

Abstract
Purpose
Amplifications of receptor tyrosine kinases (RTKS) are therapeutic targets in multiple tumor types (e.g. HER2 in breast cancer), and amplification of the chromosome 4 segment harboring the three RTKs KIT, PDGFRA, and KDR (4q12amp) may be similarly targetable. The presence of 4q12amp has been sporadically reported in small tumor specific series but a large‐scale analysis is lacking. We assess the pan‐cancer landscape of 4q12amp and provide early clinical support for the feasibility of targeting this amplicon.
Experimental Design
Tumor specimens from 132,872 patients with advanced cancer were assayed with hybrid capture based comprehensive genomic profiling which assays 186–315 genes for all classes of genomic alterations, including amplifications. Baseline demographic data were abstracted, and presence of 4q12amp was defined as 6 or more copies of KIT/KDR/PDGFRA. Concurrent alterations and treatment outcomes with matched therapies were explored in a subset of cases.
Results
Overall 0.65% of cases harbored 4q12amp at a median copy number of 10 (range 6–344). Among cancers with >100 cases in this series, glioblastomas, angiosarcomas, and osteosarcomas were enriched for 4q12amp at 4.7%, 4.8%, and 6.4%, respectively (all p < 0.001), giving an overall sarcoma (n = 6,885) incidence of 1.9%. Among 99 pulmonary adenocarcinoma cases harboring 4q12amp, 50 (50%) lacked any other known driver of NSLCC. Four index cases plus a previously reported case on treatment with empirical TKIs monotherapy had stable disease on average exceeding 20 months.
Conclusion
We define 4q12amp as a significant event across the pan‐cancer landscape, comparable to known pan‐cancer targets such as NTRK and microsatellite instability, with notable enrichment in several cancers such as osteosarcoma where standard treatment is limited. The responses to available TKIs observed in index cases strongly suggest 4q12amp is a druggable oncogenic target across cancers that warrants a focused drug development strategy.
Implications for Practice
Coamplification of the receptor tyrosine kinases (rtks) KIT/KDR/PDGFRA (4q12amp) is present broadly across cancers (0.65%), with enrichment in osteosarcoma and gliomas. Evidence for this amplicon having an oncogenic role is the mutual exclusivity of 4q12amp to other known drivers in 50% of pulmonary adenocarcinoma cases. Furthermore, preliminary clinical evidence for driver status comes from four index cases of patients empirically treated with commercially available tyrosine kinase inhibitors with activity against KIT/KDR/PDGFRA who had stable disease for 20 months on average. The sum of these lines of evidence suggests further clinical and preclinical investigation of 4q12amp is warranted as the possible basis for a pan‐cancer drug development strategy.
Keywords: amplification, tyrosine kinase inhibitor, KIT, PDGFRA, genomic profiling, sarcoma
Short abstract
Matching treatment strategy to tumor biology is central to precision medicine. The co‐amplification of three distinct RTK encoding genes (KIT, KDR, and PDGFRA) on chromosome 4q12 (4q12amp) may be an oncogenic driver and thus a target for therapy. This article assesses the pan‐cancer landscape of 4q12amp and provides clinical support for the feasibility of targeting this amplicon.
Introduction
Matching treatment strategy to tumor biology is the central tenet of precision medicine, and ectopic activation of receptor tyrosine kinases (RTKs) is a pervasive theme in oncogenesis 1. Oncogenic fusions involving the anaplastic lymphoma kinase (ALK) and ROS proto‐oncogene1 (ROS1) RTKs and small deletions in epidermal growth factor receptor (EGFR) are well established as predicting patient benefit from target‐matched tyrosine kinase inhibitors (TKIs), particularly in the context of non‐small cell lung cancer (NSCLC) 1, 2. Amplification of wildtype RTK genes, such as ERBB2 (encoding HER2) in breast and gastroesophageal cancers, also serve as oncogenic drivers; however, the underlying oncogenic mechanism is less well understood 3, 4.
The coamplification of three distinct RTK encoding genes—KIT, KDR, and PDGFRA—on chromosome 4q12 (4q12amp) may similarly be an oncogenic driver and thus target for therapy. Limited series in glioblastoma, lung cancer, and malignant peripheral nerve sheath tumor identified 4q12amp by fluorescence in situ hybridization or immunohistochemistry 5, 6, 7. Preclinical studies support the druggability of this amplicon with the use of approved, but promiscuous, TKIs that are variously active against some of the encoded genes 5, 8. A recent clinical trial of axitinib in treatment described the best responding patient as harboring 4q12amp as also profiled with the platform in this study 9. Based on the preclinical rationale and pilot clinical data, we sought to further delineate the pan‐cancer landscape of 4q12amp in a large series of advanced cancer patients assayed with genomic profiling during clinical care. Herein, we present the identification of recurrent 4q12amp across tumor types and provide additional data supporting clinical actionability.
Subjects, Methods, and Methods
Comprehensive genomic profiling (CGP) was performed in a Clinical Laboratory Improvement Amendments‐certified, College of American Pathologists‐accredited laboratory (Foundation Medicine, Inc., Cambridge, MA). Approval for this study, including a waiver of informed consent and a HIPAA (Health Insurance Portability and Accountability Act) waiver of authorization, was obtained from the Western Institutional Review Board (protocol no. 20152817). The pathologic diagnosis of each case was confirmed on routine H&E‐stained slides, and all samples forwarded for DNA extraction contained a minimum of 20% tumor nuclear area, compared with benign nuclear area. In brief, ≥50ng DNA was extracted from 40 microns of 132,872 cancer cases in formalin‐fixed, paraffin‐embedded tissue blocks. The samples were assayed by CGP using adaptor ligation, and hybrid capture was performed for all coding exons from 186 (version 1) to 287 (version 2) to 315 (version 3) cancer‐related genes plus select introns from 14 (version 1) to 19 (version 2) to 28 (version 3) genes frequently rearranged in cancer 10. A subset of samples also were interrogated for gene rearrangements in 265 genes via RNA sequencing.
Sequencing of captured libraries was performed using the Illumina HiSeq technology to a mean exon coverage depth of >500×, and resultant sequences were analyzed for genomic alterations, including short variant alterations (base substitutions, insertions, and deletions), copy number alterations (focal amplifications and homozygous deletions), and select gene fusions or rearrangements, as previously described 10, 11. Germline variants documented in the dbSNP database (dbSNP142; http://www.ncbi.nlm.nih.gov/SNP/), with two or more counts in the ExAC database (http://exac.broadinstitute.org/), or recurrent variants of unknown significance that were predicted by an internally developed algorithm to be germline were removed, except for known driver germline events (e.g., documented hereditary BRCA1/2 and deleterious TP53 mutations) 12. Known confirmed somatic alterations deposited in the Catalog of Somatic Mutations in Cancer (COSMIC, v.62) were highlighted as biologically significant. All inactivating events (i.e., truncations and deletions) in known tumor suppressor genes were also called as significant. To maximize mutation detection accuracy (sensitivity and specificity) in impure clinical specimens, the test was previously optimized and validated to detect base substitutions at a ≥5% mutant allele frequency (MAF), indels with a ≥10% MAF with ≥99% accuracy, and fusions occurring within baited introns/exons with >99% sensitivity 10, 11.
Each tumor sample was analyzed alongside an internally validated mixture of 10 heterozygous diploid HapMap control samples, which use custom algorithms to normalize the sequence coverage distribution across baited targets. Normalized coverage data for exonic, intronic, and single‐nucleotide polymorphism (SNP) targets accounting for stromal admixture are plotted on a logarithmic scale and minor allele SNP frequencies are concordantly plotted across the genome. Further cluster groupings of targets and minor allele SNPs were used to define upper and lower bounds of genomic segments. Empirical Bayesian algorithms employ a distribution of parameters including purity and base ploidy, and probability matrices are derived using different statistical sampling methodologies to fit these data and generate copy number alteration variant calls; all computational models were reviewed by expert analysts for each sample. Given that each copy number model was dynamically generated for each individual sample, credibility and confidence intervals vary with sample data. As previously described, copy number calling achieves high performance (sensitivity was 99% with positive predictive value >99%) within a range of 30%–75% tumor content and for high level amplifications (copy number ≥8), with slight reduction in sensitivity (>80%) at lower sample purities (20%–30%) and low level amplifications but still called above threshold of 6 (copy number 6–7). We used a prespecified cutoff of 6 or more predicted copies for all three genes of the 4q12 amplicon (KIT, KDR, PDGFRA) as being on a chromosomal segment less than 20Mb in length to define 4q12amp. Supplemental online Table 5 describes the number of cases that harbor two or one but not three amplified genes on the 4q12 amplicon. Descriptive statistics and Fisher exact test with false discovery rate correction were used to compare 4q12amp among tumor types.
To explore the frequency of KIT, KDR, and PDGFRA amplification co‐occurrence, we mined the publicly available The Cancer Genome Atlas (TCGA) using established methods on the cBioPortal (http://www.cbioprtal.org) 13, 14. Specifically, from samples with available mutation and copy number data (n = 44,697), we identified all cases harboring concurrent amplification of the 4q12 kinases KIT, KDR, and PDGFRA using an iterative search process. Anatomic tumor type and clinicopathologic features were also abstracted 13, 14.
Results
Among 132,872 consecutive advanced cancer specimens analyzed, 0.65% (857) harbored 4q12amp of KIT, KDR, and PDGFRA. The median 4q12 copy number was 10 (range, 6–344) within the 4q12amp population (Fig. 1A). Patients with tumors harboring 4q12amp were almost evenly divided between women (48.5%, n = 416) and men (51.5%, n = 441) and had a median age of 60 years (range, 4–87). The cases harboring 4q12amp had a low median tumor mutation burden (TMB; defined as mutations [mut] per Mb) of 5.00 mut per Mb, with the 75th percentile being 10.81 mut per Mb (supplemental online Fig. 1, supplemental online Table 1). Across the cancer landscape, recurrent (>100 cases in our series) 4q12amp was observed primarily in central nervous system (CNS) neoplasms and sarcomas (supplemental online Table 1). Specifically, 4q12amp was detected in 3.7% (211 of 5,689 cases) of primary intracranial neoplasms and enriched in glioblastomas relative to the occurrence of this alteration across cancers (4.7%, 172 of 3,620 cases; p < .001; Fig. 2A, supplemental online Table 1). 4q12amp was also detected in 1.9% (129 of 6,885; p < .001) sarcoma cases, with angiosarcoma 4.8% (10/208; p < .001) and osteosarcoma (6.4%; p < .001) demonstrating enrichment relative to all cancers (Fig. 2B). In addition, 4q12amp is present in cancers that can arise in multiple anatomic sites, including 3.3% of mucosal melanomas (6/182 cases; p < .05) and 2.7% of adenoid cystic carcinoma (AdCC; 21 of 767 cases; p < .001; supplemental online Table 1). Cases with one or two of the three 4q12amp genes were also observed. Cases harboring a single gene amplification were most prevalent, whereas cases harboring two of the three RTKs were less common than 4q12amp (supplemental online Fig. 2). Landscapes for these permutations were unique from the 4q12amp landscape suggesting amplification of all three genes occurs in a unique biological context (supplemental online Table 2).
Figure 1.
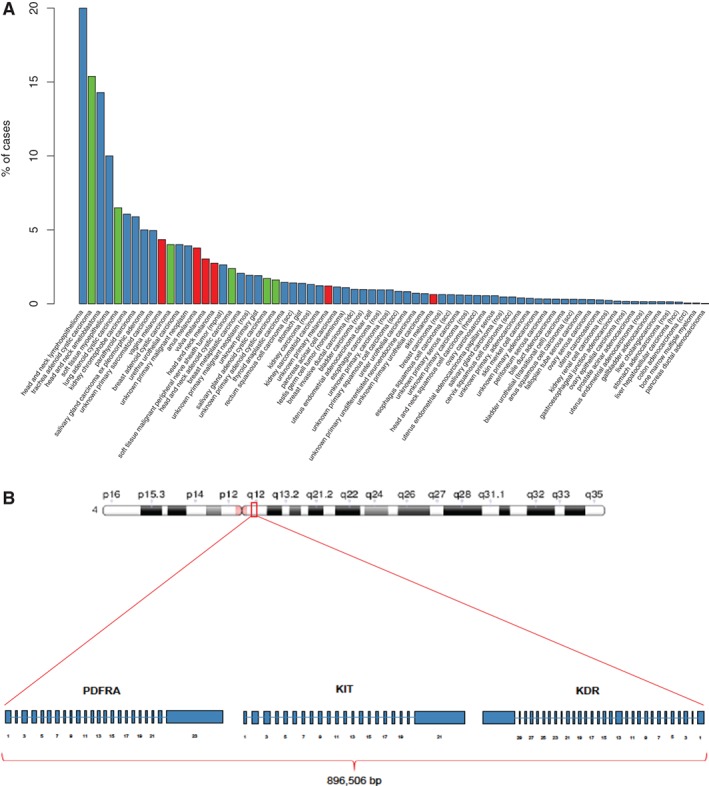
Frequency of 4q12 amp in advanced cancer cases. (A): Frequency in overall series of cancer cases excluding lung carcinoma, primary brain tumors, and sarcomas. Adenoid cystic carcinoma is in green and melanoma is in red. (B): Schematic of 4q12 on chromosome with megabase demarcation and graph.
Abbreviation: NOS, not otherwise specified.
Figure 2.
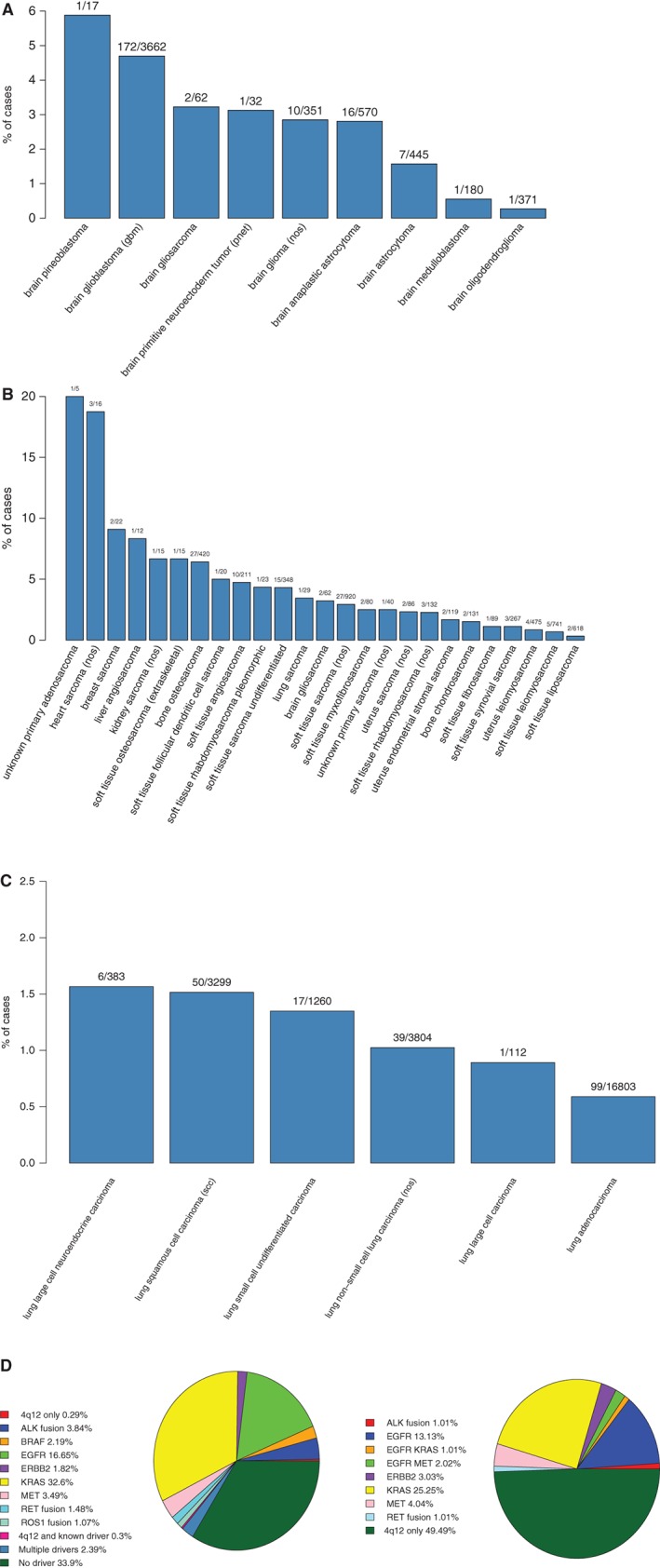
Frequency of 4q12amp in diseases that display a notable enrichment. (A): Frequency in primary brain tumors. (B): Frequency in sarcomas. (C): Frequency in lung carcinoma. (D): Pie chart of 4q12amp and known drivers in lung adeno. (E): Pie chart of co‐occurring drivers in lung adenocarcinoma cases harboring 4q12amp.
Abbreviation: NOS, not otherwise specified.
Among the major lung cancer types, 4q12amp was enriched in squamous cell carcinoma and present in small cell carcinoma, non‐small cell carcinoma not otherwise specified, and lung adenocarcinoma at frequencies of 1.5% (p < .001), 1.4%, 1.0%, and 0.6%, respectively (Fig. 2C). To assess the co‐occurrence of 4q12amp with known oncogenic drivers in NSCLC, the genomic profiles of 99 cases of adenocarcinoma harboring 4q12amp (0.6%) were reviewed (Fig. 2C). Within the 4q12amp lung cancers, 50% of cases did not have other oncogenic drivers consistent with established driver alterations in NSCLC (Fig. 2D and E). Of the 50% of cases with coexisting likely drivers, half (25/50) harbored driver alterations as follows: EGFR sensitizing alterations L858R, G779F, and G719S (16.2%, 16/99); ALK fusions (1.01%, 1/99); RET fusions (1.02%, 1/99); ERBB2‐activating alterations including amplification and S310F (3.03%, 3/99); MET exon 14 skipping mutations (1/99, 1.01%); and MET amplification (5/99, 5.1%); but not ROS1 fusions or BRAF V600E substitutions (Fig. 2E). It should be noted that three of the cases with EGFR‐sensitizing alterations also harbored mutations in either KRAS or MET (codon 12, exon 14 skipping, and amplification, respectively). The other half of such driver co‐occurring 4q12amp cases (26/50) harbored KRAS mutations but no other drivers, >95% of which occurred as KRAS G12x or G13x (total greater than 50 because of the three cases with other drivers). Complete clinical histories of these cases for assessing treatment with prior therapies was not available.
We further explored the TCGA database inclusive of cell line data and identified KDR amplification in 459/44,697 (1%) of cases, KIT amplification in 542 cases (1.2%), and PDGFRA amplification in 568 or 1.3% of TCGA cases. Among PDGFRA‐amplified cases, 80.5% (457/568) harbored concurrent KIT amplification, and among cases with both PDGFRA and KIT amplification, 84.5% (386/457) harbored amplification of KDR (TCGA sample list can be found in supplemental online Table X). Thus, we identified concurrent KDR, KIT, and PDGFRA in 0.86% of sampled TCGA cases. Recurrent 4q12‐contained kinase amplification was seen in glioblastoma, thoracic neoplasms, and sarcomas at frequencies similar to those in the series here (data not shown).
To explore the targetability of 4q12amp in advanced cancer by treatment with a TKI, we explored several lines of evidence. First, to rule out a prognostic effect of harboring 4q12amp, we analyzed glioblastoma, sarcoma, and head and neck squamous cell carcinoma TCGA datasets for cases harboring 4q12amp to compare to the remaining cases of the same tumor type and observed no difference in survival (data not shown). A study of axitinib in incurable AdCC by Ho et al. reported stable disease in 75% of patients and partial responses in 9.1% of patients. Notably, the patient harboring 14 copies of 4q12 (also profiled by this platform used in the series) had the longest duration of benefit from axitinib at 21.8 months of disease control (progression‐free survival; PFS), which is four times greater than the median of 5.7 months PFS for the overall study 9. We also sought to find other TKI‐treated cases from this series of patients in this study. On obtaining treatment histories of a subset of patient within this series, two patients were found to be treated with imatinib, and five patients did not receive a TKI. These two treated 4q12amp cases both originated in the head and neck area and diagnosed as ameloblastoma and adnexal tumors. Both were treated with imatinib, the ameloblastoma had stable disease exceeding 3 years (Fig. 4), and the adnexal tumor had an initial response but passed because of non‐neoplastic causes at 6 weeks (supplemental online Fig. 3). A TKI‐treated 4q12amp case was identified in an independent retrospective pan‐cancer research study of exceptional responders to pazopanib that had stable disease exceeding 24 months (Fig. 3). Combined analysis with the prior published case supported benefit from TKI therapy with an average duration of response exceeding 21 months in 3 of 4 cases (adnexal carcinoma case excluded because of non‐neoplastic mortality).
Figure 4.
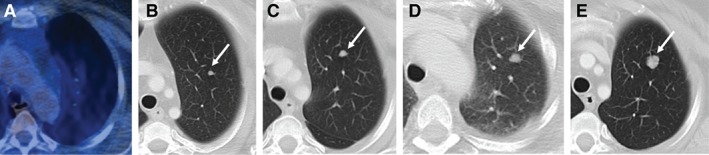
Index case no. 2, ameloblastoma with response to imatinib. (A): FDG‐PET CT. (B): Three months later, chest CT. (C): Nine months later, chest CT. (D): One year later, chest CT. (E): One year and 7 months. Chest CT demonstrating left upper lobe pulmonary nodule (white arrows), not visible on initial study, and enlarging over subsequent follow‐up studies.
Abbreviations: CT, computed tomography; FDG, fluorodeoxyglucose; PET, positron emission tomography.
Figure 3.
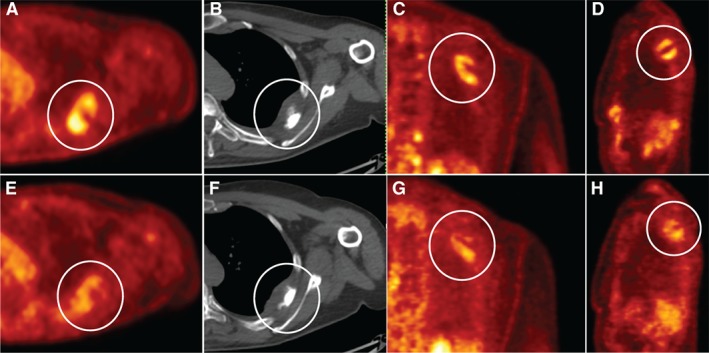
Index case no. 1, MPNST with durable clinical benefit to pazopanib. Baseline axial (A), coronal (C), and sagittal (D) FDG‐PET CT attenuation corrected and axial CT (B) images demonstrate left intercostal muscle FDG AVID mass with corresponding decreased FDG activity on corresponding 3‐month follow‐up (E–H), white circles.
Abbreviations: CT, computed tomography; FDG, fluorodeoxyglucose; MPNST, malignant peripheral nerve sheath tumor; PET, positron emission tomography.
Discussion
The histology agnostic antitumor activity of TKIs directed against genomic altered tyrosine kinases confirms the feasibility of pan‐cancer genomic biomarkers such as the recent clinical successes of the anti‐NTRK larotrectinib for NTRK fusion positive and anti‐HER2 neratinib for ERBB2/3 mutated cancers 15, 16. Here we present evidence of recurrent 4q12amp across anatomically and morphologically disparate tumor types and provide pilot clinical data suggesting actionability regardless of histology. Although rare in absolute terms, the frequencies observed for 4q12amp are aligned with other uncommon but highly targetable oncogenic kinase alterations like NTRK fusions 15. In addition to confirming recurrent 4q12 in glioblastoma, we significantly expand upon the available data and identify recurrent 4q12amp across sarcomas, particularly osteosarcoma and angiosarcoma 5, 6, 7.
Sarcomas represent a heterogenous mix of tumors with diverse anatomic origin with few well established and rationally matched therapies. In this series, 4q12amp was enriched at 6.4% in osteosarcoma, in contrast to previous whole exome and whole genome sequencing studies that defined mutated PI3K and mTOR pathway genes as a possible therapeutic targets but not 4q12amp 17. The discrepancy may be due to the lower depth of coverage (100× or less) of the whole exome and whole genome sequencing platform, which makes detection of amplifications difficult 10. Among the 4q12amp osteosarcoma cases (n = 27), the median age and TMB were similar relative to the overall osteosarcoma population, and nonspecific genomic features were enriched (data not shown). Similarly, angiosarcomas lack standard evidence‐based therapies for recurrent disease and are enriched for 4q12amp 18. Clinical history was not available for our angiosarcoma cases, but orthogonal support for 4q12amp actionability has been seen in a previously reported case treated with pazopanib on the basis of KDR and FLT4 (VEGFR3) amplification and subsequent durable response 19. Given the lack of rationally matched therapies for advanced sarcoma of all stripes, we believe that defining 4q12amp subset of sarcomas is an initial step in evaluating the targetability of this alteration for the benefit of these patients.
Beyond sarcomas, 4q12amp was enriched in some types of carcinoma, tumors of epithelial origin, and other cancers of other histogenetic origins. We observed 4q12amp in in AdCC, a slow‐growing but ultimately lethal carcinoma pathognomonically associated with fusions of MYB or MYB1 20, 21. The previously reported 4q12amp AdCC case with durable benefit from axitinib was also positive for MYB rearrangement and demonstrates that even within tumors with well‐known oncogenic drivers, co‐occurring 4q12amp can be targeted for clinical benefit 9. Similarly, our finding of 4q12amp in noncutaneous melanoma is broadly consistent with previous work describing the presence of amplified KIT in melanoma, although these studies did not assess KDR and PDGFRA and as such could not definitively presence of 4q12amp 22, 23, 24, 25, 26. This might suggest that examination of 4q12amp by CGP in KIT‐overexpressing melanoma is warranted as to best delineate which TKIs, beyond anti‐KIT TKIs, may best benefit these patients 22, 23, 24, 25, 26.
In NSCLC, oncogenic drivers predominantly occur mutually exclusive fashion (i.e., an EGFR mutant lung cancer will not also harbor an ALK fusion) 27, 28. Interestingly, however, 50% lung adenocarcinomas harboring 4q12amp have no other drivers, but the remaining 50% do (Fig. 2). Similarly, within the TCGA lung adenocarcinoma data set, three specimens (1.3%) harbored concurrent KIT/KDR/PDGFRA amplification, and among these cases, one of three had ERBB2 amplification, with no putative drivers in two of three (data now shown). Moreover, the frequency of 4q12amp is lowest in lung adenocarcinoma and higher in other lung carcinoma types, which is the opposite of known oncogenic drivers in NSCLC which are typically most frequent in adenocarcinoma.
However, the index cases presented here and previously reported suggest that despite not neatly fitting our conception of an oncogenic driver, 4q12amp in advanced cancers can be targeted for patient benefit. In the trial of axitinib for recurrent AdCC, the single longest responder (almost four times longer than the median PFS for the rest of the study cohort) was the 4q12amp patient profiled on the platform used in this study 9. Axtinib has IC50 in the nanomolar range for VEGFR1–3, PDGFRA, and KIT 29. This specific benefit of axitinib for this patient suggests that matching a TKI to 4q12amp can benefit the patient. The index cases here had similar durable clinical benefit exceeding 20 months of stable disease from TKIs with known anti‐PDGFRA and KIT activity. The aggregate of these observations and the lack of impact of 4q12amp on prognosis per analysis of the TCGA suggest strongly that 4q12amp can be targeted for clinical benefit.
Which TKI Would Be Most Effective for Patients with Advanced Cancer Harboring 4q12amp?
Imatinib, pazopanib and axitinib have all benefitted the patients in this series, but each has different potencies against KIT, KDR, and PDGFRA (supplemental online Table 5). In tandem with clinical investigation, laboratory studies are needed to develop an understanding of which TKI may be superior for targeting 4q12amp and, equally importantly, which kinase, if one can be singled out, may be the critical oncogenic driver. A recent laboratory study of 4q12amp in a thyroid cancer derived cell line suggested that levatinib, recently approved in hepatocellular carcinoma, may also be an effective agent 30.
Importantly, reviewing the therapeutic strategies for metastatic breast cancer gives a relevant insight as to optimizing therapy for patients with harboring 4q12amp and suggests that monotherapy with a TKI alone, no matter how well matched, may be insufficient. For HER2‐amplified breast cancer, trastuzumab alone, an anti‐HER2 targeted monoclonal antibody, yielded modest benefit for HER2‐amplified metastatic breast cancer, but the combination of trastuzumab and a chemotherapy backbone was very beneficial to patients and is approved in the metastatic setting 31, 32. By analogy, the combination of a TKI and a chemotherapy backbone may be more effective for 4q12amp patients than a TKI alone and increase magnitude of response. Posing this question in a clinical trial is an important next step for this class of patients with cancer.
The size of the database of patient genomic profiles (n = 132,872 cases) is a clear strength of our study and allows reliable detection of rare variants such as 4q12amp, beyond that observed in the TCGA. However, we acknowledge limitations of our findings, largely owing to lack of complete clinical annotation. In this vein, we acknowledge that broad conclusions on clinical activity cannot be definitively drawn from our limited cases. However, a genomic biomarker‐driven treatment paradigm is both feasible and associated with improved patient outcomes, and potentially lower toxicities, across multiple basket trials and meta‐analyses 15, 33, 34, 35, 36. Large collaborative efforts including the NCI‐MATCH and ASCO TAPUR trials may be the optimal forum to explore 4q12amp clinical actionability, although 4q12 is not currently included as a drug match 37.
Conclusion
We identify recurrent 4q12amp across tumor types with specific enrichment in sarcomas as a whole, several sarcoma types, CNS neoplasms, and several cancers that arise in multiple anatomic sites. Beyond this demonstration of significant frequency and selective enrichment, the index cases with durable patient benefit presented here further buttress the targetability of this amplicon by matched TKIs. However, the identification of optimally effective TKI remains to be systematically investigated, both clinically and in the laboratory setting. As such, 4q12amp can be characterized as an oncogenic contributor either in conjunction with other oncogenic drivers or by itself, as suggested by studies in lung adenocarcinoma. Ongoing clinical trials with diverse TKIs may further define the extent of 4q12amp targetability and should be linked to an assessment of whether TKI treatment with a chemotherapy backbone may maximize patient benefit.
Author Contributions
Conception/design: Umut Disel, Russell Madison, Kumar Abhishek, Jon H. Chung, Alexa B. Schrock, Jeffrey S. Ross, Vincent A. Miller, Siraj M. Ali
Provision of study material or patients: Umut Disel, Russell Madison, Kumar Abhishek, Garrett M. Frampton, Lee A. Albacker, Adam Benson, Jennifer Webster, Chaitali Nangia, M.A. Oturk, Sumanta K. Pal, Breelyn Wilky, Samuel J. Klempner
Collection and/or assembly of data: Umut Disel, Russell Madison, Kumar Abhishek, Jon H. Chung, Sally E. Trabucco, Asli O. Matos, Garrett M. Frampton, Lee A. Albacker, Venkataprasanth Reddy, Nuri Karadurmus, Sumanta K. Pal, Breelyn Wilky, Ethan S. Sokol, Laurie M. Gay, Salil Soman, Shridar Ganesan, Phil J. Stephens, Alexa B. Schrock, Jeffrey S. Ross, Vincent A. Miller, Siraj M. Ali
Data analysis and interpretation: Umut Disel, Russell Madison, Jon H. Chung, Sally E. Trabucco, Asli O. Matos, Garrett M. Frampton, Lee A. Albacker, Venkataprasanth Reddy, Nuri Karadurmus, Semra Paydas, Sherri Z. Milus, Sumanta K. Pal, Breelyn Wilky, Ethan S. Sokol, Laurie M. Gay, Salil Soman, Shridar Ganesan, Katherine Janeway, Phil J. Stephens, Viola W. Zhu, Sai‐Hong Ignatius Ou, Christine M. Lovly, Mrinal Gounder, Alexa B. Schrock, Jeffrey S. Ross, Vincent A. Miller
Manuscript writing: Umut Disel, Russell Madison, Jon H. Chung, Venkataprasanth Reddy, Semra Paydas, Ruben Cabanillas, Ethan S. Sokol, Laurie M. Gay, Shridar Ganesan, Phil J. Stephens, Sai‐Hong Ignatius Ou, Alexa B. Schrock, Jeffrey S. Ross, Vincent A. Miller
Final approval of manuscript: Umut Disel, Russell Madison, Kumar Abhishek, Jon H. Chung, Sally E. Trabucco, Asli O. Matos, Garrett M. Frampton, Lee A. Albacker, Venkataprasanth Reddy, Nuri Karadurmus, Adam Benson, Jennifer Webster, Semra Paydas, Ruben Cabanillas, Chaitali Nangia, M.A. Ozturk, Sherri Z. Millis, Sumanta K. Pal, Breelyn Wilky, Ethan S. Sokol, Laurie M. Gay, Salil Soman, Shridar Ganesan, Katherine Janeway, Phil J. Stephens, Viola W. Zhu, Sai‐Hong Ignatius Ou, Christine M. Lovly, Mrinal Gounder, Alexa B. Schrock, Jeffrey S. Ross, Vincent A. Miller, Samuel J. Klempner, Siraj M. Ali
Disclosures
Umut Disel: Foundation Medicine, Inc. (RF); Russell Madison: Foundation Medicine, Inc. (E); Jon H. Chung: Foundation Medicine, Inc. (E); Sally E. Trabucco: Foundation Medicine, Inc. (E); Asli O. Matos: Foundation Medicine (E, OI); Garrett M. Frampton: Foundation Medicine, Inc. (E); Lee A. Albacker: Foundation Medicine, Inc. (E); Venkataprasanth Reddy: Foundation Medicine (E); Jennifer Webster: Foundation Medicine, Inc. (E); Ruben Cabanillas: Foundation Medicine, Inc. (H); Breelyn Wilky: Merck, Pfizer, Agenus (RF), Springworks, Eli Lilly & Co, Immune Design (C/A); Ethan S. Sokol: Foundation Medicine, (E); Laurie M. Gay: Foundation Medicine, Inc. (E); Shridar Ganesan: Merck (E‐spouse), Inspirata (C/A, IP, SAB), Roche, Foundation Medicine (C/A); Katherine Janeway: Amgen (RF), Bayer (C/A); Phil J. Stephens: Foundation Medicine, Inc. (E‐former, stock holder); Christine M. Lovly: Pfizer, Astra Zeneca, Novartis, Genoptix, Sequenom, Clovis, ARIAD, Takeda, Foundation Medicine, Abbott, Qiagen (C/A, H); Astra Zeneca, Novartis, Xcovery (RF); Mirinal Gounder: Bayer, Epizyme, Karyopharm, Daiichi‐Sankyo, GSK, Springwork Therapeutics (C/A); Alexa B. Schrock: Foundation Medicine, Inc. (E, OI); Jeffrey S. Ross: Foundation Medicine (E); Vincent A. Miller: Foundation Medicine, Inc. (E, Other‐equity interest); Samuel J. Klempner: Astellas, Lilly Oncology, Merck (C/A), Merck, Leap Therapeutics (RF), Foundation Medicine, Inc (H); Siraj M. Ali: Foundation Medicine, Inc. (E, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figures
Supplemental Tables
Acknowledgments
The authors wish to acknowledge all the investigators whose work could not be cited in this format. This work was supported in part by a research grant of in kind testing to U.D. from Foundation Medicine, Inc, for an exceptional responder study. M.A. Ozturk is currently affiliated with Bahcesehir University, Faculty of Medicine, Department of Medical Oncology, Istanbul, Turkey. Breelyn Wilky is currently affiliated with University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA. Data presented in part at ESMO 2017 Madrid as Abstract #1633PD.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Shaw AT, Hsu PP, Awad MM et al. Tyrosine kinase gene rearrangements in epithelial malignancies. Nat Rev Cancer 2013;13:772–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garraway LA, Verweij J, Ballman KV. Precision oncology: An overview. J Clin Oncol 2013;31:1803–1805. [DOI] [PubMed] [Google Scholar]
- 3. Bagci O, Kurtgöz S. Amplification of cellular oncogenes in solid tumors. North Am J Med Sci 2015;7:341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Santarius T, Shipley J, Brewer D et al. A census of amplified and overexpressed human cancer genes. Nat Rev Cancer 2010;10:59–64. [DOI] [PubMed] [Google Scholar]
- 5. Ramos AH, Dutt A, Mermel C et al. Amplification of chromosomal segment 4q12 in non‐small cell lung cancer. Cancer Biol Ther 2009;8:2042–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Holtkamp N, Ziegenhagen N, Malzer E et al. Characterization of the amplicon on chromosomal segment 4q12 in glioblastoma multiforme. Neuro Oncol 2007;9:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zietsch J, Ziegenhagen N, Heppner FL et al. The 4q12 amplicon in malignant peripheral nerve sheath tumors: Consequences on gene expression and implications for sunitinib treatment. PloS One 2010;5:e11858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nilsson MB, Giri U, Gudikote J et al. KDR Amplification is associated with VEGF‐induced activation of the mTOR and invasion pathways but does not predict clinical benefit to the VEGFR TKI vandetanib. Clin Cancer Res 2016;22:1940–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ho AL, Dunn L, Sherman EJ et al. A phase II study of axitinib (AG‐013736) in patients with incurable adenoid cystic carcinoma. Ann 2016;27:1902–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He J, Abdel‐Wahab O, Nahas MK et al. Integrated genomic DNA/RNA profiling of hematologic malignancies in the clinical setting. Blood 2016;127:3004–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sun JX, He Y, Sanford E et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput Biol 2018;14:e1005965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cerami E, Gao J, Dogrusoz U et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gao J, Aksoy BA, Dogrusoz U et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Drilon A, Laetsch TW, Kummar S et al. Efficacy of larotrectinib in TRK fusion‐positive cancers in adults and children. N Engl J Med 2018;378:731–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hyman DM, Piha‐Paul SA, Won H et al. HER kinase inhibition in patients with HER2‐ and HER3‐mutant cancers. Nature 2018;554:189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Perry JA, Kiezun A, Tonzi P et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc Natl Acad Sci USA 2014;111:E5564–E5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khan JA, Maki RG, Ravi V. Pathologic angiogenesis of malignant vascular sarcomas: Implications for treatment. J Clin Oncol 2018;36:194–201. [DOI] [PubMed] [Google Scholar]
- 19. Ravi V, Sanford EM, Wang WL, et al. Antitumor response of VEGFR2‐ and VEGFR3‐amplified angiosarcoma to pazopanib. J Natl Compr Cancer Netw 2016;14:499–502. [DOI] [PubMed] [Google Scholar]
- 20. Bradley PJ. Adenoid cystic carcinoma evaluation and management: Progress with optimism! Curr Opin Otolaryngol Head Neck Surg 2017;25:147–153. [DOI] [PubMed] [Google Scholar]
- 21. Morris LGT, Chandramohan R, West L et al. The molecular landscape of recurrent and metastatic head and neck cancers: Insights from a precision oncology sequencing platform. JAMA Oncol 2017;3:244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hodi FS, Corless CL, Giobbie‐Hurder A et al. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun‐damaged skin. J Clin Oncol 2013;31:3182–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee SJ, Kim TM, Kim YJ et al. Phase II trial of nilotinib in patients with metastatic malignant melanoma harboring KIT gene aberration: A multicenter trial of Korean Cancer Study Group (UN10‐06). The Oncologist 2015;20:1312–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo J, Si L, Kong Y et al. Phase II, open‐label, single‐arm trial of imatinib mesylate in patients with metastatic melanoma harboring c‐Kit mutation or amplification. J Clin Oncol 2011;29:2904–2909. [DOI] [PubMed] [Google Scholar]
- 25. Carvajal RD, Antonescu CR, Wolchok JD et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011;305:2327–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guo J, Carvajal RD, Dummer R et al. Efficacy and safety of nilotinib in patients with KIT‐mutated metastatic or inoperable melanoma: Final results from the global, single‐arm, phase II TEAM trial. Ann Oncol 2017;28:1380–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Imielinski M, Berger AH, Hammerman PS et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 2012;150:1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Suh JH, Johnson A, Albacker L et al. Comprehensive genomic profiling facilitates implementation of the National Comprehensive Cancer Network guidelines for lung cancer biomarker testing and identifies patients who may benefit from enrollment in mechanism‐driven clinical trials. The Oncologist 2016;21:684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hu‐Lowe DD, Zou HY, Grazzini ML et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG‐013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res 2008;14:7272–7283. [DOI] [PubMed] [Google Scholar]
- 30. Pozdeyev N, Gay L, Sokol ES et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 2018;24:3059–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vogel CL, Cobleigh MA, Tripathy D et al. First‐line Herceptin monotherapy in metastatic breast cancer. Oncology 2001;61(suppl 2):37–42. [DOI] [PubMed] [Google Scholar]
- 32. Slamon DJ, Leyland‐Jones B, Shak S et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783–792. [DOI] [PubMed] [Google Scholar]
- 33. Hyman DM, Puzanov I, Subbiah V et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tredan O, Corset V, Wang Q et al. Routine molecular screening of advanced refractory cancer patients: An analysis of the first 2490 patients of the ProfiLER study. J Clin Oncol 2017;35:LBA100a. [Google Scholar]
- 35. Tourneau CL, Delord JP, Gonçalves A et al. Molecularly targeted therapy based on tumour molecular profiling versus conventional therapy for advanced cancer (SHIVA): A multicentre, open‐label, proof‐of‐concept, randomised, controlled phase 2 trial. Lancet Oncol 2015;16:1324–1334. [DOI] [PubMed] [Google Scholar]
- 36. Goodman AM, Kato S, Bazhenova L et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther 2017;16:2598–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jhaveri KL, Makker V, Wang XV et al. Ado‐trastuzumab emtansine (T‐DM1) in patients (pts) with HER2 amplified (amp) tumors excluding breast and gastric/gastro‐esophageal junction (GEJ) adenocarcinomas: Results from the National Cancer Institute (NCI) Molecular Analysis for Therapy Choice (MATCH) trial. J Clin Oncol 2018;36:100a. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Figures
Supplemental Tables