

Abstract
Treatment with immune checkpoint inhibitors (ICPIs) extends survival in a proportion of patients across multiple cancers. Tumor mutational burden (TMB)—the number of somatic mutations per DNA megabase (Mb)—has emerged as a proxy for neoantigen burden that is an independent biomarker associated with ICPI outcomes. Based on findings from recent studies, TMB can be reliably estimated using validated algorithms from next‐generation sequencing assays that interrogate a sufficiently large subset of the exome as an alternative to whole‐exome sequencing. Biological processes contributing to elevated TMB can result from exposure to cigarette smoke and ultraviolet radiation, from deleterious mutations in mismatch repair leading to microsatellite instability, or from mutations in the DNA repair machinery. A variety of clinical studies have shown that patients with higher TMB experience longer survival and greater response rates following treatment with ICPIs compared with those who have lower TMB levels; this includes a prospective randomized clinical trial that found a TMB threshold of ≥10 mutations per Mb to be predictive of longer progression‐free survival in patients with non‐small cell lung cancer. Multiple trials are underway to validate the predictive values of TMB across cancer types and in patients treated with other immunotherapies. Here we review the rationale, algorithm development methodology, and existing clinical data supporting the use of TMB as a predictive biomarker for treatment with ICPIs. We discuss emerging roles for TMB and its potential future value for stratifying patients according to their likelihood of ICPI treatment response.
Implications for Practice
Tumor mutational burden (TMB) is a newly established independent predictor of immune checkpoint inhibitor (ICPI) treatment outcome across multiple tumor types. Certain next‐generation sequencing‐based techniques allow TMB to be reliably estimated from a subset of the exome without the use of whole‐exome sequencing, thus facilitating the adoption of TMB assessment in community oncology settings. Analyses of multiple clinical trials across several cancer types have demonstrated that TMB stratifies patients who are receiving ICPIs by response rate and survival. TMB, alongside other genomic biomarkers, may provide complementary information in selecting patients for ICPI‐based therapies.
Keywords: Programmed cell death 1 receptor, Antibodies/therapeutic use, DNA mutational analysis, Genes, Neoplasm, Sequence analysis, DNA
Short abstract
Optimization of treatment with immune checkpoint inhibitors requires additional predictive biomarkers to establish which patients are most likely to benefit from such therapies. This review summarizes methodology and clinical data supporting tumor mutational burden as immunotherapy biomarker and complement to treatment selection.
Background
The development and therapeutic potential of immune checkpoint inhibitors (ICPIs) has fundamentally changed cancer treatment paradigms across multiple tumor types. However, only a subset of patients treated with ICPIs experience durable clinical responses 1, 2. Ongoing optimization of ICPI treatment requires additional predictive biomarkers that further establish which patients are most likely to benefit from such therapies. Expression of programmed death‐ligand 1 (PD‐L1) assessed by immunohistochemistry (IHC) has been established as one biomarker that is predictive of ICPI response in many cancer types including non‐small cell lung cancer (NSCLC), urothelial carcinoma, and most recently triple‐negative breast cancer 3, 4, 5, 6. However, stratification by PD‐L1 IHC alone is insufficient to identify the patient population most likely to respond, and approved companion and complementary diagnostics for PD‐L1 expression are variable with respect to performance and reporting thresholds 7, 8, 9. Therefore, additional independent biomarkers that predict ICPI response are needed.
Broad genomic sequencing approaches that have an established role in identifying oncogenic alterations—whole‐exome sequencing (WES) and whole genome sequencing—have been applied to samples from ICPI clinical trials. These initial studies clearly suggested that patients with a higher number of somatic tumor mutations derived more benefit from ICPI therapy compared with patients who had fewer mutations 10. This has paved the way for the use of targeted next‐generation sequencing (NGS) panels to derive the same information while sequencing fewer DNA base pairs. When properly designed and sufficient in size, these assays can assess tumor mutational burden (TMB; the number of somatic mutations per megabase [Mb] of sequenced DNA), which is a novel biomarker and a newly established independent predictor of ICPI treatment outcome 11, 12. Targeted yet comprehensive NGS panels can accurately recapitulate the TMB assessment from WES, allowing for broader clinical use of this biomarker 12, 13.
In this review, we summarize the development of currently available methods for assessing TMB, the rationale for TMB to be used as an independent biomarker for ICPI treatment outcome across tumor types, the association between TMB and other aspects of the tumor microenvironment, efforts to prospectively evaluate TMB as a predictive biomarker, and the potential implications for utilizing TMB as a selection tool for ICPI treatment.
Cancer Immunotherapy
Therapeutic manipulation of either the innate or adaptive arms of the immune system, or both, to optimize recognition and elimination of tumors is broadly known as cancer immunotherapy. The role of the immune system in cancer has been recognized for decades, and anticancer immunotherapy can be divided into three general categories: cytokine‐targeting therapies (such as interleukins, interferons, and colony‐stimulating factors), cell‐based therapies (such as chimeric antigen receptor technologies), and ICPIs 14. ICPIs, one of the most rapidly growing categories of immune‐related agents developed for the treatment of cancer, are monoclonal antibodies that block key molecules in immune checkpoint pathways such as programmed cell death protein 1 (PD‐1) and cytotoxic T‐lymphocyte–associated protein 4 (CTLA‐4) 14. PD‐L1, which is expressed on both tumor cells and immune cells and binds PD‐1 to attenuate T‐cell activity, is also an immunotherapy target 14.
At the most basic level, tumor cells differ from normal cells because of pathogenic changes in cellular function, which are often a result of genomic alterations that are considered to be “driver” mutations 15. By contrast, the vast majority of alterations in tumoral DNA are considered “passenger” mutations, neither contributing to nor detracting from tumor growth 16. However, a subset of these passenger alterations will generate neoantigens at the protein level, which may be recognized by the patient's immune system as non‐self or foreign. Although only a portion of overall immune response, neoantigenicity, correlated with response to checkpoint blockade and augmentation of the immune response, is thought to be the mechanism by which ICPIs act on tumor tissue 17, 18, 19. TMB is a surrogate measure of neoantigenicity, which allows it to serve as a predictive biomarker in this context.
Measurement and Analytical Validation of TMB
TMB was originally measured using WES, and several studies demonstrated an association between WES‐derived TMB and ICPI outcomes 10, 20, 21, 22, 23, 24, 25, 26, 27, 28. Although these were encouraging results for patient selection, WES‐derived TMB currently has limited clinical utility in many patient care settings owing to a 6–8‐week sequencing time, the requirement for a matched normal sample, and associated costs. Estimating TMB using clinically validated, commercially available, targeted NGS‐based panels that sequence a sufficient subset of the exome presents an attractive alternate method for calculating TMB 12, 13, 29 (Fig. 1; 30). In general, WES methods typically count only nonsynonymous base substitutions that alter the amino acid sequence of a protein, inferring that there is a direct link between protein coding changes and the number of potential neoantigens within a tumor genome 10, 20, 21, 22, 23, 24, 25, 26, 27. However, targeted NGS panels for estimating TMB have taken more sophisticated approaches, including the incorporation of nonsynonymous and synonymous base substitutions and short insertion and deletion alterations in the calculation 13. Synonymous variants, which are variants that do not alter the amino acid sequence of a protein, are not assumed to generate neoantigens. Their presence, however, is indicative of a mutational process also likely to result in nonsynonymous variants, and their inclusion in the TMB algorithm effectively improves assay sensitivity by increasing the number of qualifying variants into the calculation 13.
Figure 1.
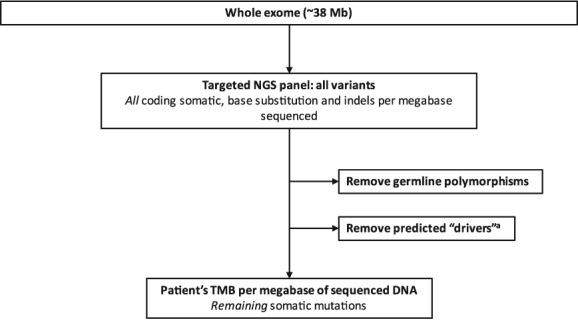
Example of targeted NGS panel‐based TMB calculation. Adapted from Spigel et al. 30. aPredicted drivers are mutations thought to be responsible for oncogenesis in a tumor.
Abbreviations: Mb, megabase; NGS, next‐generation sequencing; TMB, tumor mutational burden.
Although there are multiple platforms that have published data using TMB as a biomarker 31, 32, 33, to date only two products have gone through regulatory pathways: the FoundationOne CDx assay (Foundation Medicine, Inc., Cambridge, MA), which has been approved by the U.S. Food and Drug Administration (FDA) for the analytic calculation of TMB, and MSK‐IMPACT (Memorial Sloan Kettering Cancer Center, New York, NY), which has been authorized by the 510K pathway 31, 33. These panels have been optimized to identify all types of molecular alterations (i.e., single nucleotide variants, small and large insertion‐deletion alterations, copy number alterations, and structural variants) in cancer‐related genes, as well as genomic signatures such as microsatellite instability (MSI), loss of heterozygosity, and TMB, in a single test; this approach is collectively referred to as comprehensive genomic profiling (CGP) 34. As a result of the broad and deep coverage of targeted NGS panels across several hundred tumor genes and nontumor tissue, studies have demonstrated that TMB measurement using a CGP approach has high statistical concordance with TMB measured from WES 13. The two quality control metrics that should be considered when evaluating samples for TMB are median depth of sequencing coverage and coverage uniformity, as coverage is directly related to the sensitivity of calling both single nucleotide variants and indels, the two components that contribute to the TMB calculation 35. Foundation Medicine utilizes a minimum sequencing coverage metric of 250× median exon coverage and a uniformity metric of ≥95% of exons with at least 100× coverage 36.
Chalmers and colleagues 13 reported that a key determinant for the accuracy of the targeted NGS‐based TMB measurement is the number of megabases sequenced in the genome and that sequencing approximately 1.1 Mb over 315 genes resulted in a TMB estimate that was similar to the reference standard of WES. In this study, samples with 300× median exon coverage or greater were included. The study also estimated that sampling approximately 0.5 Mb or less resulted in an unacceptable degree of difference from the WES reference standard, suggesting that more limited assays may result in an inaccurate TMB calculation 13. Additional in silico analyses demonstrated acceptable agreement between targeted NGS‐derived and WES‐derived TMB data 37, 38, 39. In addition to establishing in silico accuracy against WES, additional key performance metrics such as reproducibility and repeatability of the TMB classifier, limit of detection according to the minimum tumor purity, and empirical accuracy against WES should be established to validate any TMB measurement currently being reported as a biomarker upon which therapeutic decisions will be based.
TMB as a Predictive Biomarker
There are several known carcinogenic processes linked to elevated TMB 10, 13. Exposure to environmental carcinogens, such as cigarette smoke and ultraviolet radiation, has been shown to cause cancers with the highest number of somatic mutations 13, 40, 41. Alterations in DNA mismatch repair (MMR) pathway–associated genes such as MSH2, MSH6, MLH1, and PMS2, which typically result in MSI, and alterations in DNA polymerase genes (POLE/POLD1) contribute to high TMB in some cancers 13. Chalmers and colleagues 13 have reported that a proportion of tumors across multiple cancer types have high TMB, showing that TMB has broad clinical validity as a biomarker. Figure 2 10, 11, 13, 21, 22, 31, 37, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 illustrates a timeline of the emergence of TMB as a biomarker.
Figure 2.
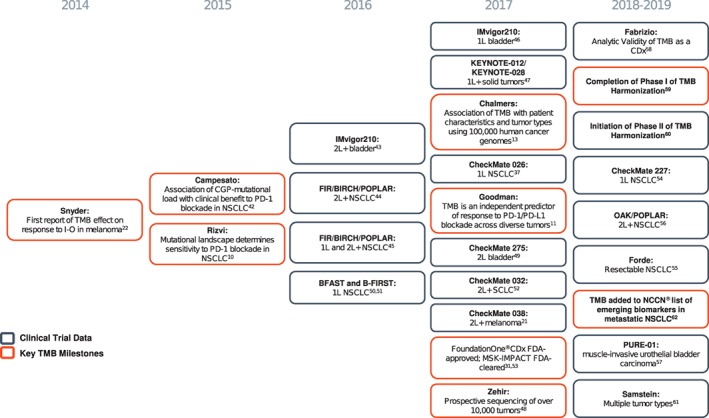
Timeline of TMB biomarker development.
Abbreviations: 1L, first‐line; 2L, second‐line; CGP, comprehensive genomic profiling; FDA, U.S. Food and Drug Administration; I‐O, immune‐oncology; NCCN, National Comprehensive Cancer Network; NSCLC, non‐small cell lung cancer; PD‐1, programmed cell death protein 1; PD‐L1, programmed death‐ligand 1; SCLC, small cell lung cancer; TMB, tumor mutational burden.
Clinical, pathologic, and molecular features influence the likely course for a given patient with cancer and predict the chance of response to a given therapy. An optimal therapeutic biomarker has analytic validity (accurate, reproducible, and reliable), clinical utility, economic feasibility, and a biologic basis 63. Retrospective analyses have demonstrated that a greater clinical benefit following treatment with ICPIs has been observed in patients with high TMB compared with those without high TMB; however, these studies used a variety of testing platforms and differing TMB cutoffs to define “high” levels rather than standardized, prospectively defined cutoffs 11, 25, 46, 54, 64, 65, 66. Early reporting of TMB included both the quantitative metric as mutations per Mb and qualitative description of high, intermediate, and low. Discrete cutoffs that define the biological subtype of cancer are currently being pursued, and such cutoffs will likely vary based on tumor type and therapeutic intervention 59, 60, 67, 68. In an analytic validation of FoundationOne CDx in patients with NSCLC, reproducibility and repeatability for TMB were shown to be 97.3% and 95.3% 58. Although the reproducibility and standardization of TMB calculations via NGS have yet to be thoroughly validated across other tumor types, efforts are currently underway to do so 60, 69.
Clinical Validation of TMB as an Independent Predictive Biomarker
Establishing whether any biomarker test, including a test for TMB, accurately and reliably separates patients into groups with distinct clinical or biological outcomes or differences (i.e., clinical validation) is an important factor for defining clinical use of a test 63.
The evidence of ICPI efficacy in MSI‐high disease led to the first tumor‐agnostic FDA approval for pembrolizumab for MSI‐high or MMR‐deficient tumors independent of anatomic origin 27, 28, 70. Additionally, the same study also found that high TMB was independently associated with longer progression‐free survival (PFS) 27. Across cancers, MSI‐high tumors are rare (∼1% as measured retrospectively by NGS, 5% in late stage colorectal cancer [CRC], 15% in endometrial/uterine cancer), and MSI assessment alone will fail to identify all patients who may benefit from ICPIs 13, 71, 72. Nearly all MSI‐high samples have high TMB and represent a subset of high TMB tumors across anatomic sites 13. However, MSI‐high is not sufficient to explain all instances of high TMB, even in tumor types where MSI is a well‐established biomarker such as CRC 73. For example, a TMB score of 12 mutations per Mb was shown to include 99.7% of all MSI‐high cases in patients with CRC, while also identifying an additional 3% of the much larger microsatellite‐stable population 73. As such, reclassification of CRC according to TMB effectively increased the number of patients eligible for ICPI therapy by more than 50% compared with MSI status alone 73. Although PD‐L1 overexpression is associated with improved ICPI response and is common among patients with high TMB levels, several studies have concluded that TMB and PD‐L1 expression are independent predictive biomarkers 12, 25, 26, 74.
The culmination of these clinical validation efforts is the identification of TMB cutoffs that predict ICPI outcome, demonstrating that TMB augments both MSI and PD‐L1 as an independent predictive biomarker for ICPI treatment. This was demonstrated in the NSCLC CheckMate‐227 trial, which found that TMB was clearly predictive of the PFS benefit observed in the combination nivolumab/ipilimumab arm and was not associated with improved PFS among patients receiving only chemotherapy 54. Elevated TMB was also observed to be predictive of improved survival in patients with any tumor type receiving ICPIs, although the TMB cutoffs varied markedly between cancer types 61, 73.
Additional Clinically Relevant Genomic Biomarkers Associated with TMB
TMB is an important treatment selection tool that complements existing molecular testing methodologies, PD‐L1, and other established and emerging oncogenes. Recurrent genomic alterations associated with elevated TMB, which together can be identified using a CGP approach, may provide additional biologic insights and inform therapy in select scenarios. For example, mutations in POLE are an emerging immunotherapy‐related biomarker that have been associated with very high TMB in multiple solid tumor types, including endometrial, CRC, gastric, melanoma, lung, and pediatric cancers 75, 76, 77, 78. POLE‐mutated, MSI‐high, and DNA MMR‐deficient CRC have each been associated with high TMB and favorable outcomes following treatment with ICPIs 27, 73, 79, 80. Therefore, tumors with pathogenic POLE mutations leading to elevated TMB may be good candidates for ICPI therapy independent of tumor type. Furthermore, as with MSI‐high, POLE‐mutated cancers represent only a subset of high TMB cancers, emphasizing the need to evaluate a broad set of biomarkers in order to capture all mechanisms of hypermutation.
Advanced NSCLC provides a particularly strong case for the use of CGP given that well‐established biomarkers, such as epidermal growth factor receptor (EGFR) mutations, anaplastic lymphoma kinase (ALK) alterations, and PD‐L1 expression, are present at different rates based on TMB levels. Each one of these biomarkers should be considered independently as well as together when making treatment decisions. In a sample of 9,347 NSCLC samples that underwent CGP (FoundationOne and FoundationOne CDx) and concurrent PD‐L1 testing, 18.0% were shown to be positive for ALK or EGFR alterations, 37.4% were TMB‐high (≥10 mutations/Mb), and 6.4% were PD‐L1 positive (data on file). However, there was minimal overlap between these molecular markers (Figs. 3 and 4). Because EGFR and ALK mutations are associated with low TMB and attenuated response rates to ICPIs, patients with tumors that are EGRF or ALK positive are ineligible for ICPI therapy in the first‐line setting according to FDA‐approved labeling. As discussed above, PD‐L1 and TMB are not mutually inclusive; thus both are needed to identify all patients who are likely to respond to ICPIs, whereas EGRF/ALK biomarker status will be needed to rule out those less likely to respond in the first‐line setting 12, 81, 82, 83.
Figure 3.
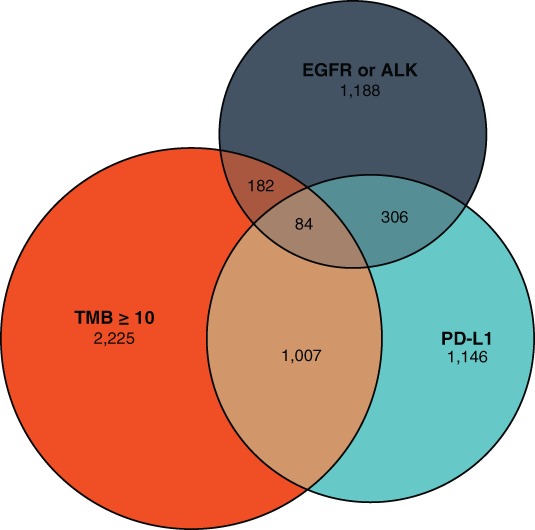
Interaction of high TMB with other cancer biomarkers. An analysis of Foundation Medicine's FoundationCore database (data on file) was undertaken to understand the relative prevalence of biomarkers that play a predictive role in immunotherapy decisions for patients with non‐small cell lung cancer (NSCLC). Through September 2018, there were 9,347 NSCLC samples with Foundation Medicine testing (FoundationOne and FoundationOne CDx) that also underwent PD‐L1 testing. The relative distribution of EGFR and/or ALK alterations, TMB ≥10 mutations per megabase, and PD‐L1 positive is shown here. Prevalence of each of the biomarkers in all patients with NSCLC (n = 35,370), regardless of PD‐L1 testing, was determined with EGFR alterations found in 14.1% and ALK alterations in 2.9%; this appears similar to the rates observed in the smaller subset of patients with concurrent PD‐L1 assessment. Overall, the overlap is limited, indicating a need to assess each of these biomarkers when making immunotherapy decisions in the NSCLC setting.
Abbreviations: ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; PD‐L1, programmed death‐ligand 1; TMB, tumor mutational burden.
Figure 4.

Degree of overlap between high TMB and PD‐L1 varies based on the presence of other alterations among patients with non‐small cell lung cancer (NSCLC). Among NSCLC samples with Foundation Medicine testing that also underwent PD‐L1 testing (n = 9,347; described in Fig. 3), the relative overlap between TMB ≥10 mutations per megabase and PD‐L1 is highest in patients with multiple genomic alterations as well as KRAS, BRAF, and MET alterations and lowest in patients with ALK and RET alterations.
Abbreviations: ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor; PD‐L1, programmed death‐ligand 1; TMB, tumor mutational burden.
Additionally, KRAS mutations have been associated with improved treatment outcomes in NSCLC 30, 82, 84, 85, and certain classes of alterations in JAK1, MDM2/MDM4, ARID1A, and STK11 have predicted a lack of response to ICPIs in a high TMB setting 12, 85, 86, 87, 88, 89, 90, 91. Initial data from studies utilizing targeted NGS panels have also suggested that certain BRAF and MET alterations are associated with longer duration of ICPI treatment, regardless of TMB status 88.
Overall, appropriate treatment selection in the era of genomically targeted therapies and immunotherapies will require insight into PD‐L1, TMB, MSI, as well as alterations in several individual genes. Looking at only a subset of these biomarkers could potentially result in suboptimal treatment among a considerable portion of patients and lead to inefficiencies in our health care ecosystem. Utilizing a CGP approach has the advantage of providing the data to generate composite biomarkers, which can be used collectively to further stratify patient populations most likely to derive maximal clinical benefit from both ICPIs and other genomically matched targeted treatments.
Utilizing a CGP approach has the advantage of providing the data to generate composite biomarkers, which can be used collectively to further stratify patient populations most likely to derive maximal clinical benefit from both ICPIs and other genomically matched targeted treatments.
Summary of Outcomes Associated with TMB
Clinical studies and observational data have further reinforced the notion of TMB as an independent biomarker that is predictive of ICPI outcomes 92. Increased nonsynonymous TMB from WES was first demonstrated as a predictor for ICPI treatment outcome by Snyder and colleagues 22 and Rizvi and colleagues 10 for CTLA‐4 and PD‐1/PD‐L1 inhibition, respectively. The relationship between WES‐derived TMB was further explored in the CheckMate‐032 study, wherein patients with tumors in the top tertile of TMB experienced 46.2% objective response rate (ORR) with nivolumab plus ipilimumab compared with 16.0% and 22.2% in the medium and low tertiles, respectively 26. As mentioned above, PD‐L1 expression and TMB have not been significantly correlated in most ICPI studies (Table 1). Reported and ongoing clinical trials and observational studies that have analyzed or will analyze TMB outcomes are summarized in Table 1 25, 27, 37, 45, 46, 54, 57, 61, 64, 74, 93, 94, 95, Table 2 10, 11, 12, 20, 22, 24, 65, 66, 96, and Table 3 97, 98, 99, 100, 101, 102.
Table 1.
ICPI clinical trials that have evaluated TMB as a biomarker
| Trial [reference]/study design/population | Intervention(s) | Type of sequencing for TMB | TMB cutpoint or highest threshold | TMB‐related results |
|---|---|---|---|---|
|
CheckMate‐227 54 Open‐label, randomized, phase III trial Advanced NSCLC n = 1,739 |
1st‐line nivolumab plus ipilimumab vs. platinum‐doublet chemotherapy | CGP | ≥10 mutations per Mb |
Nivolumab plus ipilimumab vs. chemotherapy—with TMB ≥10 mutations/Mb:
Nivolumab plus ipilimumab vs. chemotherapy—with TMB <10 mutations/Mb:
Treatment difference in TMB ≥10 mutations/Mb was consistent across PD‐L1 expression subgroups (<1% vs. ≥1%) |
|
CheckMate‐012 25 Prospective study Advanced NSCLC n = 75 |
1st‐line nivolumab plus ipilimumab | WES | > median of 158 mutations |
TMB > median vs. TMB < median:
TMB was independent of PD‐L1 expression (r = .087; p = .48) and most strongly associated with ORR (p = .001) and PFS (p = .002) (multivariable analysis). |
|
Open‐label, randomized, phase III trial Advanced NSCLC with PD‐L1 ≥ 1% n = 541 |
1st‐line nivolumab vs. doublet platinum chemotherapy | CGP | ≥243 somatic missense mutations per sample |
Nivolumab vs. chemotherapy—TMB ≥243 mutations subgroup:
Nivolumab vs. chemotherapy—TMB <243 mutations subgroup:
There was no association between PD‐L1 and TMB (all patients had PD‐L1 ≥ 1%). |
|
CheckMate‐032 25 Single‐arm, randomized phase I/II trial Advanced SCLC n = 211 |
Nivolumab plus ipilimumab, nivolumab alone | WES | ≥248 mutations (high tertile) |
The high vs. low TMB tertile were compared:
PD‐L1 expression ≥1% was rare and evenly distributed among the TMB tertiles. There was no association between PD‐L1 expression and TMB. |
|
CheckMate‐568 93 Single‐arm, phase II trial Advanced NSCLC n = 288 |
1st‐line nivolumab plus ipilimumab vs. platinum‐doublet chemotherapy | CGP | ≥10 mutations per Mb |
ORR at TMB cutpoints for nivolumab plus ipilimumab:
TMB ≥10 mutations/Mb was associated with enhanced response to nivolumab plus ipilimumab regardless of PD‐L1 expression |
|
BIRCH/FIR/POPLAR/OAK, IMvigor 210/211, PCD4989g 94 Retrospective study of tumor tissue samples from 7 monotherapy studies NSCLC (n = 342), advanced UC (n = 400), advanced solid tumors (n = 245) |
Atezolizumab | CGP | ≥16 mutations per Mb | Biomarker‐evaluable population vs. TMB ≥16 mutations/Mb vs. <16 mutations/Mb:
|
|
PURE‐01 57 Single‐arm, open‐label, phase II trial Muscle‐invasive urothelial bladder cancer n = 43 |
Neoadjuvant pembrolizumab before radical cystectomy | CGP | NA | Mean TMB in transurethral resection of the bladder resection samples was identical between patients with and without pathologic complete response 11.2 mutations/Mb vs. 11.2 mutations/Mb. |
|
Apache 95 Open‐label, randomized, 3‐stage, phase II trial Advanced germ cell tumors n = 18 |
Durvalumab alone or with tremelimumab | CGP | NA | Median TMB was 4 mutations/Mb and was not related to efficacy. |
|
Single‐arm, phase II trial Advanced UC n = 310 |
1st‐line atezolizumab | CGP | ≥16 mutations per Mba |
There was a greater proportion of TMB patients with ≥16 mutations/Mb among responders vs. nonresponders.
TMB ≥16 mutations/Mb was associated with increased expression of
Responders exhibited higher mean APOBEC3 expression. |
|
Le et al. (2015) 27 Single‐arm, phase II study Advanced pan‐tumor N = 41 |
Pembrolizumab | WES | NA |
Tumors with MSI‐high had high TMB levels vs non‐MSI tumors (p = .007). High TMB was associated with
|
|
POPLAR and OAK 56 Randomized, phase II trial Previously treated NSCLC OAK, n = 425; POPLAR, n = 287 |
2nd + line atezolizumab vs. docetaxel | Blood‐based CGP | ≥10 mutations per Mb | PFS and OS in patients with bTMB ≥10 mutations/Mb were higher than in the overall population. |
|
BIRCH/FIR, POPLAR 45 Single‐arm (BIRCH/FIR) and randomized (POPLAR) phase II trials NSCLC OAK, n = 425; POPLAR, n = 287; FIR, n = 138 |
1st/2nd + line atezolizumab (single‐arm) in BIRCH/FIR, 2nd‐line atezolizumab vs. docetaxel in POPLAR | CGP |
BIRCH/FIR: In 1st line, ≥13.5 mutations/Mb; in 2nd line or later, ≥17.1 mutations/Mb POPLAR: ≥15.8 mutations/Mb |
RR and OS were higher in patients with both TMB ≥9 mutations/Mb (median) and ≥ 13.5 mutations/Mb (high) vs <9 and <13.5 mutations/Mb, respectively. |
|
Samstein et al. (2019) 61 Retrospective study Multiple tumor types Treated with ICPI, n = 1,662; Non‐ICPI treated, n = 5,371 |
Atezolizumab, avelumab, durvalumab, ipilimumab, nivolumab, pembrolizumab or tremelimumab (monotherapy or in combination) | CGP | NA |
OS was higher in patients with high TMB (highest 20% in each cancer type), across entire cohort. TMB cutoff associated with the top 20% varied markedly between cancer types. |
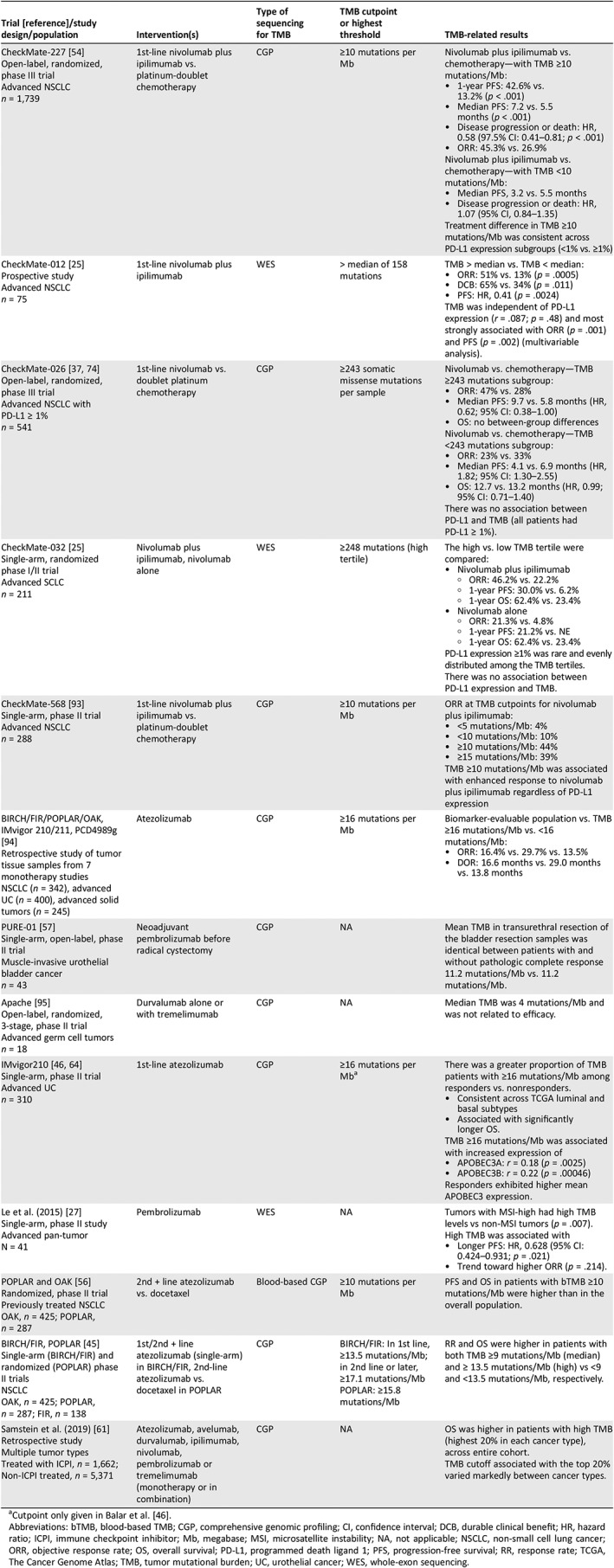
Cutpoint only given in Balar et al. 46.
Abbreviations: bTMB, blood‐based TMB; CGP, comprehensive genomic profiling; CI, confidence interval; DCB, durable clinical benefit; HR, hazard ratio; ICPI, immune checkpoint inhibitor; Mb, megabase; MSI, microsatellite instability; NA, not applicable; NSCLC, non‐small cell lung cancer; ORR, objective response rate; OS, overall survival; PD‐L1, programmed death ligand 1; PFS, progression‐free survival; RR, response rate; TCGA, The Cancer Genome Atlas; TMB, tumor mutational burden; UC, urothelial cancer; WES, whole‐exon sequencing.
Table 2.
Studies demonstrating the relationship between TMB and treatment outcome in patients with cancer
| Trial [reference]/study design/population | Intervention(s) | Type of sequencing for TMB | TMB cutpoint or highest threshold | TMB‐related results |
|---|---|---|---|---|
|
Eroglu et al. (2018) 20 Retrospective review of pathology reports Advanced desmoplastic melanoma n = 60 |
PD‐1 or PD‐L1 blockade therapy | WES | NA | Patients with desmoplastic melanoma had substantial clinical benefit from PD‐1 or PD‐L1 immune checkpoint blockade therapy likely resulting from high TMB, increased CD8 density, and high expression of PD‐L1 in tumor invasive margin (median follow‐up of 22 months).
|
|
Rizvi et al. (2018) 12 Prospective and retrospective study Advanced NSCLC n = 240 |
1st/2nd/3rd + line immunotherapy: Anti–PD‐1 or anti–PD‐L1 | CGP and WES | NA |
Median TMB:
TMB was stratified into increasing thresholds above vs. below the 50th percentile in patients treated with immunotherapy:
TMB was independent of PD‐L1 expression (r = .1915; p = .08). |
|
Greally et al. (2018) 65 Retrospective study of tumor tissue samples Esophagogastric cancer n = 120 |
Various immune checkpoint inhibitors | CGP | ≥7.4 mutations per Mb | High TMB vs. low TMB, OS: 27.1 months vs. 8.4 months (p = .063) |
|
Goodman et al. (2017) 11 Retrospective study of clinical records Locally advanced or metastatic pan‐tumor n = 151 |
Various immune checkpoint inhibitors | CGP | ≥20 mutations per Mb |
TMB ≥20 mutations/Mb vs. <20 mutations/Mb:
High TMB was independently associated with better outcome parameters (multivariable analysis). |
|
Rozenblum et al. (2017) 96 Retrospective cohort study Advanced lung cancer n = 33 |
Nivolumab or pembrolizumab | CGP | NA |
Response rate in patients treated with immunotherapy:
Patients who were not carrying any treatment‐associated driver (n = 17) had the highest mean TMB (11.8 ± 5 mutations/Mb) and the highest ORR to immunotherapy (33%). |
|
Johnson et al. (2016) 66 Retrospective study of tumor tissue samples Metastatic melanoma Initial cohort: n = 32; validation cohort: n = 33 |
2nd‐line immune checkpoint inhibitors | CGP | >23.1 mutations per Mb (high) | High TMB vs. intermediate vs. low:
|
|
Rizvi et al. (2015) 10 Prospective study NSCLC Discovery cohort: n = 16; validation cohort: n = 18 |
Pembrolizumab | CGP | >median mutations per sample within cohort |
TMB > median vs. others, both cohorts:
TMB > median vs. others, discovery cohort:
|
|
Van Allen et al. (2015) 24 Retrospective study of tumor tissue samples Metastatic melanoma n = 110 |
Ipilimumab | WES | NA | TMB was significantly associated with CB from ipilimumab (p = .0076). |
|
Snyder et al. (2014) 22 Retrospective study of tumor tissue samples Malignant melanoma Discovery cohort: n = 25; validation cohort: n = 39 |
Ipilimumab or tremelimumab | WES | NA |
Higher TMB in long‐term benefit subgroup vs. minimal benefit subgroup (p = .009) OS correlated with higher TMB, discovery cohort (p = .04) |
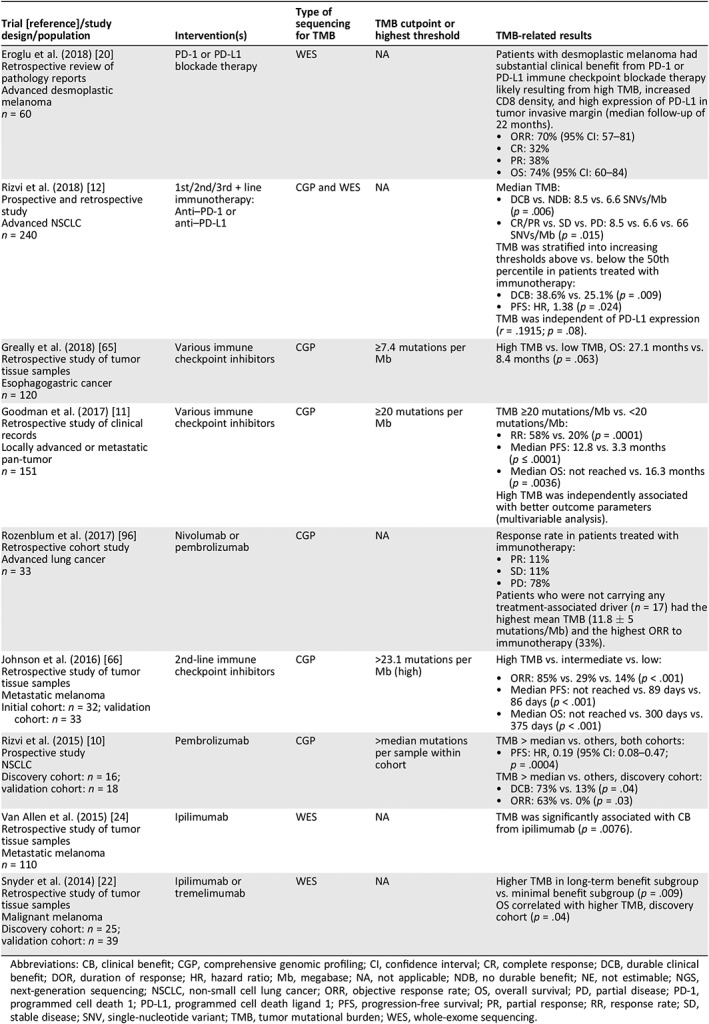
Abbreviations: CB, clinical benefit; CGP, comprehensive genomic profiling; CI, confidence interval; CR, complete response; DCB, durable clinical benefit; DOR, duration of response; HR, hazard ratio; Mb, megabase; NA, not applicable; NDB, no durable benefit; NE, not estimable; NGS, next‐generation sequencing; NSCLC, non‐small cell lung cancer; ORR, objective response rate; OS, overall survival; PD, partial disease; PD‐1, programmed cell death 1; PD‐L1, programmed cell death ligand 1; PFS, progression‐free survival; PR, partial response; RR, response rate; SD, stable disease; SNV, single‐nucleotide variant; TMB, tumor mutational burden; WES, whole‐exome sequencing.
Table 3.
Examples of planned or ongoing clinical trials evaluating TMB as a biomarker
| Trial | Study design | TMB‐related design elements | Completion date, estimated |
|---|---|---|---|
|
B‐F1RST 97 |
Phase II, single‐arm trial of atezolizumab in advanced NSCLC | Primary biomarker endpoint is bTMB; clinical outcomes (OS, PFS, OR) will be evaluated according to bTMB | 2018 |
|
B‐FAST 98 |
Phase II/III nonrandomized multiple cohort trial of atezolizumab, alectinib, pemetrexed, or gemcitabine in advanced NSCLC | Enrollment by actionable genomic alterations or positive bTMB |
2020 (primary) 2022 (study) |
|
CAPTUR 99 |
Phase II basket trial of various targeted therapies in advanced cancer (pan‐tumor) | Study group: Nivolumab ± ipilimumab in patients with high TMB and/or alterations in POLE/POLD1 | 2021 |
|
Javelin Parp Medley 100 |
Phase Ib/II dose‐finding trial of avelumab plus talazoparib in advanced cancer (pan‐tumor) | Secondary endpoint: TMB at baseline | 2020 |
|
My Pathway 101 |
Phase II basket trial of various targeted therapies in advanced cancer (pan‐tumor) | Study group: Atezolizumab in patients with high TMB/MSI‐high and/or alterations in PD‐L1, POLE, or POLD1 | 2019 |
| PECULIAR 102 | Phase II, single‐arm trial of neoadjuvant pembrolizumab and epacadostat in muscle‐invasive urothelial bladder cancer | Endpoint: TMB among biomarkers to be assessed | Unknown |
|
TAPUR [109] |
Phase II basket trial of various targeted therapies in advanced cancer (pan‐tumor) | Study group: Pembrolizumab or nivolumab + ipilimumab in patients with high TMB and/or alterations in POLE/POLD1 | 2019 (primary) |
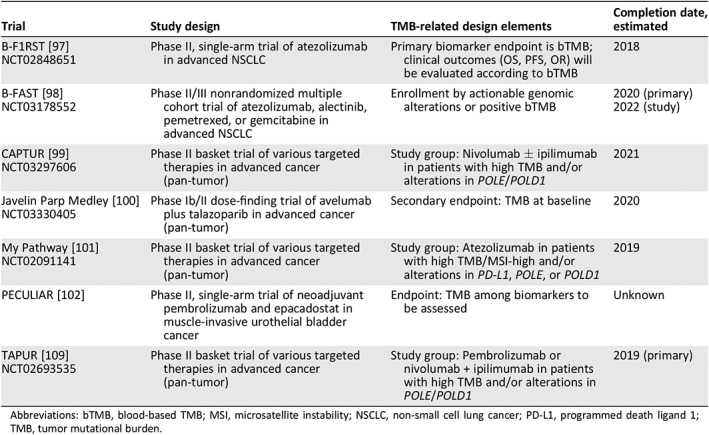
Abbreviations: bTMB, blood‐based TMB; MSI, microsatellite instability; NSCLC, non‐small cell lung cancer; PD‐L1, programmed death ligand 1; TMB, tumor mutational burden.
It should be noted that TMB cutoffs have been defined differently across studies, testing platforms, and in various patient populations, and it is also important to acknowledge that cutoffs might differ by tumor type and ICPI agent (e.g., >16 mutations/Mb for atezolizumab in urothelial carcinoma; >23.1 mutations/Mb for pembrolizumab in NSCLC; and ≥13.5, ≥15.8, or ≥17.1 mutations/Mb for atezolizumab in NSCLC; Tables 1, 2) 45, 46, 61, 66. Goodman and colleagues 11 suggested a pan‐tumor cutoff of 20 mutations per Mb, and Yarchoan and colleagues 103 have reported a nearly linear relationship between TMB and ORR. A TMB cutoff of 10 mutations per Mb for treatment outcome among patients with advanced NSCLC treated with nivolumab plus ipilimumab was recently validated in the CheckMate‐568 trial 93, which demonstrated ORRs of 4% and 10% at cutoffs of <5 and <10 mutations per Mb, respectively, compared with 44% at ≥10 mutations per Mb. These findings informed the cutoff for the phase III, randomized, placebo‐controlled CheckMate‐227 study, in which the treatment group with TMB ≥10 mutations per Mb experienced 1‐year PFS of 42.6% compared with 13.2% in the chemotherapy group 54. It remains to be seen how TMB cutoffs will be applied broadly across tumor types in a clinical setting, but the possibility exists for TMB to redefine therapeutic approaches agnostic of tumor type, similar to MSI.
Current Value of TMB to the Oncology Community
The value of TMB is intrinsically tied to the value of identifying patients who are likely to have a clinical benefit from ICPIs given that the magnitude of such benefit is often considerable. As discussed above, immuno‐oncology biomarkers are not mutually inclusive. Comprehensive assays capable of measuring TMB are likely to identify information about other biomarkers and alterations associated with targeted therapies, allowing care providers to make fully informed therapeutic decisions. By developing and refining this stratification, certain patients with low TMB or other alterations predictive of lack of response or hyper‐progression may avoid costly ineffective treatment, whereas others who are strong candidates for ICPIs may become eligible to receive these agents at earlier lines of therapy.
There is a recognized need for standardization of clinically valid TMB assays across testing platforms. Lessons can be learned from the challenges of validation for PD‐L1 testing 7, which led to an acknowledgment of the need for improved harmonization for biomarker testing. International studies and a coalition of organizations are currently working on methods to ensure the standardization of TMB across assays in order to confirm that accurate clinical decisions are being made for patients with cancer 60. At the time of this writing, the TMB Harmonization Working Group is reviewing the current methods of TMB calculation and reporting as well as developing a consensus on how best to standardize these measurements (phase I has been completed; phase II is underway; Fig. 2) 59, 60, 68.
Improved patient selection for immunotherapy is also likely to enhance the economic value of ICPIs. In particular, health‐related quality of life outcomes among patients with NSCLC and urothelial cancer who were treated with pembrolizumab have demonstrated a substantial improvement compared with chemotherapy, thus showing the potential of TMB to increase incremental quality‐adjusted life‐years in economic analyses 104, 105. Although there is limited direct evidence evaluating the economic value of using TMB as a biomarker for treatment stratification, the potential impact of incorporating TMB testing into routine clinical practice might lead to improved outcomes and greater stratification of patients who undergo effective immunotherapy for a longer duration. In this scenario, testing costs are likely to remain stable when TMB is provided in the context of an existing CGP panel, while clinical value improves.
Although there is limited direct evidence evaluating the economic value of using TMB as a biomarker for treatment stratification, the potential impact of incorporating TMB testing into routine clinical practice might lead to improved outcomes and greater stratification of patients who undergo effective immunotherapy for a longer duration.
Future Directions
Collectively, the current data suggest that measuring TMB for all patients with cancer has the potential to increase access to life‐extending therapies while improving the overall clinical and economic value of ICPIs. Several ongoing or planned ICPI studies are using TMB to enroll patients and thereby increase the proportion of patients who are likely to benefit (Table 3). Additionally, TMB measurement from circulating tumor DNA, derived from blood specimens (bTMB), was recently shown to be predictive of survival in patients with NSCLC who were treated with atezolizumab, and several ongoing trials are evaluating the prospective efficacy of bTMB in a first‐line NSCLC setting 56, 97, 98, 106 (Table 3).
There are many more avenues of research to be explored in order to better understand the relationship between TMB and ICPI outcomes. For example, recent findings have suggested that TMB could be useful for patient stratification in trials assessing ICPI use at earlier stages of cancer 55, 107. Important questions regarding the role of concurrent genomic alterations in high TMB tumors that may negatively predict the impact ICPI responsiveness, such as pre‐existing STK11, JAK1/2, MDM2 or B2M alterations, remain unclear. In addition, the potential for treatment to affect a patient's TMB status, and whether such a change in TMB over time has any clinical significance, is not yet known.
Finally, the role of TMB should be considered in combination therapy trials, including combinations of ICPIs as well as ICPIs with conventional therapies. As noted above, the first prospective clinical validation of a TMB cutoff was with nivolumab plus ipilimumab, which are PD‐1 and CTLA‐4 inhibitors, respectively 54. Furthermore, a benefit from chemotherapy‐based approaches was seen in patients with TMB >8 in the phase III SWOG 80405 trial 108. Although there is limited research evaluating TMB as a predictive biomarker for response to non‐ICI treatment such as chemotherapy, this represents an area of future investigation 54, 61. Utilizing TMB and other biomarkers to select patients for specific ICPI‐based combinations could be critical in subsets of patients, and the effect of such combinations on the TMB threshold for clinical benefit should be examined.
Conclusion
Here we provide an overview of the current evidence for TMB as a clinically relevant predictive biomarker of ICPI outcomes in several tumor types. Targeted NGS assays have been validated against WES for accurate TMB measurement. Current research to establish appropriate TMB cutoffs is ongoing, and these cutoffs are likely to be ICPI‐ and tumor type–specific. The standardization of TMB and clinical studies of its use in varying disease states and drug regimens are expected to result in the approval of TMB as a companion diagnostic for ICPIs.
Collectively, current and future trials utilizing CGP to inform enrollment will further elucidate the value of TMB as a biomarker alone and in context with other biomarkers and genomic data. With ongoing study, TMB alongside other genomic biomarkers can direct appropriate patients to ICPI or other targeted therapies at earlier lines of treatment and potentially identify those likely to continue to have durable responses after short‐term treatment, while simultaneously sparing those unlikely to benefit. Overall, the measurement of and appropriate use of TMB has the potential to add substantially to both the clinical and economic value of ICPI agents in oncology.
Author Contributions
Conception/design: Samuel J. Klempner, David Fabrizio, Shalmali Bane, Marcia Reinhart, Tim Peoples, Siraj M. Ali, Ethan S. Sokol, Garrett Frampton, Alexa B. Schrock, Rachel Anhorn, Prasanth Reddy
Data analysis and interpretation: Samuel J. Klempner, David Fabrizio, Shalmali Bane, Marcia Reinhart, Tim Peoples, Siraj M. Ali, Ethan S. Sokol, Garrett Frampton, Alexa B. Schrock, Rachel Anhorn, Prasanth Reddy
Manuscript writing: Samuel J. Klempner, David Fabrizio, Shalmali Bane, Marcia Reinhart, Tim Peoples
Final approval of manuscript: Samuel J. Klempner, David Fabrizio, Shalmali Bane, Marcia Reinhart, Tim Peoples, Siraj M. Ali, Ethan S. Sokol, Garrett Frampton, Alexa B. Schrock, Rachel Anhorn, Prasanth Reddy
Disclosures
Samuel J. Klempner: Foundation Medicine, Inc., Astellas, Lilly Oncology, Boston Biomedical (C/A), Leap Therapeutics (RF), Foundation Medicine, Inc. (H), TP Therapeutics (SAB); David Fabrizio: Foundation Medicine, Inc. (E, OI); Shalmali Bane: Analysis Group, Inc. (E), Foundation Medicine, Inc. (C/A—employer); Marcia Reinhart: Analysis Group, Inc. (E), Foundation Medicine, Inc. (C/A—employer); Tim Peoples: Amgen (E), Analysis Group, Inc. (E—former); Siraj M. Ali: Foundation Medicine, Inc. (E, OI); Ethan Sokol: Foundation Medicine, Inc. (E, OI); Garrett Frampton: Foundation Medicine, Inc. (E, OI); Alexa B. Schrock: Foundation Medicine, Inc. (E, OI); Rachel Anhorn: Foundation Medicine, Inc. (E, OI); Prasanth Reddy: Foundation Medicine, Inc. (E, OI).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Acknowledgments
The authors take full responsibility for this work and thank Bethany Sawchyn of Foundation Medicine, Inc., for providing a critical review of the manuscript. This study was funded by Foundation Medicine, Inc.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Wang C, Yu X, Wang W. A meta‐analysis of efficacy and safety of antibodies targeting PD‐1/PD‐L1 in treatment of advanced nonsmall cell lung cancer. Medicine (Baltimore) 2016;95:e5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin Z, Chen X, Li Z et al. PD‐1 antibody monotherapy for malignant melanoma: A systematic review and meta‐analysis. PLoS One 2016;11:e0160485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Garon EB, Rizvi NA, Hui R et al. Pembrolizumab for the treatment of non‐small‐cell lung cancer. N Engl J Med 2015;372:2018–2028. [DOI] [PubMed] [Google Scholar]
- 4. Herbst RS, Soria J‐C, Kowanetz M et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fehrenbacher L, Spira A, Ballinger M et al. Atezolizumab versus docetaxel for patients with previously treated non‐small‐cell lung cancer (POPLAR): A multicentre, open‐label, phase 2 randomised controlled trial. Lancet 2016;387:1837–1846. [DOI] [PubMed] [Google Scholar]
- 6. Taube JM, Klein A, Brahmer JR et al. Association of PD‐1, PD‐1 ligands, and other features of the tumor immune microenvironment with response to anti–PD‐1 therapy. Clin Cancer Res 2014;20:5064–5074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirsch FR, McElhinny A, Stanforth D et al. PD‐L1 immunohistochemistry assays for lung cancer: Results from phase 1 of the blueprint PD‐L1 IHC assay comparison project. J Thorac Oncol 2017;12:208–222. [DOI] [PubMed] [Google Scholar]
- 8. Patel SP, Kurzrock R. PD‐L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther 2015;14:847–856. [DOI] [PubMed] [Google Scholar]
- 9. Topalian SL, Taube JM, Anders RA et al. Mechanism‐driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rizvi NA, Hellmann MD, Snyder A et al. Cancer immunology. Mutational landscape determines sensitivity to PD‐1 blockade in non‐small cell lung cancer. Science 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Goodman AM, Kato S, Bazhenova L et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 2017;16:2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rizvi H, Sanchez‐Vega F, La K et al. Molecular determinants of response to anti‐programmed cell death (PD)‐1 and anti‐programmed death‐ligand 1 (PD‐L1) blockade in patients with non‐small‐cell lung cancer profiled with targeted next‐generation sequencing. J Clin Oncol 2018;36:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chalmers ZR, Connelly CF, Fabrizio D et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Martin‐Liberal J, Ochoa de Olza M, Hierro C et al. The expanding role of immunotherapy. Cancer Treat Rev 2017;54:74–86. [DOI] [PubMed] [Google Scholar]
- 15. Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell 2011;144:646–674. [DOI] [PubMed] [Google Scholar]
- 16. McFarland CD, Yaglom JA, Wojtkowiak JW et al. The damaging effect of passenger mutations on cancer progression. Cancer Res 2017;77:4763–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yi M, Qin S, Zhao W et al. The role of neoantigen in immune checkpoint blockade therapy. Exp Hematol Oncol 2018;7:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ott PA, Hu Z, Keskin DB et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017;547:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Keskin DB, Anandappa AJ, Sun J et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature 2019;565:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eroglu Z, Zaretsky JM, Hu‐Lieskovan S et al. High response rate to PD‐1 blockade in desmoplastic melanomas. Nature 2018;553:347–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Riaz N, Havel JJ, Makarov V et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–949.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Snyder A, Makarov V, Merghoub T et al. Genetic basis for clinical response to CTLA‐4 blockade in melanoma. N Engl J Med 2014;371:2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Snyder A, Nathanson T, Funt SA et al. Contribution of systemic and somatic factors to clinical response and resistance to PD‐L1 blockade in urothelial cancer: an exploratory multi‐omic analysis. PLoS Med 2017;14:e1002309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Van Allen EM, Miao D, Schilling B et al. Genomic correlates of response to CTLA‐4 blockade in metastatic melanoma. Science 2015;350:207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hellmann MD, Nathanson T, Rizvi H et al. Genomic features of response to combination immunotherapy in patients with advanced non‐small‐cell lung cancer. Cancer Cell 2018;33:843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hellmann MD, Callahan MK, Awad MM et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small‐cell lung cancer. Cancer Cell 2018;33:853–861.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le DT, Uram JN, Wang H et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015;372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Le DT, Durham JN, Smith KN et al. Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 2017;357:409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Endris V, Buchhalter I, Allgäuer M et al. Measurement of tumor mutational burden (TMB) in routine molecular diagnostics: In‐silico and real‐life analysis of three larger gene panels. Int J Cancer 2019;144:2303–2312. [DOI] [PubMed] [Google Scholar]
- 30. Spigel DR, Schrock AB, Fabrizio D et al. Tumor mutation burden (TMB) in lung cancer (LC) and relationship with response to PD‐1/PD‐L1 targeted therapies. J Clin Oncol 2016;34(suppl 15):9017A. [Google Scholar]
- 31. U.S. Food and Drug Administration . FDA unveils a streamlined path for the authorization of tumor profiling tests alongside its latest product action [press release]. November 15, 2017. Available at https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm585347.htm. Accessed September 19, 2018.
- 32. Rothschild S, Jermann P, Savic S et al. Tumor mutational burden assessed by a targeted NGS assay to predict benefit from immune checkpoint inhibitors in non‐small cell lung cancer. J Clin Oncol 2019;37(suppl 15):e14266A. [Google Scholar]
- 33. U.S. Food and Drug Administration . FDA announces approval, CMS proposes coverage of first breakthrough‐designated test to detect extensive number of cancer biomarkers [press release]. November 30, 2017. Available at https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm587273.htm. Accessed September 21, 2018.
- 34. Palmetto GBA. Next Generation Sequencing Coding and Billing Guidelines (M00127, V3). 2017. Available at https://palmettogba.com/palmetto/MolDX.nsf/DocsCat/MolDx%20Website~MolDx~Browse%20By%20Topic~Technical%20Assessment~9NKP9T2602?open. Accessed September 21, 2018.
- 35. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foundation Medicine, Inc. FoundationOne CDx technical information. 2017. Available at https://www.foundationmedicine.com/genomic-testing/foundation-one-cdx. Accessed December 18, 2017.
- 37. Carbone DP, Reck M, Paz‐Ares L et al. First‐line nivolumab in stage IV or recurrent non‐small‐cell lung cancer. N Engl J Med 2017;376:2415–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szustakowski JD, Green G, Geese WJ et al. Evaluation of tumor mutation burden as a biomarker for immune checkpoint inhibitor efficacy: A calibration study of whole exome sequencing with FoundationOne. Cancer Res 2018;78(suppl 13):5528A. [Google Scholar]
- 39. Buchhalter I, Rempel E, Endris V et al. Size matters: Dissecting key parameters for panel‐based tumor mutational burden analysis. Int J Cancer 2019;144:848–858. [DOI] [PubMed] [Google Scholar]
- 40. Alexandrov LB, Ju YS, Haase K et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016;354:618–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cancer Genome Atlas Network. Genomic classification of cutaneous melanoma. Cell 2015;161:1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Campesato LF, Barroso‐Sousa R, Jimenez L et al. Comprehensive cancer‐gene panels can be used to estimate mutational load and predict clinical benefit to PD‐1 blockade in clinical practice. Oncotarget 2015;6:34221–34227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rosenberg JE, Hoffman‐Censits J, Powles T et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum‐based chemotherapy: A single‐arm, multicentre, phase 2 trial. Lancet 2016;387:1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kowanetz M, Zou W, Shames DS et al. Tumor mutation load assessed by FoundationOne (FM1) is associated with improved efficacy of atezolizumab (atezo) in patients with advanced NSCLC. Ann Oncol 2016;27(suppl 6):77PA. [Google Scholar]
- 45. Kowanetz M, Zou W, Shames D et al. Tumor mutation burden (TMB) is associated with improved efficacy of atezolizumab in 1L and 2L+ NSCLC patients. J Thorac Oncol 2017;12(suppl 1):OA20.01A. [Google Scholar]
- 46. Balar AV, Galsky MD, Rosenberg JE et al. Atezolizumab as first‐line treatment in cisplatin‐ineligible patients with locally advanced and metastatic urothelial carcinoma: a single‐arm, multicentre, phase 2 trial. Lancet 2017;389:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Seiwert TY, Cristescu R, Mogg R et al. Genomic biomarkers in relation to PD‐1 checkpoint blockade response. J Clin Oncol 2018;36(suppl 25):25A.29035642 [Google Scholar]
- 48. Zehir A, Benayed R, Shah RH et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature Med 2017;23:703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Galsky MD, Saci A, Szabo PM et al. Impact of tumor mutation burden on nivolumab efficacy in second‐line urothelial carcinoma patients: Exploratory analysis of the phase II CheckMate 275 study. Ann Oncol 2017;28(suppl 5):848PDA. [Google Scholar]
- 50. Fabrizio DA, Malboeuf C, Lieber D et al. Analytic validation of a next generation sequencing assay to identify tumor mutational burden from blood (bTMB) to support investigation of an anti‐PD‐L1 agent, atezolizumab, in a first line non‐small cell lung cancer trial (BFAST). Ann Oncol 2017;28(suppl 5):102PA. [Google Scholar]
- 51. Mok TSK, Gadgeel S, Kim ES et al. Blood first line ready screening trial (B‐F1RST) and blood first assay screening trial (BFAST) enable clinical development of novel blood‐based biomarker assays for tumor mutational burden (TMB) and somatic mutations in 1L advanced or metastatic NSCLC. Ann Oncol 2017;28(suppl 5):1383TiPA. [Google Scholar]
- 52. Antonia S, Callahan MK, Awad MM, Calvo E et al. OA 07.03a ‐ Impact of tumor mutation burden on the efficacy of nivolumab or nivolumab + ipilimumab in small cell lung cancer: An exploratory analysis of CheckMate 032 (ID 659). Presented at IASLC 18th World Conference on Lung Cancer, Yokohama, Japan, 2017.
- 53.Foundation Medicine, Inc. FDA approves Foundation Medicine's FoundationOne CDx, the first and only comprehensive genomic profiling test for all solid tumors incorporating multiple companion diagnostics [press release]. November 30, 2017. Available at http://investors.foundationmedicine.com/news-releases/news-release-details/fda-approves-foundation-medicines-foundationone-cdxtm-first-and. Accessed September 19, 2018.
- 54. Hellmann MD, Ciuleanu TE, Pluzanski A et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 2018;378:2093–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Forde PM, Chaft JE, Smith KN et al. Neoadjuvant PD‐1 blockade in resectable lung cancer. N Engl J Med 2018;378:1976–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gandara DR, Paul SM, Kowanetz M et al. Blood‐based tumor mutational burden as a predictor of clinical benefit in non‐small‐cell lung cancer patients treated with atezolizumab. Nature Med 2018;24:1441–1448. [DOI] [PubMed] [Google Scholar]
- 57. Necchi A, Briganti A, Bianchi M et al. Preoperative pembrolizumab (pembro) before radical cystectomy (RC) for muscle‐invasive urothelial bladder carcinoma (MIUC): Interim clinical and biomarker findings from the phase 2 PURE‐01 study. J Clin Oncol 2018;36(suppl 15):4507A. [DOI] [PubMed] [Google Scholar]
- 58. Fabrizio DA, Milbury C, Yip WK et al. Analytic validation of tumor mutational burden as a companion diagnostic for combination immunotherapy in non‐small cell lung cancer. Ann Oncol 2018;29(suppl 8):56PDA. [Google Scholar]
- 59. Fabrizio DA, Chen SJ, Xie M et al. In silico assessment of variation in TMB quantification across diagnostic platforms: Phase 1 of the Friends of Cancer Research Harmonization Project. J Immunother Cancer 2018;6(suppl 2):O48A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Friends of Cancer Research . Tumor mutational burden (TMB). Available at https://www.focr.org/tmb. Updated 2018. Accessed June 13, 2018.
- 61. Samstein R, Lee C, Shoushtari A et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nature Genet 2019;51:202–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. National Comprehensive Cancer Network . NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) for Non‐Small Cell Lung Cancer. Version 3.2019. Plymouth Meeting, PA: National Comprehensive Cancer Network, Inc.; 2019. Accessed February 19, 2019. To view the most recent and complete version of the guideline, go online to http://nccn.org. [Google Scholar]
- 63. Hayes DF. Biomarker validation and testing. Mol Oncol 2015;9:960–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mariathasan S, Turley SJ, Nickles D et al. TGFbeta attenuates tumour response to PD‐L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Greally M, Chatila WK, Margolis M et al. Tumor mutation burden (TMB) and immune‐related adverse events (irAEs) compared to antibiotic (abx) use to predict for response to immune checkpoint inhibitors in esophagogastric cancer (EGC). J Clin Oncol 2018;36(suppl 15):4056A. [Google Scholar]
- 66. Johnson DB, Frampton GM, Rioth MJ et al. Targeted next generation sequencing identifies markers of response to PD‐1 blockade. Cancer Immunol Res 2016;4:959–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chae YK, Davis AA, Agte S et al. Clinical implications of circulating tumor DNA tumor mutational burden (ctDNA TMB) in non‐small cell lung cancer. The Oncologist 2019;24:820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stenzinger A, Allen J, Maas J et al. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer 2019;58:578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Miao D, Margolis CA, Vokes NI et al. Genomic correlates of response to immune checkpoint blockade in microsatellite‐stable solid tumors. Nat Genetics 2018;50:1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. U.S. Food and Drug Administration . FDA grants accelerated approval to pembrolizumab for first tissue/site agnostic indication [press release]. May 30, 2017. Available at https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm560040.htm. Accessed April 22, 2018.
- 71. Hall MJ, Gowen K, Sanford EM et al. Evaluation of microsatellite instability (MSI) status in 11,573 diverse solid tumors using comprehensive genomic profiling (CGP). J Clin Oncol 2016;34(suppl 15):1523A. [Google Scholar]
- 72. Frampton GM, Fabrizio DA, Chalmers ZR et al. Assessment and comparison of tumor mutational burden and microsatellite instability status in >40,000 cancer genomes. Ann Oncol 2016;27(suppl 6):52OA. [Google Scholar]
- 73. Fabrizio DA, George TJ, Dunne RF et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointestin Oncol 2018;9:610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Peters S, Creelan B, Hellmann MD et al. Impact of tumor mutation burden on the efficacy of first‐line nivolumab in stage IV or recurrent non‐small cell lung cancer: An exploratory analysis of CheckMate 026. Cancer Res 2017;77(suppl 13):CT082A. [Google Scholar]
- 75. Voutsadakis IA. Polymerase epsilon mutations and concomitant beta2‐microglobulin mutations in cancer. Gene 2018;647:31–38. [DOI] [PubMed] [Google Scholar]
- 76. Chae YK, Anker JF, Bais P et al. Mutations in DNA repair genes are associated with increased neo‐antigen load and activated T cell infiltration in lung adenocarcinoma. Oncotarget 2018;9:7949–7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schrock AB, Fabrizio D, He Y et al. Analysis of POLE mutation and tumor mutational burden (TMB) across 80,853 tumors: Implications for immune checkpoint inhibitors (ICPIs). Ann Oncol 2017;28(suppl 5):1170PA. [Google Scholar]
- 78. Campbell BB, Light N, Fabrizio D et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017;171:1042–1056.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gargiulo P, Della Pepa C, Berardi S et al. Tumor genotype and immune microenvironment in POLE‐ultramutated and MSI‐hypermutated endometrial cancers: New candidates for checkpoint blockade immunotherapy? Cancer Treat Rev 2016;48:61–68. [DOI] [PubMed] [Google Scholar]
- 80. Gong J, Wang C, Lee P et al. Response to PD‐1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Canc Netw 2017;15:142–147. [DOI] [PubMed] [Google Scholar]
- 81. Gainor JF, Shaw AT, Sequist LV et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD‐1 pathway blockade in non‐small cell lung cancer (NSCLC): A retrospective analysis. Clin Cancer Res 2016;22:4585–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Davis AA, Chae YK, Agte S et al. Association of tumor mutational burden with smoking and mutation status in non‐small cell lung cancer (NSCLC). J Clin Oncol 2017;35(suppl 7):24A.28034071 [Google Scholar]
- 83. Chae Y, Davis A, Raparia K et al. Association of tumor mutational burden with dna repair mutations and response to Anti–PD‐1/PD‐L1 therapy in non–small‐cell lung cancer. Clin Lung Cancer 2019;20:88–96. [DOI] [PubMed] [Google Scholar]
- 84. Kim JH, Kim HS, Kim BJ. Prognostic value of KRAS mutation in advanced non‐small‐cell lung cancer treated with immune checkpoint inhibitors: A meta‐analysis and review. Oncotarget 2017;8:48248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Skoulidis F, Goldberg ME, Greenawalt DM et al. STK11/LKB1 mutations and PD‐1 inhibitor resistance in KRAS‐mutant lung adenocarcinoma. Cancer Discov 2018;8:822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Flex E, Petrangeli V, Stella L et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med 2008;205:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Albacker LA, Wu J, Smith P et al. Loss of function JAK1 mutations occur at high frequency in cancers with microsatellite instability and are suggestive of immune evasion. PLoS One 2017;12:e0176181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Ross JS, Goldberg ME, Albacker LA et al. Immune checkpoint inhibitor (ICPI) efficacy and resistance detected by comprehensive genomic profiling (CGP) in non‐small cell lung cancer (NSCLC). Ann Oncol 2017;28(suppl 5):1138PDA. [Google Scholar]
- 89. Kato S, Goodman A, Walavalkar V et al. Hyperprogressors after immunotherapy: Analysis of genomic alterations associated with accelerated growth rate. Clin Cancer Res 2017;23:4242–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Shen J, Ju Z, Zhao W et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med 2018;24:556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zaretsky JM, Garcia‐Diaz A, Shin DS et al. Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med 2016;375:819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Teng F, Meng X, Kong L et al. Progress and challenges of predictive biomarkers of anti PD‐1/PD‐L1 immunotherapy: A systematic review. Cancer Lett 2018;414:166–173. [DOI] [PubMed] [Google Scholar]
- 93. Ramalingam SS, Hellmann MD, Awad MM et al. Tumor mutation burden (TMB) as a biomarker for clinical benefit from dual immune checkpoint blockade with nivolumab (nivo) + ipilimumab (ipi) in first‐line (1L) non‐small cell lung cancer (NSCLC): Identification of TMB cutoff from CheckMate 568. Cancer Res 2018;78(suppl 13):CT078A. [Google Scholar]
- 94. Legrand FA, Gandara DR, Mariathasan S et al. Association of high tissue TMB and atezolizumab efficacy across multiple tumor types. J Clin Oncol 2018;36(suppl 15):12000A. [Google Scholar]
- 95. Raggi D, Giannatempo P, Mariani L et al. Apache: An open label, randomized, phase 2 study of durvalumab (Durva), alone or in combination with tremelimumab (Treme), in patients (pts) with advanced germ cell tumors (GCT): Results at the end of first stage. J Clin Oncol 2018;36(suppl 15):4547A. [Google Scholar]
- 96. Rozenblum AB, Ilouze M, Dudnik E et al. Clinical impact of hybrid capture‐based next‐generation sequencing on changes in treatment decisions in lung cancer. J Thorac Oncol 2017;12:258–268. [DOI] [PubMed] [Google Scholar]
- 97. http://clinicaltrials.gov. A study of atezolizumab as first‐line monotherapy for advanced or metastatic non‐small cell lung cancer (B‐F1RST). http://clinicialtrials.gov identifier: NCT02848651. Available at https://clinicaltrials.gov/ct2/show/NCT02848651. Updated 2018. Accessed April 25, 2018.
- 98. http://clinicaltrials.gov. A study to evaluate efficacy and safety of multiple targeted therapies as treatments for participants with non‐small cell lung cancer (NSCLC) (B‐FAST). http://clinicaltrials.gov identifier: NCT03178552. Available at https://clinicaltrials.gov/ct2/show/NCT03178552. Updated 2018. Accessed April 25, 2018.
- 99. http://clinicaltrials.gov. Canadian profiling and targeted agent utilization trial (CAPTUR). http://clinicaltrials.gov identifier: NCT03297606. Available at https://clinicaltrials.gov/ct2/show/NCT03297606. Updated 2018. Accessed April 25, 2018.
- 100. http://clinicaltrials.gov. Javelin Parp Medley: Avelumab plus talazoparib in locally advanced or metastatic solid tumors. http://clinicaltrials.gov identifier: NCT03330405. Available at https://clinicaltrials.gov/ct2/show/NCT03330405. Updated 2018. Accessed April 25, 2018.
- 101. http://clinicaltrials.gov. My Pathway: A study evaluating Herceptin/Perjeta, Tarceva, Zelboraf/Cotellic, Erivedge, Alecensa, and Tecentriq treatment targeted against certain molecular alterations in participants with advanced solid tumors. http://clinicaltrials.gov identifier: NCT02091141. Available at https://clinicaltrials.gov/ct2/show/NCT02091141. Updated 2018. Accessed April 25, 2018.
- 102. Necchi A, Briganti A, Bianchi M et al. PECULIAR: An open label, multicenter, single‐arm, phase 2 study of neoadjuvant pembrolizumab (PEM) and epacadostat (EPA), preceding radical cystectomy (Cy), for patients (pts) with muscle‐invasive urothelial bladder cancer (MIUBC). J Clin Oncol 2018;36(suppl 6):TPS534A. [DOI] [PubMed] [Google Scholar]
- 103. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD‐1 inhibition. N Engl J Med 2017;377:2500–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Brahmer JR, Rodriguez‐Abreu D, Robinson AG et al. Health‐related quality‐of‐life results for pembrolizumab versus chemotherapy in advanced, PD‐L1‐positive NSCLC (KEYNOTE‐024): A multicentre, international, randomised, open‐label phase 3 trial. Lancet Oncol 2017;18:1600–1609. [DOI] [PubMed] [Google Scholar]
- 105. Vaughn DJ, Bellmunt J, Fradet Y et al. Health‐related quality‐of‐life analysis from KEYNOTE‐045: A phase III study of pembrolizumab versus chemotherapy for previously treated advanced urothelial cancer. J Clin Oncol 2018;36:1579–1587. [DOI] [PubMed] [Google Scholar]
- 106. Wang Z, Duan J, Cai S et al. Assessment of blood tumor mutational burden as a potential biomarker for immunotherapy in patients with non–small cell lung cancer with use of a next‐generation sequencing cancer gene panel. JAMA Oncol 2019;5:696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Owada‐Ozaki Y, Muto S, Takagi H et al. Prognostic impact of tumor mutation burden in patients with completely resected non‐small cell lung cancer: Brief report. J Thorac Oncol 2018;13:1217–1221. [DOI] [PubMed] [Google Scholar]
- 108. Innocenti F, Ou FS, Qu X et al. Mutational analysis of patients with colorectal cancer in CALGB/SWOG 80405 identifies new roles of microsatellite instability and tumor mutational burden for patient outcome. J Clin Oncol 2019;10:1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]