

Abstract
Cytochrome P450 (CYP450) enzymes belong to a superfamily of heme-containing proteins that are involved in the metabolism of structurally diverse endogenous and exogenous compounds. Various proof-of-concept studies indicate that metabolic stability and intrinsic clearance of CYP450 substrates are linked with the respective lipophilicity (log P or log D). This necessitates the normalization of lipophilicity (log P or log D) of a given CYP450 substrate with respect to its metabolic stability (LipMetE) and intrinsic clearance (log10CLint,u). Therefore, in this article, the LipMetE values of already known substrates of CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4, including some marketed drugs, have been calculated to elucidate the relationship between lipophilicity (log D7.4) and in vitro clearance. Moreover, various drug efficiency metrics including lipophilic efficiency (LipE) and ligand efficiency (LE) have been evaluated, and the thresholds of these parameters have been defined for the CYP450 substrates exhibiting normalized LipMetE. Our results indicate that for a given range of LipMetE, greater the log D value of the substrate the more avidly it binds to a given CYP450 enzyme and shows more intrinsic clearance (log10CLint,u). Overall, the majority of the model substrates of CYP450 isoforms and already marketed drugs in our datasets exhibit log D7.4 values of ∼2.5 with LipMetE values in the range of 0–2.5 and LipE values of ≤3. Overall, consideration of these parameters in ADME profiling could aid in reducing the drug failure rate in the later stages of clinical investigations.
1. Introduction
Research and development (R&D) is the initial stage of the development process in which the pharmaceutical companies apply research-based knowledge and adopt various strategies to develop new products and bring innovation to the industry.1−3 During the past decades, huge technological advancements and access to a plethora of scientific knowledge have prognosticated an all-time high R&D output.2 However, it is challenging for the pharmaceutical companies to maintain the R&D productivity at a justifiable level.2 Moreover, the R&D productivity gap of the pharmaceutical industry has been associated with poor pharmacokinetics/ADME-Tox properties, lack of efficacy, and adverse drug–drug interactions.1
More recently, AstraZeneca implemented a revised and more focused R&D strategy to be used in the drug discovery and development projects, showing a shift from quantitative (high-volume) to qualitative strategies with a deeper understanding. To improve the overall R&D productivity, AstraZeneca proposed a decision-making five-“R” concept that is based on the right target, right tissue, right safety, right patient, and right commercial potential. At AstraZeneca, evaluation and application of the five-“R” concept to target validation and selectivity, hit and lead optimization, pharmacokinetics/pharmacodynamics (PK/PD) modeling, safety/toxicology, and efficacy have improved the success rate at phase III from 4% in 2005–2010 to 19% in 2012–2016.2,3
Furthermore, it has been demonstrated that understanding of PK, PK/PD, and ADME properties of new chemical entities is crucial for improving quality in lead and drug candidate selection. Overall, drug metabolism has been recognized as one of the most important factors in pharmacokinetics and hence modulates the behavior of a drug.4 Among several human metabolizing enzymes, cytochrome P450s are the heme-containing enzymes that account for ∼75% of the drug metabolism.5 Among these, the CYP1, CYP2, and CYP3 families mediate 70–80% of all phase I metabolic reactions of clinically relevant drugs with CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP3A5, and CYP2E1 performing 90% of the drug metabolism.5
Induction of metabolic enzymes by various chemical entities may produce a suboptimal effect that results in high clearance due to a high metabolic rate, whereas enzyme inhibition by some other chemical scaffolds might produce an effect longer than that required to achieve the desired therapeutic effect, thus resulting in undesired side effects.4,6 Moreover, the cytochrome P450 family of enzymes has an inherent affinity for lipophilic substrates due to their lipophilic nature.7,8 Yet, highly lipophilic compounds might also possess a greater CYP inhibition potential depending on their ionization states.9 However, in the drug discovery and design programs, lipophilicity evaluates the permeability of a drug through the biomembrane and, thus, affects its bioavailability.10 Therefore, the need to probe toxicological profiles of new chemical entities (NCEs) during early stages of investigations is highly demanded.11
Toward this goal, various authors in the past used several ligand-12 as well as structure-based in silico approaches13,14 and hybrid methods15,16 for toxicological profiling of lead candidates. These include QSAR,17,18 machine-learning methods,19,20 pharmacophore-based methods,21,22 shape-focused approaches, molecular interaction fields (MIFs),23 reactivity-focused techniques,24 docking25,26 and molecular dynamics (MD) simulation studies on different classes of modulators of CYP450, ABC transporters, and the hERG K+ ion channel.27−30
Additionally, the impact of lipophilicity on membrane permeability, bioavailability, promiscuity, drug metabolism by CYP450s, and overall ADME-Tox properties has also been reported by several authors in the past.9−11,31−34 Various studies have reported a trend of increase in lipophilicity during lead optimization protocols that results in low solubility and poor absorption, and thus, might lead to rapid metabolic turnover by CYP450 enzymes.35,36 The recently established lipophilic metabolic efficiency (LipMetE) parameter ensures adequate metabolic stability at the required lipophilicity level, even for compounds with high lipophilic efficiency (LipE).37 It has been observed that a compound with metabolic flaws and consistently very low LipMetE might fail to yield a quality clinical candidate even if high LipE against the respective CYP450 isoform was achieved.38
To probe the therapeutic activities and metabolism-related effects of compounds, screening of large chemical libraries against specific antitargets including CYP450s and on-targets is crucial during very early phases of drug discovery, mainly during hit identification and prior to lead optimization. Therefore, ligand optimization in the context of targets as well as antitargets is highly demanded.39 Overall, CYP450s are known to metabolize a diverse set of substrates depending on the nature of their binding sites, thus representing diverse substrate properties for each CYP isoform.40 Therefore, in the present investigation, we utilized the LipMetE parameter to elucidate the relationship between lipophilicity and in vitro clearance of the CYP substrates. Additionally, hit-to-lead efficiency metrics including lipophilic efficiency (LipE) and ligand efficiency (LE) were calculated to probe the metabolic attributes of CYP450 substrates.
2. Results and Discussion
2.1. Lipophilic Metabolic Efficiency (LipMetE)
Monitoring compound lipophilicity and maintaining it at a lower level form an integral part of drug design/discovery criteria because highly lipophilic chemical entities are recurrently associated with greater risks.41 It is well established that compounds with higher lipophilicity (c log P > 3) and lower polar surface area (TPSA < 75 Å2) pose a 6-fold greater risk during the preclinical toxicology testing.42 Moreover, the oxidative liability of the highly lipophilic compounds leads to high clearance, poor bioavailability, and high dosage-dependent targeted efficacy, making it amenable to severe toxicological outcomes.43 Therefore, due to the importance of lipophilicity as a design parameter and to establish meaningful relationships (i.e., lipophilicity vs clearance), many design indices take into account compound lipophilicity.37
The lipophilic metabolic efficiency (LipMetE) is one parameter that depicts the relationship between lipophilicity and clearance (in vitro HLM) in a similar manner to LipE, where LipE describes the relationship of lipophilicity with potency.37 Graphing the relationship between lipophilicity (log D7.4) and metabolic stability (log10CLint,u) can also be used to understand the contribution of lipophilicity toward metabolic stability through other factors (i.e., a compound’s intrinsic chemical stability). For the clusters of related compounds with variable lipophilicity values and the same LipMetE values, the differences in clearance are mainly associated with changes in lipophilicity. However, for compounds with similar lipophilicity values traversing the LipMetE lines, the clearance might be modulated due to other factors (i.e., difference in the chemical stability, blockage of the metabolic site, or the alteration in a substrate’s intrinsic affinity for a particular CYP450 isozyme).37
Previously, it has been reported that drug-like compounds usually show LipMetE values between −2.0 and 2.0, and higher LipMetE values (>2.5) are indicative of greater metabolic stability in comparison to lower LipMetE values.37 The compounds with higher LipMetEs provide a wide range of log D7.4 values and, thus, can be used as important starting points for the optimization/improvement of properties, such as potency and permeability, in combination with low clearance.37 Various studies advocating the LipMetE concept have been reported in the literature. Pettersson et al. demonstrated LipMetE values of 0.9–2.0 for a series of pyridopyrazine-1,6-dione γ-secretase modulators (GSMs) designed for the treatment of Alzheimer’s disease38 and a LipMetE of 1.5 for the optimized cyclopropyl chromane-derived pyridopyrazine-1,6-dione-type γ-secretase modulator.44 Similarly, the optimization of the LipMetE parameter to achieve better ADME profiles of a drug by 11 hydrogen-to-fluorine matched molecular pair (MMP) transformations has been established elsewhere, and it is shown that the OCH3-to-OCF3 transformation corresponded to an increase in LipMetE from −0.5 to 2.0 in the MDR modulators.45
Optimization of LipMetE values with respect to lipophilicity and in vitro clearance advocates a promising concept to estimate the CYP450 substrate properties of new chemical entities (NCEs). Therefore, here the LipMetE parameter has been calculated to decipher the relationship between lipophilicity and in vitro clearance of the CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 substrate datasets, as described by Stepan et al.37 (data shown in Table S2 of the Supporting Information), to probe the threshold values of lipophilicity, in vitro clearance, and LipMetE for better metabolic profiles of drug-like CYP450 substrates. Overall, the LipMetE and log D7.4 distribution profiles of the collated datasets display a wide range of LipMetE and log D7.4 values (i.e., <0 to >3), as shown in Figures S1 and S2. The LipMetE, log D7.4, and log10CLint,u values within −4.54 to 4.35, −1.19 to 4.24, and −3.08 to 4.7 (Figure 1a, Table S2) have been calculated for 43 CYP1A2 substrates, including 31 FDA-approved drugs (for drug classes refer to Table S1).
Figure 1.

LipMetE profiling of (a) CYP1A2, (b) CYP2C9, (c) CYP2C19, (d) CYP2D6, and (e) CYP3A4 substrates. Data points labeled in red in all LipMetE profiling plots indicate the CYP450 substrates with metabolic stability values between 0 and 4 (LipMetE).
Interestingly, only one compound, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), exhibits a LipMetE value of 4.35, which might be correlated with its very low clearance value (log10CLint,u −3.08, Table S2), whereas the model substrate, 7-ethoxycoumarin, showed a LipMetE value of 2.99, log D7.4 of 2.22, and an in vitro clearance of −0.77 (log10CLint,u) (Table S2). The remaining compounds in our CYP1A2 substrate dataset span the LipMetE range of −4.54 to 1.86 with the marketed drugs (amitriptyline, mexiletine, zolpidem, mirtazapine, clozapine, nortriptyline, pimozide, and acetaminophen) exhibiting LipMetE values within 0.11–1.86 (Figure 1a). Overall, for the CYP1A2 substrates, it has been observed that marketed drugs and model substrates with suitable metabolic profiles (LipMetE 0–2.5) show average LipMetE, log D7.4, and log10CLint,u values of 0.94, 2.25, and 1.28, respectively, as shown in Table 1. This is further supported by the similar LipMetE profiles of the CYP2C9 substrates.
Table 1. Table Summarizing the LipMetE, LipE, log D7.4, and log10CLint,u Ranges for the CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 Substrate datasets That Were Used for LipMetE Calculationsa.
| known
drugs and model substrates data
(LipMetE 0–2.5) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CYP450 | LipE range | LipMetE range | log D7.4 range | log10CLint,u range | no of substrates with LipMetE between 0 and 2.5 | LipE range | average LipE | average LipMetE | average log D7.4 | average log10CLint,u |
| CYP1A2 | –1.96 to 7.24 | –4.54 to 4.35 | –1.19 to 4.24 | –3.08 to 4.72 | 8 | –1.96 to 3.03 | 0.88 | 0.94 | 2.25 | 1.28 |
| CYP2C9 | –1.02 to 5.01 | –3.72 to 3.63 | –0.98 to 5.92 | –0.015 to 5.18 | 6 | –0.83 to 2.73 | 1.00 | 0.69 | 2.63 | 1.94 |
| CYP2C19 | –4.20 to 5.13 | –6.40 to 4.70 | –1.29 to 5.68 | –1.87 to 6.49 | 3 | –2.18 to 0.95 | –0.15 | 0.70 | 2.73 | 2.02 |
| CYP2D6 | –1.18 to 4.73 | –6.35 to 2.93 | –2.01 to 5.09 | –0.53 to 6.72 | 11 | –1.14 to 2.17 | 0.50 | 1.03 | 2.48 | 1.46 |
| CYP3A4 | –2.36 to 6.66 | –6.19 to 3.85 | –1.26 to 5.16 | –3.08 to 7.27 | 23 | –1.98 to 2.72 | 0.85 | 0.75 | 2.64 | 1.89 |
The average values for LipMetE, LipE, log D7.4, and log10CLint,u have been derived using drugs and model substrates from each dataset with better metabolic profiles (LipMetE 0–2.5).
A CYP2C9 substrate dataset of 43 compounds, including flavonoids, marker substrates, insecticides, experimental agents, and various FDA-approved drugs (Table S1), with Km values in the range 0.4–2250 μM was used for the estimation of log D7.4 (−0.98 to 5.92), log10CLint,u (−0.015 to 5.18), and LipMetE values (−3.72 to 3.63), as shown in Table S2. The already known drugs metabolized by CYP2C9, namely, sertraline, zolpidem, mirtazapine, meloxicam, and perphenazine along with one model substrate 7-ethoxycoumarin, one flavonoid derivative, one organosulfur analogue, and two pesticides (fenthion and sulprofos) displayed positive metabolic stability (LipMetE 0.066–1.56), as shown in Figure 1b. However, only desogestrel exhibits a very high positive LipMetE of 3.62, which might be linked with its high log D7.4 value (5.92) (Figure 1b) as compared to the log D7.4 values of the rest of the data (−0.98 to 4.88). For the metabolically stable (LipMetE between 0 and 2.5) marketed drugs and model substrates of CYP2C9, average LipMetE, log D7.4, and log10CLint,u values of 0.69, 2.63, and 1.94 are shown (Table 1). Overall, the LipMetE values of CYP2C9 substrates lie within the range of LipMetE values established previously for different drug-like compounds,37,38 which further reflects the robustness of our LipMetE calculation model.
LipMetE estimation was also performed on a dataset of 54 CYP2C19 substrates including investigational agents, hexobarbital enantiomers, marker substrates, withdrawn drugs, insecticides, and 28 approved drugs (for drug classes refer to Table S1) with Km values in the range 0.43–89 000 μM (Table S2). The LipMetE values of the dataset range from −5.69 to 3.90 with only diazepam, flunitrazepam, and 8:2 fluorotelomer alcohol possessing LipMetE of 3.2/3.9/4.75 and log D7.4 values of 2.92/2.03/5.68, respectively (Table S2, Figure 1c). The marketed drugs and model substrates metabolized by CYP2C19 that have values within the desired LipMetE (0–2.5) range display an average LipMetE value of 0.703 with log D7.4 and log10CLint,u values of 2.73 and 2.02, respectively (Table 1), which is in line with the already established LipMetE parameters against CYP1A2 and CYP2C9.
Similar results were achieved for our dataset of CYP2D6 substrates. Briefly, CYP2D6 contributes to the metabolism of about 20–25% of clinically relevant drugs, including β-blockers, neuroleptics, antidepressants, and antiarrhythmics.46 For the calculation of the LipMetE parameter, the CYP2D6 substrate dataset was composed of 65 compounds, including marker substrates, antioxidants, plant extracts, experimental and investigational compounds, clinical candidates,47 active drug metabolites, and a large number of FDA-approved drugs (Table S1), presenting LipMetE values within −6.35 to 2.93, as shown in Table S2. However, already marketed drugs displayed LipMetE values from 0.21 to 2.92 with an average value of 1.03 (Table 1). Overall, loperamide, dextrorphan, and K11777 epitomize the candidates with the highest LipMetE (2.9/2.19/2.83) and log D7.4 (3.94/1.67/3.8) values (Table S2, Figure 1d).
For CYP3A4, LipMetE values between −6.19 and 3.85 have also been calculated for 86 substrates, including clinical trial compounds and 63 FDA-approved drugs (drug classes shown in Table S1, Figure 1e, and Table S2). However, 26 CYP3A4 substrates, including 24 clinically available drugs, displayed a greater degree of metabolic stability (LipMetE 0.024–3.85). Theophylline exhibited a LipMetE of 3.05 mainly due to its poor in vitro clearance (log10Clint,u −3.08) and lower log D7.4 (−0.03) as compared to log D7.4 and in vitro clearance values of the remaining CYP3A4 substrates. Docetaxel, loperamide, and mifepristone displayed higher LipMetE values of 3.04/3.18/3.84 mainly due to higher log D7.4 (3.54/3.94/5.19) values. The CYP3A4 substrate, meloxicam, exhibited a LipMetE value of 2.5 with log D7.4 and log10CLint,u values of 1.04 and −1.51, which reflects an overall balance of the LipMetE, log D7.4, and in vitro clearance profile (Figure 1e).
Furthermore, it is also important to establish meaningful relationships between the calculated parameters presented in this study. Generally, it is shown that the metabolic clearance of lipophilic compounds increases with an increase in lipophilic character due to the lipophilic substrate binding site of cytochrome P450 enzymes.48 Similarly, a reduction in lipophilicity normally leads to reduced metabolic clearance and greater metabolic stability; yet there is considerable variation in this correlation in various investigations. For a series of dihydropyridine calcium channel blockers, a direct correlation (r = 0.87) has been observed between lipophilicity and plasma clearance.49 Similarly, Rand et al. demonstrated a considerable trend of increased metabolic clearance with an increase in log D7.4 values, while optimizing the pharmacokinetic and structural properties for a series of 16 cyclic peptides having diverse therapeutic properties.50 Additionally, a direct correlation has also been established between the log D7.4 and metabolic clearance values in humans for a set of neutral compounds with metabolic clearance values in the range 0.01–1000 mL/min/kg and log D7.4 values between −2 and 5.51 However, a weak trend toward increase in clearance with lipophilicity in humans has also been noted for 670 intravenously administered drugs and clinical candidates.52
Herein, a direct correlation between log D7.4 and intrinsic clearance has been identified in the CYP3A4 substrates, including the marketed drugs that reside in the LipMetE range between 0 and 1 as well as for those substrates that reside in the LipMetE range of >2, as shown in Figure 1e. This is in line with the previous relationship established by Smith and Waterbeemd who explicated that metabolic clearance of the CYP3A4 substrates can be reduced by lowering the lipophilicity, irrespective of the structure or reaction types through which metabolism occurs. Moreover, they observed a very good correlation (r2 = 0.877) between log D7.4 and metabolic clearance values of 14 substrates of CYP3A4, mainly drug compounds.51,53 Overall, in the present investigation, a poor or no correlation has been observed between intrinsic clearance (log10CLint,u) and log D7.4 of the entire dataset of CYP450 substrates, as shown in Figure S3. However, known drugs and model substrates of the respective CYP450 subtypes show a direct correlation between intrinsic clearance (log10CLint,u) and log D7.4 within a given range of LipMetE values, as shown in Figure 2.
Figure 2.

Plot showing a direct correlation between intrinsic clearance (log10Clint,u) and log D7.4 values for the particular range of LipMetE for the marketed drugs and model substrates metabolized by CYP450s (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4).
Previously, several investigations have also reported the dependence of ADME properties on lipophilicity. Yoshida et al. analyzed a dataset of 232 drugs with human pharmacokinetics data and revealed higher bioavailability for compounds having log D7.4 values between 2 and 3.54 In contrast to this, a study by Paul Gleeson reported that no clear relationship exists between lipophilicity and oral bioavailability in rat for more than 4400 preclinical compounds. Moreover, a weak relationship between lipophilicity (c log P) and clearance has been observed with more lipophilic compounds clearing more rapidly, thus showing lower metabolic stability although some dependence on the ionization state of the compounds has been observed.9 A similar trend has been observed in our study, where an increase in metabolic stability (LipMetE) has been observed with a decrease in intrinsic clearance (log10CLint,u) of the CYP450 substrates, as shown in Figure 3. This may reflect that high lipophilicity is associated, in part, with increased metabolic rate by the CYP450 enzymes in liver microsomes, although many other factors are involved in clearance mechanisms.9
Figure 3.
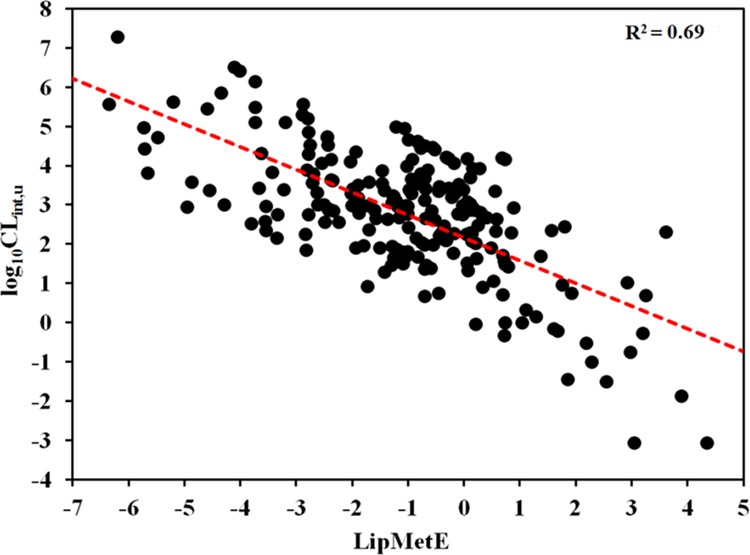
Relationship between metabolic stability (LipMetE) and in vitro clearance of the CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 substrate datasets.
2.2. Lipophilic Efficiency (LipE)
Lipophilic efficiency (LipE) profiling of the CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 substrates (Figure 4a–e) has been performed to normalize the lipophilicity with respect to the Michaelis–Menten constant (Km) values of the datasets (Table S2). The LipE and c log P distribution profiles of the respective CYP450 isoforms are provided in Figures S4 and S5, and the detailed LipE analysis of 58 CYP1A2 substrates is discussed in Section S1 of the Supporting Information. It has been observed that most of the drugs metabolized by CYP1A2 span the LipE range of −0.11 to 3.84 with Km values of 0.87–3440 μM and c log P values in the range −0.07 to 5.3 (Figure 4a, Table S2). This observation is retained with the exception of a few highly lipophilic marketed drugs, including amitriptyline and pimozide, that exhibit higher c log P (4.85/6.4), positive LipMetE (0.108/1.81) but negative LipE (−0.78/–1.96) values, which might be due to their higher c log P values. Overall, the difference in LipE values of the CYP1A2 substrate dataset, mainly drugs (−0.11 to 3.84) from the LipE value range (5–7), for an average oral drug defined by Leeson and Springthorpe11 against a drug therapeutic target might be due to the difference in potency (Ki, IC50) values of the drugs against the true therapeutic target as compared to Km values against CYP1A2. Thus, it reflects that the CYP1A2 substrates (drugs) exhibiting LipE values between 1 and 3 and c log P values in the range −0.07 to 5.3 may offer better metabolic reactions and metabolic stability.
Figure 4.

LipE profiling of (a) CYP1A2, (b) CYP2C9, (c) CYP2C19, (d) CYP2D6, and (e) CYP3A4 substrates. Data points labeled in red in LipE profiling plots indicate CYP450 substrates with metabolic stability between 0–4 (LipMetE) and optimal LipE values.
Our results for 55 CYP2C9 substrates indicate that about 27% of substrates that are principally metabolized by CYP2C9 exhibit LipE values ≥2, as shown in Figure 4b, with only three substrates (a dietary flavonoid, lornoxicam, and sildenafil) displaying LipE ≥3 (details in Section S1). However, the LipE values of metabolically stable CYP2C9 substrates (LipMetE: 0.066–2) vary from −0.83 to 2.72. The CYP2C9 substrates including meloxicam, 7-ethoxycoumarin, and kaempferide display LipMetE/LipE values of 1.05/2.73, 0.74/1.65, and 0.69/2.39, respectively. Generally, it is observed that the CYP2C9 substrates exhibiting LipMetE values from 0.0 to 2.5 lie within the LipE range of 0–2.7, whereas with an increase in LipMetE values above 3, a negative LipE value has been observed due to low intrinsic clearance. Thus, a compound with higher LipE might indicate more affinity toward the substrate binding sites of CYP2C9 that in turn shows more intrinsic clearance, lower metabolic stability, and rapid metabolism turnover, which may represent a clinically inadequate candidate.
The LipE profiling of the CYP2C19 substrates (Section S1, Table S2, and Figure 4c) revealed only one hexobarbital enantiomer with a LipE value of 5.13, c log P value of −0.78, Km value of 45 μM and a very low LipMetE value (−3.79). Similarly, three other hexobarbital enantiomeric CYP2C19 substrates exhibit LipE values >4 and LipMetE values between −5.69 and −4.86 (Figure 4c). Thus, this also reflects a trend of increase in LipE with decrease in metabolic stability, which is also observed for the substrates of CYP1A2 and CYP2C9. However, for the CYP2C19 substrates with higher LipMetE, lower LipE values were observed as shown for the low-affinity substrates flunitrazepam and diazepam (Km values of 89 000/74 000 μM) that exhibit exceptionally high LipMetE values (3.90/3.2), but the respective LipE values turn out to be −0.72 and −1.83. Considering only metabolically stable CYP2C19 substrates, zolpidem reflects a good balance of LipMetE (1.37), LipE (0.79), and c log P (3.03), which might display an overall better substrate profile.
For the CYP2D6 dataset of 73 substrates only one compound β-carboline harmaline presented a LipE of 5.14 (Figure 4d, Section S1, and Table S2). Debrisoquine exhibited a higher LipE of 4.73 (Km: 13.4, c log P: 0.14) and a negative LipMetE of −4.70. However, for other substrates including chlorpheniramine and the experimental N-substituted amphetamine analogues ((+) MDMA) and ((-) MDMA), LipMetE values of −1.8/–5.72/–5.47, LipE values of 3.65/4.0/3.64, and c log P values of 0.77/1.85/1.85 have been observed. A similar trend of increase in metabolic stability with decrease in LipE has been observed in the case of dextrorphan and loperamide, which show LipMetE values in the range of 2.19–2.92 and LipE values between −1.14 and 0.89. Overall, for the CYP2D6 substrates with better metabolic stability (LipMetE: 1–2), it was observed that the LipE values lie between 0.91 and 2.17 with the exception of imipramine, which shows a LipMetE of 1.94 and a negative LipE (−0.15), mainly due to its high c log P (5.04) and high affinity Km of 12.9 μM.
The detailed LipE analysis of the CYP3A4 substrates is discussed in Section S1 and Table S2 of the Supporting Information. The CYP3A4 substrate, etoposide, exhibits a LipE of 4.08 and poor metabolic stability (LipMetE: −1.18). Additionally, loperamide and theophylline display very high metabolic stability (LipMetE: 3.26/3.05) and low LipE (0.54/1.63) values. Thus, it further strengthens our observation of increase in metabolic stability with decrease in LipE of the CYP1A2, 2C9, 2C19, 2D6, and CYP3A4 substrates, as shown in Figure S6. Therefore, the optimization of a compound’s lipophilicity may guide to improve bioavailability- and clearance-associated problems. Toward this goal, Nassar et al. reported different strategies for enhancing metabolic stability, which includes reduction of the overall lipophilicity of a compound and addition or modification of metabolically labile groups.55 However, metabolic stability problems solved by applying structural modifications might not necessarily lead to a compound with enhanced pharmacokinetic properties. Therefore, optimization of lipophilicity that supports good bioavailability, metabolic stability, clearance, and binding affinity of a substrate with the respective enzyme (Km) might assist in achieving the highest quality clinical candidate.
2.3. Ligand Efficiency (LE)
Finally, the ligand efficiency (LE) values for substrates with suitable metabolic stability and clearance have been evaluated to estimate the binding free energies of compounds within the substrate binding sites of the respective CYP450 subtypes. Binding free energies (ΔG) and LE ranges of the respective CYP450 substrates, including marketed drugs, are summarized in Table 2. Overall, for our CYP450 substrate datasets, LE values from 0.065 to 1.42 (Figure S7 and Table S2) and ΔG values from −10.45 to −1.49 (Table S2) have been observed. Hopkins et al. reported an LE value of 0.29 kcal/mol/HA for an average oral drug, exhibiting an optimal fit within the binding site of the respective therapeutic target.36 However, the metabolically stable substrates of the respective CYP450 isoforms in our datasets show LE values between 0.064 and 0.6 with ΔG values from −8.23 to −1.48, which depicts their conducive fit within the substrate binding site of CYP450.
Table 2. Table Summarizing LipE, ΔG, LE, and c log P Ranges for the CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 Substrate Datasets That Were Used for LipE and LE Profilinga.
| known
drugs and model substrates data |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| CYP450 | no of substrates | LipE range | ΔG range | LE range | c log P range | no of substrates | average LipE | average LE | average c log P |
| CYP1A2 | 58 | –1.96 to 7.24 | –9.09 to −3.48 | 0.185–1.42 | –2.8 to 6.4 | 35 | 1.62 | 0.35 | 2.68 |
| CYP2C9 | 55 | –1.02 to 5.00 | –9.04 to −3.74 | 0.187–0.75 | 0.23 to 5.68 | 39 | 1.68 | 0.32 | 3.06 |
| CYP2C19 | 60 | –4.21 to 5.13 | –10.0 to −1.49 | 0.064–0.50 | –0.78 to 5.59 | 35 | 1.08 | 0.29 | 3.19 |
| CYP2D6 | 73 | –1.18 to 5.14 | –11.25 to −2.86 | 0.12–0.63 | –0.04 to 7.41 | 46 | 1.57 | 0.32 | 3.02 |
| CYP3A4 | 101 | –2.52 to 6.66 | –10.45 to −2.26 | 0.1–1.28 | –2.8 to 7.41 | 75 | 1.13 | 0.24 | 3.23 |
The average values of LipE, ΔG, LE, and c log P against each CYP isoform were calculated using all marketed drugs and model substrates included in each CYP substrate dataset.
The analysis of successful drugs that are efficiently metabolized by CYP450 subtypes facilitated the favorable thresholds of LipMetE, LipE, and LE for an average oral drug with a suitable metabolic profile. Generally, for the CYP450 substrates, the LipMetE of 0–2.5, LipE of −0.50 to 3, LE >0.25, and c log P of 1–3 might reflect important thresholds for the suitable metabolic parameters of a new chemical entity. The CYP450 substrates from all datasets are presented in Figure 5 as points in the 3D space mapped according to the respective LipE, LipMetE, LE, and c log P properties. Figure 6 shows our CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 datasets, where compounds fulfilling the established thresholds are color-coded in green. However, a deviation for the substrates of each CYP450 subtype from these thresholds is depicted by red color. Most prominently, the study identifies the CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 substrates representing optimal metabolic properties. The identified substrates with suitable metabolic attributes for the selected CYP450 subtypes are presented in Table 3 (Figure 6). The current study estimates the overall properties of CYP450 substrates through the calculation of efficiency metrics. Therefore, these parameters might serve as valuable tools to aid the selection of high-quality or drug-like clinical candidates through the optimization of lipophilicity, which supports the proposed metabolic stability, clearance, and binding affinity of a substrate with the respective enzyme (Km).
Figure 5.
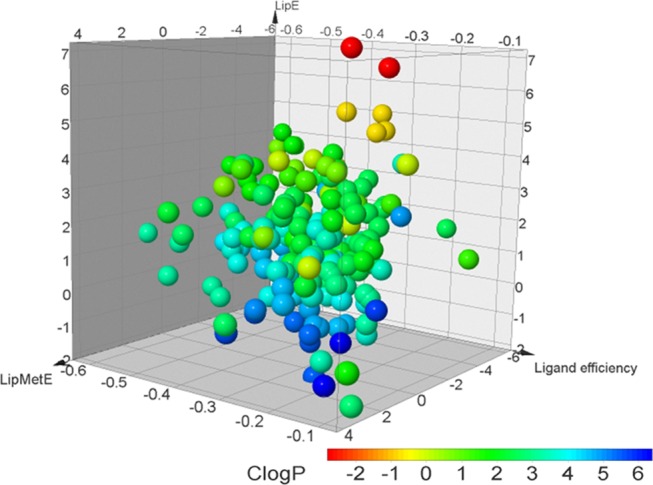
CYP450 substrates represented as points in the 3D space mapped according to the respective LipE, LipMetE, ligand efficiency values and points color-coded according to the c log P values.
Figure 6.

CYP450 substrates represented in the 2D heat map according to the thresholds derived from marketed drugs and model substrates for c log P, LipE, LipMetE, and LE values. Green lines depict the compounds exhibiting our established threshold values of LipMetE (0–2.5), LipE (−0.50 to 3), LE (>0.25), and c log P (1–3). Red color indicates compounds that show deviation from the optimized thresholds of LipMetE, LipE, LE, and c log P.
Table 3. CYP1A2, CYP2C9, CYP2D6, and CYP3A4 Substrates Representing Optimal Metabolic Properties.
| CYP subtype | substrates | LipE | LipMetE | LE | c log P |
|---|---|---|---|---|---|
| CYP1A2 | galangin, kaempferide, mexiletine, and mirtazapine | 1.10–2.84 | 0.08–0.70 | 0.27–0.52 | 2.57–2.81 |
| CYP2C9 | 7-ethoxycoumarin, kaempferide, meloxicam, and mirtazapine | 1.09–2.72 | 0.50–1.05 | 0.28–0.39 | 2.27–2.81 |
| CYP2C19 | zolpidem and 7-ethoxycoumarin | 0.79–0.95 | 0.15–1.38 | 0.23–0.32 | 2.27–3.03 |
| CYP2D6 | 7-ethoxycoumarin, dextromethorphan, promethazine, zolpidem, desipramine, R-PPF, S-PPF, imipramine, and dextrorphan | 0.21–2.19 | 0.03–2.17 | 0.14–0.39 | 2.27–4.47 |
| CYP3A4 | meloxicam, eplerenone, simvastatin lactone, dexamethasone, midazolam, valspodar, atorvastatin lactone, K11777, fenthion, nefazodone, 7-ethoxycoumarin, acetaminophen, perphenazine, laquinimod, clopidogrel, clozapine, triazolam, erythromycin, zolpidem, tacrolimus, azelastine, etoperidone, mirtazapine, carbamazepine, and naphthalene | 0.002–2.55 | 0.023– 3.06 | 0.09–0.59 | 0.49–5.78 |
3. Conclusions
Drug efficiency metrics have commonly been applied to compounds with potency values (Ki, IC50) against one or multiple therapeutic targets. Herein, these efficiency metrics have been applied to the antitargets, CYP450 enzymes, to probe the overall metabolic efficiency in terms of metabolic stability (LipMetE), in vitro clearance, and log P/log D7.4. The optimization of lipophilic metabolic stability (LipMetE) of a new chemical entity (NCE) with respect to lipophilicity and in vitro clearance may advocate a reasonable concept to estimate the CYP450 substrate properties. Therefore, LipMetE calculations have been performed to provide a LipMetE threshold for the CYP450 substrates with suitable metabolism, intrinsic clearance, and lipophilicity that may offer better metabolic properties of new chemical entities. Here, we propose a LipE threshold of ≤3, LipMetE of 0.0–2.5, and c log P of 1–3 for metabolically suitable compounds. Substrate binding sites of CYP450s are considered lipophilic in nature, and thus, within a given range of LipMetE, an increase in metabolic clearance has been achieved with an increase in lipophilic character of substrates. Our results also demonstrate that if a compound shows the already established threshold of lipophilic efficiency (5–7) against an antitarget, such as CYP450, it may show more affinity toward the substrate binding site that in turn shows lower metabolic stability and rapid metabolism turnover, which may not be suitable for clinical investigations. Overall, our study estimated an approximate drug metabolic stability from its log D7.4 and intrinsic clearance values, although it is important to emphasize that other factors might also be involved in binding affinity/metabolic stability, such as polarity, hydrogen bond donor and/or acceptor properties, and the number of aromatic rings present in the molecule. Nevertheless, the consideration of the calculated parameters could facilitate the process of NCE candidate selection during drug discovery and, thus, aid in the overall right safety of the five-“R” concept.
4. Materials and Methods
4.1. Database Collection
A dataset of 291 CYP450 substrates, including 43 CYP1A2, 43 CYP2C9, 54 CYP2C19, 65 CYP2D6, and 86 CYP3A4 substrates, with known Km (0.011–89 000 μM), Vmax values (0.973–5.791 × 10+14 pmol/min/mg), and protein concentration (Cprot (1.2 × 10–11–2 mg/mL)) was used for LipMetE calculations (Table S2). As the LipE and LE calculations do not require Vmax and Cprot values, more compounds exhibiting Km values were added to the existing datasets for LipE and LE profiling. Figure S8 gives an overall representation of the datasets, including marketed drugs, model substrates and other substrates, used for LipMetE, LipE, and LE calculations. The overall distribution of Km values against each CYP450 subtype indicates that a greater number of high-affinity substrates (Km 0–100 μM) are present in each dataset in comparison to low-affinity substrates (Km > 100 μM), as shown in Figure S9.
4.2. Lipophilic Metabolic Efficiency (LipMetE)
LipMetE is a novel parameter that takes into account the intrinsic clearance of a compound to decipher the relationship between compound lipophilicity and metabolic stability. Since datasets for LipMetE profiling were extracted from various studies, unit conversion calculations have been performed to homogenize our datasets (Km (μM), Vmax (pmol/min/mg), and Cprot (mg/mL)). The intrinsic clearance (CLint,app) was calculated for each CYP450 substrate dataset using eq 1, as described by Rane et al.56
| 1 |
To correct the intrinsic clearance parameter for nonspecific binding (CLint,u), the unbound fraction in microsomal incubations (Fuinc) was calculated using eq 2 retrieved from Halifax and Houston’s empirical model based on physicochemical properties.57 Finally, CLint,u was calculated for the entire datasets using eq 3.37
| 2 |
| 3 |
Log D7.4 values were obtained from ChemSpider (http://www.chemspider.com/), which were determined using ACD/Labs Percepta Platforms PhysChem module.58 The lipophilic metabolic efficiency (LipMetE) parameter was calculated as described by Stepan et al.37 (eq 4)
| 4 |
4.3. Lipophilic Efficiency (LipE)
Lipophilicity plays an important role in determining the ADME-Tox properties and binding affinity of compounds to their targets. Therefore, lipophilic efficiency (LipE) metric takes into account both potency and lipophilicity for the evaluation of a compound’s drug-likeness.36 Herein, this concept was applied to substrates of the selected CYP450 subtypes (antitargets) having major contribution in drug metabolism. LipE profiling was performed to identify better substrates of the CYP450 family of enzymes with best Km and lipophilicity ratio (eq 5).
| 5 |
The Bio-Loom software package59 was used for calculating the c log P values of the entire datasets, whereas the LipE calculations were performed using the Excel spreadsheet.
4.4. Ligand Efficiency (LE)
Ligand efficiency (LE) metric has been originally used to decipher a ligand’s affinity toward its target and is measured as the ratio of binding free energy (ΔG) to the number of heavy atoms (HA).60 Binding affinity of the CYP450 substrates has been associated with the metabolic turnover and body clearance.61 Therefore, to normalize binding affinities of the CYP450 substrates with respect to heavy atom count, the LE values for the entire substrate datasets have been computed, as explained by Hopkins et al. and Jabeen et al.60,62Equation 6 was used to calculate the free energies (ΔG) for the CYP450 substrates, as explained by Kuntz et al.63 The equation is based on the assumption that enzyme–substrate dissociation constant (Kd) is approximately equal to the kinetic parameter, Km. Therefore, the Km values can be substituted for the Kd values, as described by Lewis et al. and Bauer et al.8,64
| 6 |
A temperature of 310 K was used to compute the ligand efficiencies in kcal/mol/heavy atom. LE profiling was performed using the expression (eq 7)
| 7 |
ΔG and LE values for the substrates of each CYP450 subtype are shown in the Supporting Information (Table S2). The Excel spreadsheet was used to perform all LE calculations.
Acknowledgments
The authors would like to acknowledge the Higher Education Commission (HEC), Pakistan, for their support and assistance.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b02344.
LipMetE, LipE, and LE calculations (Table S2) (XLSX)
Table S1: Drug classes of the CYP450 substrates used for the LipMetE calculations; Section S1: Lipophilic efficiency (LipE) profiling of the CYP450 substrate datasets; Figure S1: LipMetE distribution of the CYP450 substrate datasets; Figure S2: log D7.4 distribution across the CYP450 substrate datasets; Figure S3: Correlation between intrinsic clearance (log10CLint,u) and log D7.4 of the entire dataset of the CYP450 substrates; Figure S4: Distribution of LipE values across each dataset of the CYP450 substrates; Figure S5: Distribution of c log P values across each dataset of the CYP450 substrates; Figure S6: Relationship between metabolic stability (LipMetE) and lipophilic efficiency (LipE) of the CYP450 substrate datasets; Figure S7: Distribution of LE values across each dataset of the CYP450 substrates; Figure S8: Bar chart representation of marketed drugs, model substrates, and other substrates included in the CYP substrate datasets used for LipMetE, LipE, and LE profiling; and Figure S9: Distribution of Km values across each dataset of the CYP450 substrates (PDF)
Author Contributions
Y.S.K. and I.J. conceived and designed the project. Y.S.K. and I.J. carried out all work, analyzed the results, and wrote the manuscript. Both authors have reviewed the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Guengerich F. P.; MacDonald J. S. Applying mechanisms of chemical toxicity to predict drug safety. Chem. Res. Toxicol. 2007, 20, 344–369. 10.1021/tx600260a. [DOI] [PubMed] [Google Scholar]
- Cook D.; Brown D.; Alexander R.; March R.; Morgan P.; Satterthwaite G.; Pangalos M. N. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat. Rev. Drug Discovery 2014, 13, 419. 10.1038/nrd4309. [DOI] [PubMed] [Google Scholar]
- Morgan P.; Brown D. G.; Lennard S.; Anderton M. J.; Barrett J. C.; Eriksson U.; Fidock M.; Hamrén B.; Johnson A.; March R. E.; et al. Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat. Rev. Drug Discovery 2018, 167–181. 10.1038/nrd.2017.244. [DOI] [PubMed] [Google Scholar]
- Livezey M.; Nagy L. D.; Diffenderfer L. E.; Arthur E. J.; Hsi D. J.; Holton J. M.; Lowe Furge L. Molecular analysis and modeling of inactivation of human CYP2D6 by four mechanism based inactivators. Drug Metab. Lett. 2012, 6, 7–14. 10.2174/187231212800229318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. 10.1021/tx700079z. [DOI] [PubMed] [Google Scholar]
- Kumar G. N.; Surapaneni S. Role of drug metabolism in drug discovery and development. Med. Res. Rev. 2001, 21, 397–411. 10.1002/med.1016. [DOI] [PubMed] [Google Scholar]
- Nassar A.-E. F.; Kamel A. M.; Clarimont C. Improvingthe decision-making process in structural modification of drug candidates: reducing toxicity. Drug Discovery Today 2004, 9, 1055–1064. 10.1016/S1359-6446(04)03297-0. [DOI] [PubMed] [Google Scholar]
- Lewis D. F.; Dickins M. Baseline lipophilicity relationships in human cytochromes P450 associated with drug metabolism. Drug Metab. Rev. 2003, 35, 1–18. 10.1081/DMR-120018245. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- Arnott J. A.; Planey S. L. The influence of lipophilicity in drug discovery and design. Expert Opin. Drug Discovery 2012, 7, 863–875. 10.1517/17460441.2012.714363. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Dudek A. Z.; Arodz T.; Galvez J. Computational methods in developing quantitative structure-activity relationships (QSAR): a review. Comb. Chem. High Throughput Screening 2006, 9, 213–228. 10.2174/138620706776055539. [DOI] [PubMed] [Google Scholar]
- Liu X.; Shen Q.; Li J.; Li S.; Luo C.; Zhu W.; Luo X.; Zheng M.; Jiang H. In silico prediction of cytochrome P450-mediated site of metabolism (SOM). Protein Pept. Lett. 2013, 20, 279–289. 10.2174/0929866511320030006. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Chen Q.; Li L.; Angela Liu L.; Wei D.-Q. In silico prediction of cytochrome P450-mediated drug metabolism. Comb. Chem. High Throughput Screening 2011, 14, 388–395. 10.2174/138620711795508412. [DOI] [PubMed] [Google Scholar]
- de Groot M. J.; Ackland M. J.; Horne V. A.; Alex A. A.; Jones B. C. Novel approach to predicting P450-mediated drug metabolism: development of a combined protein and pharmacophore model for CYP2D6. J. Med. Chem. 1999, 42, 1515–1524. 10.1021/jm981118h. [DOI] [PubMed] [Google Scholar]
- Zamora I.; Afzelius L.; Cruciani G. Predicting drug metabolism: a site of metabolism prediction tool applied to the cytochrome P450 2C9. J. Med. Chem. 2003, 46, 2313–2324. 10.1021/jm021104i. [DOI] [PubMed] [Google Scholar]
- Handa K.; Nakagome I.; Yamaotsu N.; Gouda H.; Hirono S. Three-Dimensional Quantitative Structure–Activity Relationship Analysis for Human Pregnane X Receptor for the Prediction of CYP3A4 Induction in Human Hepatocytes: Structure-Based Comparative Molecular Field Analysis. J. Pharm. Sci. 2015, 104, 223–232. 10.1002/jps.24235. [DOI] [PubMed] [Google Scholar]
- Jónsdóttir S. Ó.; Ringsted T.; Nikolov N. G.; Dybdahl M.; Wedebye E. B.; Niemelä J. R. Identification of cytochrome P450 2D6 and 2C9 substrates and inhibitors by QSAR analysis. Bioorg. Med. Chem. 2012, 20, 2042–2053. 10.1016/j.bmc.2012.01.049. [DOI] [PubMed] [Google Scholar]
- Cheng F.; Yu Y.; Shen J.; Yang L.; Li W.; Liu G.; Lee P. W.; Tang Y. Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J. Chem. Inf. Model. 2011, 51, 996–1011. 10.1021/ci200028n. [DOI] [PubMed] [Google Scholar]
- Hammann F.; Gutmann H.; Baumann U.; Helma C.; Drewe J. Classification of cytochrome P450 activities using machine learning methods. Mol. Pharmaceutics 2009, 6, 1920–1926. 10.1021/mp900217x. [DOI] [PubMed] [Google Scholar]
- Schuster D.; Laggner C.; Steindl T. M.; Langer T. Development and validation of an in silico P450 profiler based on pharmacophore models. Curr. Drug Discovery Technol. 2006, 3, 1–48. 10.2174/157016306776637609. [DOI] [PubMed] [Google Scholar]
- Yu R.; Wang J.; Wang R.; Lin Y.; Hu Y.; Wang Y.; Shu M.; Lin Z. Combined Pharmacophore Modeling, 3D-QSAR, Homology Modeling and Docking Studies on CYP11B1 Inhibitors. Molecules 2015, 20, 1014–1030. 10.3390/molecules20011014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haji-Momenian S.; Rieger J. M.; Macdonald T. L.; Brown M. L. Comparative molecular field analysis and QSAR on substrates binding to cytochrome P450 2D6. Bioorg. Med. Chem. 2003, 11, 5545–5554. 10.1016/S0968-0896(03)00525-X. [DOI] [PubMed] [Google Scholar]
- Andrade C. H.; Silva D. C.; Braga R. C. In silico Prediction of Drug Metabolism by P450. Curr. Drug Metab. 2014, 15, 514–525. 10.2174/1389200215666140908102530. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Wang Y.; Hu T.; Or P. M.; Wong J.; Kwan Y. W.; Wan D. C.; Hoi P. M.; Lai P. B.; Yeung J. H. Enzyme kinetic and molecular docking studies for the inhibitions of miltirone on major human cytochrome P450 isozymes. Phytomedicine 2013, 20, 367–374. 10.1016/j.phymed.2012.09.021. [DOI] [PubMed] [Google Scholar]
- Teixeira V. H.; Ribeiro V.; Martel P. J. Analysis of binding modes of ligands to multiple conformations of CYP3A4. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 2036–2045. 10.1016/j.bbapap.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Rouse S. L.; Sansom M. S.; Mulholland A. J. A multiscale approach to modelling drug metabolism by membrane-bound cytochrome P450 enzymes. PLoS Comput. Biol. 2014, 10, e1003714 10.1371/journal.pcbi.1003714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrychova T.; Berka K.; Navratilova V.; Anzenbacher P.; Otyepka M. Dynamics and hydration of the active sites of mammalian cytochromes P450 probed by molecular dynamics simulations. Curr. Drug Metab. 2012, 13, 177–189. 10.2174/138920012798918408. [DOI] [PubMed] [Google Scholar]
- Masetti M.; Cavalli A.; Recanatini M. Modeling the hERG potassium channel in a phospholipid bilayer: Molecular dynamics and drug docking studies. J. Comput. Chem. 2008, 29, 795–808. 10.1002/jcc.20842. [DOI] [PubMed] [Google Scholar]
- Jones P. M.; George A. M. Mechanism of ABC transporters: a molecular dynamics simulation of a well characterized nucleotide-binding subunit. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 12639–12644. 10.1073/pnas.152439599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Testa B.; Fahr A. Lipophilicity and its relationship with passive drug permeation. Pharm. Res. 2011, 28, 962–977. 10.1007/s11095-010-0303-7. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Defining optimum lipophilicity and molecular weight ranges for drug candidates—molecular weight dependent lower logD limits based on permeability. Bioorg. Med. Chem. Lett. 2009, 19, 2844–2851. 10.1016/j.bmcl.2009.03.109. [DOI] [PubMed] [Google Scholar]
- Rutkowska E.; Pajak K.; Jóźwiak K. Lipophilicity--methods of determination and its role in medicinal chemistry. Acta Pol. Pharm. 2012, 70, 3–18. [PubMed] [Google Scholar]
- Gleeson M. P.; Hersey A.; Montanari D.; Overington J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discovery 2011, 10, 197–208. 10.1038/nrd3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meanwell N. A. Improving drug design: an update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem. Res. Toxicol. 2016, 29, 564–616. 10.1021/acs.chemrestox.6b00043. [DOI] [PubMed] [Google Scholar]
- Hopkins A. L.; Keserü G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Kauffman G. W.; Keefer C. E.; Verhoest P. R.; Edwards M. Evaluating the differences in cycloalkyl ether metabolism using the design parameter “lipophilic metabolism efficiency”(LipMetE) and a matched molecular pairs analysis. J. Med. Chem. 2013, 56, 6985–6990. 10.1021/jm4008642. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Johnson D. S.; Humphrey J. M.; Butler T. W.; am Ende C. W.; Fish B. A.; Green M. E.; Kauffman G. W.; Mullins P. B.; O’Donnell C. J.; et al. Design of pyridopyrazine-1, 6-dione γ-secretase modulators that align potency, MDR efflux ratio, and metabolic stability. ACS Med. Chem. Lett. 2015, 6, 596–601. 10.1021/acsmedchemlett.5b00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Sosa A. T.; Sild S.; Takkis K.; Maran U. Combined approach using ligand efficiency, cross-docking, and antitarget hits for wild-type and drug-resistant Y181C HIV-1 reverse transcriptase. J. Chem. Inf. Model. 2011, 51, 2595–2611. 10.1021/ci200203h. [DOI] [PubMed] [Google Scholar]
- Conner K. P.; Woods C. M.; Atkins W. M. Interactions of cytochrome P450s with their ligands. Arch. Biochem. Biophys. 2011, 507, 56–65. 10.1016/j.abb.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M. P.; Price D. A.. Role of Physicochemical Properties and Ligand Lipophilicity Efficiency in Addressing Drug Safety Risks. In Annual Reports in Medicinal Chemistry; Elsevier, 2010; Vol. 45, pp 380–391. [Google Scholar]
- Hughes J. D.; Blagg J.; Price D. A.; Bailey S.; DeCrescenzo G. A.; Devraj R. V.; Ellsworth E.; Fobian Y. M.; Gibbs M. E.; Gilles R. W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- Stepan A. F.; Walker D. P.; Bauman J.; Price D. A.; Baillie T. A.; Kalgutkar A. S.; Aleo M. D. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 2011, 24, 1345–1410. 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- Pettersson M.; Johnson D. S.; Rankic D. A.; Kauffman G. W.; am Ende C. W.; Butler T. W.; Boscoe B.; Evrard E.; Helal C. J.; Humphrey J. M.; et al. Discovery of cyclopropyl chromane-derived pyridopyrazine-1, 6-dione γ-secretase modulators with robust central efficacy. MedChemComm 2017, 730–743. 10.1039/C6MD00406G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersson M.; Hou X.; Kuhn M.; Wager T. T.; Kauffman G. W.; Verhoest P. R. Quantitative Assessment of the Impact of Fluorine Substitution on P-Glycoprotein (P-gp) Mediated Efflux, Permeability, Lipophilicity, and Metabolic Stability. J. Med. Chem. 2016, 59, 5284–5296. 10.1021/acs.jmedchem.6b00027. [DOI] [PubMed] [Google Scholar]
- Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, baevolutionary aspects and functional diversity. Pharmacogenomics J. 2005, 5, 6–13. 10.1038/sj.tpj.6500285. [DOI] [PubMed] [Google Scholar]
- Leonardi A.; Motta G.; Testa R.. Adrenergic receptor antagonists selective for both alpha1A-and alpha1D-subtypes and uses therefor. Google Patents: US20020183290A12002.
- Van De Waterbeemd H.; Smith D. A.; Beaumont K.; Walker D. K. Property-based design: optimization of drug absorption and pharmacokinetics. J. Med. Chem. 2001, 44, 1313–1333. 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]
- Kato̅ R.; Estabrook R. W.; Cayen M. In Xenobiotic Metabolism and Disposition, Proceedings of the 2nd International ISSX Meeting, Kobe, Japan, May 16–20, 1988; Taylor & Francis, 1989; Vol. 2.
- Rand A. C.; Leung S. S.; Eng H.; Rotter C. J.; Sharma R.; Kalgutkar A. S.; Zhang Y.; Varma M. V.; Farley K. A.; Khunte B.; et al. Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance. MedChemComm 2012, 3, 1282–1289. 10.1039/c2md20203d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. A.; Van de Waterbeemd H.. Pharmacokinetics and Metabolism in Drug Design; John Wiley & Sons, 2012. [Google Scholar]
- Obach R. S.; Lombardo F.; Waters N. J. Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 670 drug compounds. Drug Metab. Dispos. 2008, 36, 1385–1405. 10.1124/dmd.108.020479. [DOI] [PubMed] [Google Scholar]
- Davis A. M.; Webborn P. J.; Salt D. W. Robust assessment of statistical significance in the use of unbound/intrinsic pharmacokinetic parameters in quantitative structure–pharmacokinetic relationships with lipophilicity. Drug Metab. Dispos. 2000, 28, 103–106. [PubMed] [Google Scholar]
- Yoshida F.; Topliss J. G. QSAR Model for Drug Human Oral Bioavailability 1. J. Med. Chem. 2000, 43, 2575–2585. 10.1021/jm0000564. [DOI] [PubMed] [Google Scholar]
- Nassar A.-E. F.; Kamel A. M.; Clarimont C. Improving the decision-making process in the structural modification of drug candidates: enhancing metabolic stability. Drug Discovery Today 2004, 9, 1020–1028. 10.1016/S1359-6446(04)03280-5. [DOI] [PubMed] [Google Scholar]
- Rane A.; Wilkinson G.; Shand D. Prediction of hepatic extraction ratio from in vitro measurement of intrinsic clearance. J. Pharmacol. Exp. Ther. 1977, 200, 420–424. [PubMed] [Google Scholar]
- Hallifax D.; Houston J. B. Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab. Dispos. 2006, 34, 724–726. 10.1124/dmd.105.007658. [DOI] [PubMed] [Google Scholar]
- Pence H. E.; Williams A. ChemSpider: an online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. 10.1021/ed100697w. [DOI] [Google Scholar]
- Bio-Loom program, t. v., by BioByte Co.
- Hopkins A. L.; Groom C. R.; Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- Smart R. C.; Hodgson E.. Molecular and Biochemical Toxicology; Wiley, 2018. [Google Scholar]
- Jabeen I.; Pleban K.; Rinner U.; Chiba P.; Ecker G. F. Structure–activity relationships, ligand efficiency, and lipophilic efficiency profiles of benzophenone-type inhibitors of the multidrug transporter P-glycoprotein. J. Med. Chem. 2012, 55, 3261–3273. 10.1021/jm201705f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz I.; Chen K.; Sharp K.; Kollman P. The maximal affinity of ligands. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9997–10002. 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer C.; Osman A. M.; Cercignani G.; Gialluca N.; Paolini M. A unified theory of enzyme kinetics based upon the systematic analysis of the variations of k cat, K M, and k cat/K M and the relevant ΔG 0≠ values—possible implications in chemotherapy and biotechnology. Biochem. Pharmacol. 2001, 61, 1049–1055. 10.1016/S0006-2952(01)00579-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.