery disease patients. Ann Transl Med. 2016;4:200. doi: 10.21037/atm.2016.05.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulte C, Molz S, Appelbaum S, Karakas M, Ojeda F, Lau DM, Hartmann T, Lackner KJ, Westermann D, Schnabel RB, Blankenberg S, Zeller T. miRNA-197 and miRNA-223 predict cardiovascular death in a cohort of patients with symptomatic coronary artery disease. PLoS One. 2015;10:e0145930. doi: 10.1371/journal.pone.0145930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Zhi H, Li Y, Ma G, Ye X, Yu X, Yang T, Jin H, Lu Z, Wei P. Polymorphism in miRNA-1 target site and circulating miRNA-1 phenotype are associated with the decreased risk and prognosis of coronary artery disease. Int J Clin Exp Pathol. 2014;7:5093–5102. [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong XD, Cho M, Cai XP, Cheng J, Jing X, Cen JM, Liu X, Yang XL, Suh Y. A common variant in pre-miR-146 is associated with coronary artery disease risk and its mature miRNA expression. Mutat Res. 2014;761:15–20. doi: 10.1016/j.mrfmmm.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Zhang R, Sui L, Hong X, Yang M, Li W. MiR-448 promotes vascular smooth muscle cell proliferation and migration in through directly targeting MEF2C. Environ Sci Pollut Res Int. 2017;24:22294–22300. doi: 10.1007/s11356-017-9771-1. [DOI] [PubMed] [Google Scholar]
- 29.Tao S, Liu YB, Zhou ZW, Lian B, Li H, Li JP, Zhou SF. miR-3646 promotes cell proliferation, migration, and invasion via regulating G2/M transition in human breast cancer cells. Am J Transl Res. 2016;8:1659–1677. [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang X, Zhong S, Xu Y, Yu D, Ma T, Chen L, Zhao Y, Chen X, Yang S, Wu Y, Tang J, Zhao J. MicroRNA-3646 contributes to docetaxel resistance in human breast cancer cells by GSK-3beta/beta-catenin signaling pathway. PLoS One. 2016;11:e0153194. doi: 10.1371/journal.pone.0153194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward J, Kanchagar C, Veksler-Lublinsky I, Lee RC, McGill MR, Jaeschke H, Curry SC, Ambros VR. Circulating microRNA profiles in human patients with acetaminophen hepatotoxicity or ischemic hepatitis. Proc Natl Acad Sci U S A. 2014;111:12169–12174. doi: 10.1073/pnas.1412608111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu X, Li H. Integrated microRNAgene analysis of coronary artery disease based on miRNA and gene expression profiles. Mol Med Rep. 2016;13:3063–3073. doi: 10.3892/mmr.2016.4936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hua L, Xia H, Zhou P, Li D, Li L. Combination of microRNA expression profiling with genome-wide SNP genotyping to construct a coronary artery disease-related miRNA-miRNA synergistic network. Biosci Trends. 2014;8:297–307. doi: 10.5582/bst.2014.01031. [DOI] [PubMed] [Google Scholar]
- 34.Tao F, Tian X, Ruan S, Shen M, Zhang Z. miR-211 sponges lncRNA MALAT1 to suppress tumor growth and progression through inhibiting PHF19 in ovarian carcinoma. FASEB J. 2018:fj201800495RR. doi: 10.1096/fj.201800495RR. [DOI] [PubMed] [Google Scholar]
- 35.Zhou J, Shao G, Chen X, Yang X, Huang X, Peng P, Ba Y, Zhang L, Jehangir T, Bu S, Liu N, Lian J. miRNA 206 and miRNA 574-5p are highly expression in coronary artery disease. Biosci Rep. 2015;36:e00295. doi: 10.1042/BSR20150206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mehta R, Otgonsuren M, Younoszai Z, Allawi H, Raybuck B, Younossi Z. Circulating miRNA in patients with non-alcoholic fatty liver disease and coronary artery disease. BMJ Open Gastroenterol. 2016;3:e000096. doi: 10.1136/bmjgast-2016-000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward-Caviness C, Haynes C, Blach C, Dowdy E, Gregory SG, Shah SH, Horne BD, Kraus WE, Hauser ER. Gene-smoking interactions in multiple Rho-GTPase pathway genes in an early-onset coronary artery disease cohort. Hum Genet. 2013;132:1371–1382. doi: 10.1007/s00439-013-1339-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aizawa K, Yasuda S, Takahashi J, Takii T, Kikuchi Y, Tsuburaya R, Ito Y, Ito K, Nakayama M, Takeda M, Shimokawa H. Involvement of rho-kinase activation in the pathogenesis of coronary hyperconstricting responses induced by drug-eluting stents in patients with coronary artery disease. Circ J. 2012;76:2552–2560. doi: 10.1253/circj.cj-12-0662. [DOI] [PubMed] [Google Scholar]
- 39.Nohria A, Grunert ME, Rikitake Y, Noma K, Prsic A, Ganz P, Liao JK, Creager MA. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ Res. 2006;99:1426–1432. doi: 10.1161/01.RES.0000251668.39526.c7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X, Bu X, Lu B, Avraham H, Flavell RA, Lim B. The hematopoiesis-specific GTP-binding protein RhoH is GTPase deficient and modulates activities of other Rho GTPases by an inhibitory function. Mol Cell Biol. 2002;22:1158–1171. doi: 10.1128/MCB.22.4.1158-1171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]