

Abstract
Background
This multicenter, open‐label, phase Ib study investigated the safety and efficacy of binimetinib (MEK inhibitor) in combination with buparlisib (phosphatidylinositol 3‐kinase [PI3K] inhibitor) in patients with advanced solid tumors with RAS/RAF alterations.
Materials and Methods
Eighty‐nine patients were enrolled in the study. Eligible patients had advanced solid tumors with disease progression after standard therapy and/or for which no standard therapy existed. Evaluable disease was mandatory, per RECIST version 1.1 and Eastern Cooperative Oncology Group performance status 0‐2. Binimetinib and buparlisib combinations were explored in patients with KRAS‐, NRAS‐, or BRAF‐mutant advanced solid tumors until the maximum tolerated dose and recommended phase II dose (RP2D) were defined. The expansion phase comprised patients with epidermal growth factor receptor (EGFR)‐mutant, advanced non‐small cell lung cancer, after progression on an EGFR inhibitor; advanced RAS‐ or BRAF‐mutant ovarian cancer; or advanced non‐small cell lung cancer with KRAS mutation.
Results
At data cutoff, 32/89 patients discontinued treatment because of adverse events. RP2D for continuous dosing was buparlisib 80 mg once daily/binimetinib 45 mg twice daily. The toxicity profile of the combination resulted in a lower dose intensity than anticipated. Six (12.0%) patients with RAS/BRAF‐mutant ovarian cancer achieved a partial response. Pharmacokinetics of binimetinib were not altered by buparlisib. Pharmacodynamic analyses revealed downregulation of pERK and pS6 in tumor biopsies.
Conclusion
Although dual inhibition of MEK and the PI3K pathways showed promising activity in RAS/BRAF ovarian cancer, continuous dosing resulted in intolerable toxicities beyond the dose‐limiting toxicity monitoring period. Alternative schedules such as pulsatile dosing may be advantageous when combining therapies.
Implications for Practice
Because dysregulation of the mitogen‐activated protein kinase (MAPK) and the phosphatidylinositol 3‐kinase (PI3K) pathways are both frequently involved in resistance to current targeted therapies, dual inhibition of both pathways may be required to overcome resistance mechanisms to single‐agent tyrosine kinase inhibitors or to treat cancers with driver mutations that cannot be directly targeted. A study investigating the safety and efficacy of combination binimetinib (MEK inhibitor) and buparlisib (PI3K inhibitor) in patients harboring alterations in the RAS/RAF pathway was conducted. The results may inform the design of future combination therapy trials in patients with tumors harboring mutations in the PI3K and MAPK pathways.
Keywords: Binimetinib, Buparlisib, RAS/RAF, Phase Ib, Ovarian cancer
Short abstract
This article reports on the safety and efficacy of binimetinib and buparlisib combination therapy in patients with advanced solid tumors harboring selected genomic alterations in the RAS/RAF pathway.
Introduction
The mitogen‐activated protein kinase (MAPK [RAS/RAF/MEK/ERK]) and phosphatidylinositol 3‐kinase (PI3K [PI3K/AKT/mTOR]) pathways are frequently dysregulated in cancer. Oncogenic mutations in RAS and its downstream signals occur in a variety of tumors, including non‐small cell lung cancer (NSCLC; 20%–30%), melanoma (30%–60%), thyroid (30%–50%), colorectal (2%–45%), and ovarian cancers (3%–11%) 1, 2, 3, 4, 5, 6, 7, 8. Furthermore, the PI3K and MAPK pathways are interconnected; inhibition of one pathway can lead to compensatory activation of the other. Therefore, dual blockade of both pathways may be required 9, 10. Synergistic antitumor efficacy by dual PI3K/MAPK pathway inhibition has been demonstrated in preclinical in vitro and in vivo models of basal‐like breast cancer and lung cancer, and has shown promise in the clinical setting 11, thus supporting this hypothesis 12, 13.
Binimetinib (MEK162) is a potent, highly selective, orally bioavailable, non‐ATP‐competitive allosteric inhibitor of MEK1 and MEK2 14, 15, 16. The maximum tolerated dose (MTD) of single‐agent binimetinib is 60 mg b.i.d. 17; single‐agent binimetinib has shown preliminary clinical activity in solid tumors such as NRAS‐ and BRAF‐mutant melanoma (objective response rate 11% and 6%, respectively) 14, 17, 18. A phase III study (NEMO) of binimetinib versus dacarbazine in unresectable or metastatic NRAS‐mutant melanoma met its primary endpoint of improvement in progression‐free survival (PFS; hazard ratio = 0.62; 95% confidence interval [CI]: 0.47–0.80; p < .001) 19.
Buparlisib (BKM120) is an oral pan‐class I PI3K inhibitor that selectively targets all four isoforms of class I PI3K 20, 21. The MTD of single‐agent buparlisib is 100 mg once daily (q.d.) 20, 22. Although only modest improvements in PFS were reported in a randomized clinical trial in estrogen receptor‐positive breast cancer, higher activity was seen in patients with PIK3CA‐activating mutations 23.
Given the preclinical rationale for dual inhibition of the PI3K and MAPK pathways, a phase Ib basket trial was conducted to evaluate the safety and MTD of binimetinib/buparlisib combination therapy in patients with advanced solid tumors harboring selected genomic alterations in the RAS/RAF pathway (http://clinicaltrials.gov, number NCT01363232). Secondary objectives of the study were evaluation of efficacy, pharmacokinetics (PK), and pharmacodynamics (PD).
Materials and Methods
Study Design
This was a multicenter, open‐label, phase Ib study to determine the MTD and/or recommended phase II dose (RP2D) for combination buparlisib/binimetinib, followed by an expansion phase to further assess safety and efficacy in patients with advanced solid tumors harboring selected genomic alterations in the RAS/RAF pathway. Dose escalation was conducted in patients with KRAS‐, NRAS‐, or BRAF‐mutant advanced solid tumors. Upon determination of RP2D, two expansion arms were then opened in order to further assess safety and preliminary antitumor activity.
Eligibility Criteria
Patients >18 years of age with advanced solid tumors and disease progression after standard therapy and/or for whom no standard anticancer therapy exists were eligible. Evaluable disease was mandatory as determined by RECIST version 1.1 and Eastern Cooperative Oncology Group performance status 0‐2. Patients with diabetes mellitus, impaired cardiovascular function, clinically significant cardiovascular diseases, history of depression, or ocular disease (with a history or current evidence of central serous retinopathy [CSR], retinal vein occlusion [RVO], or ophthalmopathy that would be considered a risk factor for CSR/RVO) were excluded.
Dose escalation was performed in patients with advanced refractory colorectal cancer harboring KRAS, NRAS, or BRAF mutations; NSCLC with KRAS mutation; cutaneous malignant melanoma with NRAS or BRAF mutations; pancreatic cancer (irrespective of RAS/RAF mutation status, because most have KRAS mutations); triple‐negative breast cancer (because tumors have high prevalence of RAS/MAPK pathway activation) 24; and other advanced solid tumors with documented KRAS, NRAS, or BRAF mutations. For evaluation of RP2D, patients were enrolled in one of two expansion arms. Arm 1 comprised patients with epidermal growth factor receptor (EGFR)‐mutant, advanced NSCLC (including NSCLC with documented T790M activating mutations), with disease progression on a prior EGFR‐inhibitor treatment. Arm 2 comprised patients with advanced ovarian cancer with documented RAS or RAF mutations, or advanced NSCLC with documented KRAS mutation.
The study was conducted in accordance with the Declaration of Helsinki and guidelines for Good Clinical Practice as defined by the International Conference on Harmonisation. Patients provided written informed consent, and regulatory approval was obtained from participating institutions.
Treatments
Starting doses for the combination were 30 mg binimetinib b.i.d. and 50 mg buparlisib q.d. (both 50% of the single‐agent MTD). Combinations of doses were explored based on an adaptive Bayesian logistic regression model (BLRM) for dose‐escalation with overdose control (EWOC) 25 until the MTD and RP2D were defined (supplemental online Table 1). At all decision time points, the adaptive BLRM permitted alterations in the dose increments based on the observed dose‐limiting toxicities (DLTs). Only one of the two combination partners could be escalated, and the maximum intercohort dose escalation for a combination partner was limited to 100%. Following determination of the RP2D, patients enrolled in Arm 1 (n ≥ 15) and Arm 2 (advanced ovarian cancer with documented RAS or RAF mutations [n ≥ 15], or advanced NSCLC with documented KRAS mutation [n ≥ 10]) received the recommended RP2D: 80 mg buparlisib q.d. and 45 mg binimetinib b.i.d.
Safety and Efficacy Assessments
A treatment cycle was defined as 28 days. Clinical and laboratory assessments were conducted at baseline, throughout the study, and up to 30 days after the last treatment dose.
Safety assessments included monitoring and recording all adverse events (AEs) with their severity and relationship to study drug, according to Common Terminology Criteria for Adverse Events (version 4.0). Given known pulmonary, ophthalmic, and psychiatric effects of the drugs, patients were monitored at baseline and during the study with pulmonary function tests, mood questionnaires (Patient Health Questionnaire [PHQ‐9] depression scale and the Generalized Anxiety Disorder 7‐item [GAD‐7] anxiety scale), and detailed ophthalmic examination 10, 20. Supportive medications to manage side effects were permitted.
Efficacy assessments were conducted at baseline and every 8 weeks and evaluated by RECIST version 1.1 criteria.
Pharmacokinetic Assessments
Pharmacokinetic assessments were performed for binimetinib, its primary active metabolite (AR00426032), and buparlisib; plasma levels were determined from samples collected before dose and 0.5, 1.5, 3, 5, 8, and 24 hours after dose on Day 1 and Day 15 of Cycle 1 from all patients in the dose‐escalation phase and expansion arm 2. The pharmacokinetics of binimetinib and buparlisib were investigated after a single dose (Day 1) and multiple doses (Day 15) in Cycle 1. The potential impact of coadministration of buparlisib on binimetinib was assessed by examining maximum plasma concentration (Cmax) and area under the concentration curve (AUCtau) at the same level of binimetinib across doses of buparlisib on Day 15.
Pharmacodynamic Assessments
Paired fresh tumor biopsy samples were collected at baseline and Day 15. Biopsies were recommended for all patients in the dose‐escalation phase and were required for patients in the dose‐expansion phase. Specimens were flash frozen and analyzed by immunohistochemistry for the PD markers pS6 and pERK (IPG, Belgium) to confirm target modulation of the MAPK and PI3K pathway, respectively. Immunohistochemistry results were semiquantitatively evaluated using the H‐score. The percentage of cells with positive staining was estimated for each intensity level (0, no staining; 1+, weak staining; 2+, moderate staining; 3+, strong staining). The H‐score is calculated using the following equation: [1 × (% cells 1+) + 2 × (% cells 2+) + 3 × (% cells 3+)]. The H‐score ranges from 0 to 300.
Statistical Analyses
A BLRM guided by EWOC principle was used to guide dose escalation of the combination treatment to its MTD and RP2D. A five‐parameter BLRM for combination treatment was fitted on Cycle 1 DLT data accumulated throughout the dose escalation to model the dose‐toxicity relationship of buparlisib and binimetinib when given in combination. Dose recommendation was based on the probability that the true DLT rate for each dose combination lies in one of the following categories: underdosing (0%, 16%), targeted toxicity (16%, 35%), or excessive toxicity (35%, 100%). The MTD and/or RP2D was expanded by enrolling additional patients eligible for the safety set to be evaluated for safety, tolerability, pharmacokinetics, and biologic activity of buparlisib and binimetinib. A sample size of 40 patients in the expansion cohort had an 87% probability of detecting an AE with a true incidence rate of ≥5%.
Results
Patient Characteristics
From August 15, 2011, to March 24, 2014 (data cutoff date), 89 patients were enrolled and received at least one dose of the buparlisib/binimetinib combination (Table 1). One patient withdrew consent before receiving the first dose of study drug; 48 and 41 patients were treated in the escalation and expansion phases, respectively. Baseline characteristics were similar across treatment groups in the escalation phase.
Table 1.
Baseline patient characteristics
| Characteristic | Buparlisib/binimetinib dose, mg q.d./b.i.d. | |||||||
|---|---|---|---|---|---|---|---|---|
| 50/30 (n = 11) | 50/45 (n = 5) | 60/45 (n = 5) | 70/30 (n = 6) | 80/30 (n = 6) | 80/45 (n = 50) | 90/45 (n = 6) | All patients (n = 89) | |
| Sex, male/female, n (%) | 5 (46)/6 (55) | 3 (60)/2 (40) | 2 (40)/3 (60) | 1 (17)/5 (83) | 3 (50)/3 (50) | 17 (34)/33 (66) | 3 (50)/3 (50) | 34 (38)/55 (62) |
| Age, median (range), years | 64 (36–77) | 57 (47–75) | 52 (52–78) | 50 (42–69) | 56 (43–73) | 54 (30–77) | 66 (54–76) | 57 (30–78) |
| WHO PS, n (%) | ||||||||
| 0 | 3 (27) | 0 | 1 (20) | 4 (67) | 4 (67) | 21 (42) | 2 (33) | 35 (39) |
| 1 | 8 (73) | 5 (100) | 4 (80) | 2 (33) | 2 (33) | 28 (56) | 3 (50) | 52 (58) |
| 2 | 0 | 0 | 0 | 0 | 0 | 1 (2) | 1 (17) | 2 (2) |
| Primary cancer site, n (%) | ||||||||
| Lung | 0 | 1 (20) | 0 | 1 (17) | 0 | 24 (48) | 1 (17) | 27 (30) |
| Ovary | 1 (9) | 0 | 0 | 0 | 0 | 18 (36) | 0 | 19 (21) |
| Colon | 4 (36) | 0 | 1 (20) | 4 (67) | 1 (17) | 2 (4) | 1 (17) | 13 (15) |
| Rectum | 4 (36) | 1 (20) | 1 (20) | 0 | 0 | 2 (4) | 1 (17) | 9 (10) |
| Othera | 0 | 2 (40) | 1 (20) | 0 | 1 (17) | 1 (2) | 2 (33) | 7 (8) |
| Breast (triple negative) | 2 (18) | 0 | 0 | 1 (17) | 2 (33) | 1 (2) | 0 | 6 (7) |
| Pancreas | 0 | 1 (20) | 2 (40) | 0 | 2 (33) | 0 | 1 (17) | 6 (7) |
| Melanoma | 0 | 0 | 0 | 0 | 0 | 2 (4) | 0 | 2 (2) |
| No. of prior antineoplastic regimens, median (range) | 3.0 (2.0–12.0) | 3.5 (1.0–6.0) | 2.0 (1.0–4.0) | 3.0 (2.0–3.0) | 2.5 (1.0–5.0) | 3.0 (1.0–12.0) | 2.0 (1.0–5.0) | 3.0 (1.0–12.0) |
Other sites included unknown/unspecified (n = 3) and cervix, head and neck, small intestine, and thyroid (n = 1 each).
Abbreviations: PS, performance status; q.d., once daily; WHO, World Health Organization.
Patient Disposition
At the cutoff date, 84 patients (94.4%) had discontinued study treatment; 5 patients in the expansion phase were still ongoing. The most common reasons for discontinuation were disease progression (43 patients, 48.3%) and AEs (32 patients, 36.0%). Other reasons included withdrawal of consent (seven patients, 7.9%), death due to disease progression (one patient, 1.1%), and administrative problems (one patient, 1.1%).
MTD and RP2D Determination
Thirteen patients (20.3%) experienced at least one DLT during the first cycle; the most frequent DLTs were diarrhea (four patients, 6.3%), CSR, and stomatitis (two patients each, 3.1%). Both CSRs were grade 1, reversible, and manageable. In the dose‐escalation phase, no DLTs were reported in the buparlisib 50‐mg/binimetinib 45‐mg group or the buparlisib 60‐mg/binimetinib 45‐mg group. The dose at which DLTs occurred in both the dose‐escalation and dose‐expansion phases are specified in supplemental online Table 2.
The MTD for the dose combination was buparlisib 90 mg q.d. and binimetinib 45 mg b.i.d. in a continuous schedule; however, a lower RP2D of buparlisib (80 mg q.d.) and binimetinib (45 mg b.i.d.) was recommended. During the dose escalation phase, there were fewer DLTs reported in the buparlisib (80 mg q.d.) and binimetinib (45 mg b.i.d.) group (1/7 reported grade 3 maculo‐papular rash) than in the buparlisib (90 mg q.d.) and binimetinib (45 mg b.i.d.) group (2/6 reported DLTs; grade 3 diarrhea [n = 1] and grade 3 anaphylactic reaction and grade 3 swelling face [n = 1]); the Bayesian model estimated the probability of excessive toxicity was higher for buparlisib 90 mg q.d. and binimetinib 45 mg b.i.d. (0.052) than for buparlisib 80 mg q.d. and binimetinib 45 mg b.i.d. (0.012), and the true DLT rate lies in the excessive toxicity category. Therefore, both dose combinations satisfied EWOC criterion (less than 25% chance that true DLT rate ≥ 35%). The lower buparlisib dose was chosen to limit DLTs and excessive toxicity while still meeting EWOC criterion.
Patient Exposure and Safety
The most common all‐grade AEs (≥10%) suspected of being study drug related included increased blood creatine phosphokinase (CPK; 59.6%), diarrhea (57.3%), and increased aspartate aminotransferase (AST; 49.4%; Table 2). The most common grade 3/4 AEs suspected of being study drug related included increased CPK (27%), increased alanine aminotransferase (ALT; 14.6%), and increased AST (13.5%; Table 2). The most common (≥10%) all‐cause all‐grade AEs and all‐cause grade 3/4 AEs are shown in supplemental online Table 3.
Table 2.
Adverse events, suspected to be related to study drug, reported by ≥10% of patients (all grades; safety set)
| Preferred term | Buparlisib/binimetinib dose, mg q.d./b.i.d. | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 50/30 (n = 11) | 50/45 (n = 5) | 60/45 (n = 5) | 70/30 (n = 6) | 80/30 (n = 6) | 80/45 (n = 49) | 90/45 (n = 7) | All patients (N = 89) | |||||||||
| All | G3/4 | All | G3/4 | All | G3/4 | All | G3/4 | All | G3/4 | All | G3/4 | All | G3/4 | All | G3/4 | |
| n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | n (%) | |
| Total | 11 (100) | 5 (45.5) | 5 (100) | 5 (100) | 5 (100) | 3 (60.0) | 5 (83.3) | 3 (50.0) | 6 (100) | 4 (66.7) | 44 (89.8) | 33 (67.3) | 7 (100) | 4 (57.1) | 83 (93.3) | 57 (64.0) |
| Blood CPK increased | 5 (45.5) | 2 (18.2) | 4 (80.0) | 2 (40.0) | 3 (60.0) | 2 (40.0) | 3 (50.0) | 2 (33.3) | 3 (50.0) | 0 | 32 (65.3) | 15 (30.6) | 3 (42.9) | 1 (14.3) | 53 (59.6) | 24 (27.0) |
| Diarrhea | 5 (45.5) | 0 | 3 (60.0) | 1 (20.0) | 2 (40.0) | 0 | 2 (33.3) | 0 | 4 (66.7) | 0 | 29 (59.2) | 5 (10.2) | 6 (85.7) | 1 (14.3) | 51 (57.3) | 7 (7.9) |
| AST increase | 3 (27.3) | 0 | 3 (60.0) | 0 | 4 (80.0) | 1 (20.0) | 3 (50.0) | 1 (16.7) | 2 (33.3) | 1 (16.7) | 25 (51.0) | 8 (16.3) | 4 (57.1) | 1 (14.3) | 44 (49.4) | 12 (13.5) |
| Stomatitis | 2 (18.2) | 1 (9.1) | 2 (40.0) | 1 (20.0) | 1 (20.0) | 0 | 2 (33.3) | 0 | 4 (66.7) | 1 (16.7) | 20 (40.8) | 3 (6.1) | 2 (28.6) | 0 | 33 (37.1) | 6 (6.7) |
| ALT increase | 2 (18.2) | 0 | 1 (20.0) | 0 | 2 (40.0) | 1 (20.0) | 2 (33.3) | 0 | 1 (16.7) | 1 (16.7) | 20 (40.8) | 10 (20.4) | 4 (57.1) | 1 (14.3) | 32 (36.0) | 13 (14.6) |
| Nausea | 2 (18.2) | 0 | 1 (20.0) | 0 | 2 (40.0) | 0 | 2 (33.3) | 0 | 1 (16.7) | 0 | 21 (42.9) | 0 | 3 (42.9) | 0 | 32 (36.0) | 0 |
| Maculopapular rash | 1 (9.1) | 0 | 1 (20.0) | 1 (20.0) | 2 (40.0) | 1 (20.0) | 2 (33.3) | 1 (16.7) | 4 (66.7) | 2 (33.3) | 17 (34.7) | 4 (8.2) | 3 (42.9) | 2 (28.6) | 30 (33.7) | 11 (12.4) |
| Rash | 4 (36.4) | 0 | 3 (60.0) | 0 | 1 (20.0) | 1 (20.0) | 1 (16.7) | 0 | 4 (66.7) | 0 | 14 (28.6) | 3 (6.1) | 1 (14.3) | 0 | 28 (31.5) | 4 (4.5) |
| Dermatitis acneiform | 1 (9.1) | 1 (9.1) | 2 (40.0) | 1 (20.0) | 3 (60.0) | 0 | 2 (33.3) | 1 (16.7) | 1 (16.7) | 0 | 15 (30.6) | 1 (2.0) | 2 (28.6) | 0 | 26 (29.2) | 4 (4.5) |
| Fatigue | 4 (36.4) | 0 | 1 (20.0) | 0 | 0 | 0 | 2 (33.3) | 0 | 2 (33.3) | 0 | 12 (24.5) | 1 (2.0) | 2 (28.6) | 0 | 23 (25.8) | 1 (1.1) |
| Vomiting | 3 (27.3) | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 3 (50.0) | 0 | 13 (26.5) | 0 | 2 (28.6) | 0 | 22 (24.7) | 0 |
| Decreased appetite | 3 (27.3) | 0 | 1 (20.0) | 0 | 1 (20.0) | 0 | 1 (16.7) | 0 | 3 (50.0) | 0 | 9 (18.4) | 0 | 2 (28.6) | 0 | 20 (22.5) | 0 |
| Amylase increased | 1 (9.1) | 0 | 1 (20.0) | 1 (20.0) | 0 | 0 | 2 (33.3) | 0 | 1 (16.7) | 1 (16.7) | 11 (22.4) | 2 (4.1) | 1 (14.3) | 0 | 17 (19.1) | 4 (4.5) |
| Hyperglycemia | 3 (27.3) | 0 | 0 | 0 | 2 (40.0) | 0 | 0 | 0 | 0 | 0 | 10 (20.4) | 0 | 2 (28.6) | 0 | 17 (19.1) | 0 |
| Chorioretinopathy | 2 (18.2) | 0 | 2 (40.0) | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 0 | 7 (14.3) | 0 | 3 (42.9) | 0 | 16 (18.0) | 0 |
| Lipase increased | 1 (9.1) | 0 | 0 | 0 | 1 (20.0) | 0 | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) | 11 (22.4) | 6 (12.2) | 1 (14.3) | 0 | 16 (18.0) | 7 (7.9) |
| Peripheral edema | 2 (18.2) | 0 | 2 (40.0) | 0 | 1 (20.0) | 0 | 0 | 0 | 2 (33.3) | 0 | 7 (14.3) | 0 | 0 | 0 | 14 (15.7) | 0 |
| Pruritus | 2 (18.2) | 0 | 1 (20.0) | 0 | 1 (20.0) | 0 | 1 (16.7) | 0 | 0 | 0 | 6 (12.2) | 1 (2.0) | 1 (14.3) | 0 | 12 (13.5) | 1 (1.1) |
| Dry skin | 0 | 0 | 2 (40.0) | 1 (20.0) | 1 (20.0) | 0 | 1 (16.7) | 0 | 0 | 0 | 3 (6.1) | 0 | 2 (28.6) | 0 | 9 (10.1) | 1 (1.1) |
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; G, grade; q.d., once daily.
Fifty‐three patients (59.6%) reported a grade 3/4 AE that required dose interruption and/or change; 54 patients (60.7%) with a grade 3/4 AE required additional therapy for diarrhea, pneumonia, nausea, and vomiting. At the RP2D dose, 26 patients (29.2%) discontinued study treatment for drug‐related AEs. The most commonly occurring AEs leading to discontinuation were maculopapular rash (7.9%), symptomatic blood CPK elevation, diarrhea, and ALT increase (3.4% each). One patient died while on treatment as a result of disease progression, and 10 patients died within 30 days after the last dose of study drug as a result of disease progression (n = 7), pneumonia (n = 2), and disseminated intravascular coagulation (n = 1). No deaths were suspected to be study drug related.
Efficacy
Sixty‐nine patients (77.5%) were evaluable for tumor response according to RECIST version 1.1 (Fig. 1A). Six patients (12.0%) from the dose‐expansion arm achieved a partial response (PR) and remained on treatment for 57 to 169 days. In patients treated with the RP2D (Fig. 1B; supplemental online Table 4), PR was most frequently observed in RAS/BRAF ovarian cancer (27.8%); one patient (7.7%) in the EGFR‐mutant NSCLC group achieved a PR. In the RP2D population, the highest disease control rate (the proportion of patients with a best overall response of complete response, PR, or stable disease) was reported for patients with RAS/BRAF ovarian cancer (61.1%; 95% CI: 35.7%–82.7%; supplemental online Table 4). The longest median PFS based on Kaplan‐Meier estimates was also reported in patients with RAS/BRAF ovarian cancer (median 3.7 months; 95% CI: 1.8 to not estimable).
Figure 1.
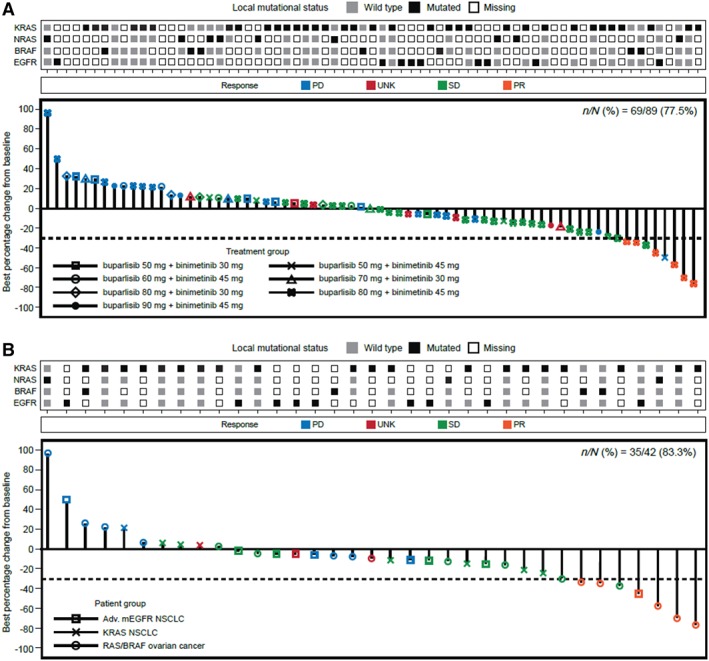
Best percentage change from baseline in sum of tumor diameters. (A): By local mutational status and treatment group (full analysis set). (B): By local mutational status (recommended phase II dose).
Abbreviations: Adv, advanced; mEGFR, mutant epidermal growth factor receptor; NSCLC, non‐small cell lung cancer; PD, progressive disease; SD, stable disease; UNK, unknown.
Pharmacokinetics
The potential impact of coadministration of buparlisib and binimetinib was assessed by examining Cmax and AUCtau at the same level of binimetinib across doses of buparlisib on Day 15 (Fig. 2).
Figure 2.
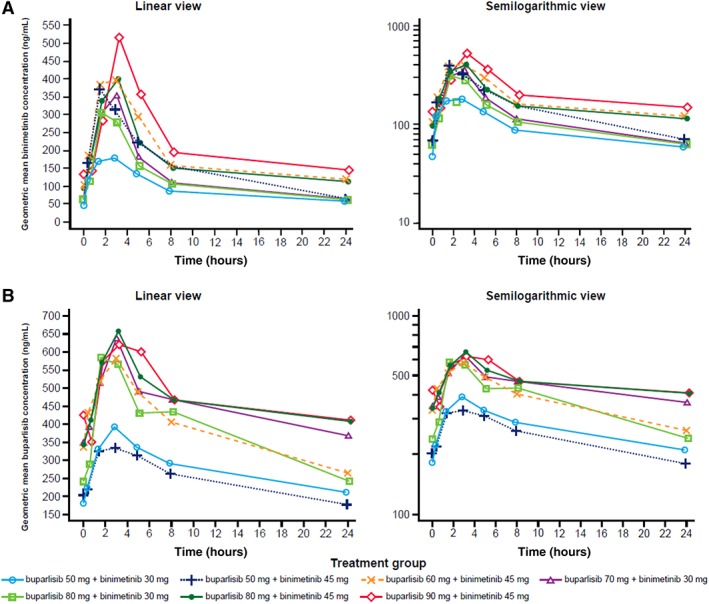
Geometric mean plasma concentration‐time profiles after repetitive (Day 15) daily combination doses of buparlisib and binimetinib by treatment group. (A): Binimetinib. (B): Buparlisib. “tau” represents the dosing interval; tau = 12 hours for binimetinib, tau = 24 hours for buparlisib.
For the combination of binimetinib 45 mg b.i.d. and buparlisib 80 mg q.d., the mean AUCtau of 2,668 ng•hour/mL (supplemental online Table 5) was comparable to or higher than the expected exposure (2,679 ng•hour/mL) derived from the population PK model developed for binimetinib monotherapy and that seen in the single‐agent phase I study (mean AUCtau [1,740 ng•hour/mL]) using the same dosing schedule 17. In the present study, the AUCtau and Cmax were 11,690.71 hour•ng/mL and 764.3 ng/mL, respectively. In the single‐agent study, AUCtau and Cmax were 15,413 ng•hour/mL and 1,098 ng/mL, respectively 22. Overall, the peak concentration of buparlisib and exposure at steady state were lower than expected from single‐agent data (supplemental online Table 5), and the PK of binimetinib was not altered by buparlisib. When combined with binimetinib, Cmax and AUCtau of buparlisib were reduced by >50% in many patients, but this reduction is within the variability of buparlisib exposure and clinical relevance is unclear.
Pharmacodynamics
Biomarker analyses on baseline and postbaseline fresh tumor samples (20 evaluable baseline and postbaseline pairs for pERK analysis; 19 evaluable baseline and postbaseline pairs for pS6 analysis) showed that inhibition of MAPK and PI3K signaling by combination therapy resulted in downregulation of pERK and pS6 (Fig. 3). At the RP2D dose combination, median cytoplasmic pERK H‐score at baseline and postbaseline was 210 (range, 0–300) and 190 (range, 60–300), respectively; the median nuclear pERK H‐score at baseline and postbaseline was 180 (range, 0–300) and 160 (range, 0–300). Median cytoplasmic pS6 (Ser235 and Ser240) H‐score at baseline and postbaseline was 120 (range, 3‐280) and 110 (range, 9–140). H‐score could not be analyzed for nuclear pS6 levels.
Figure 3.
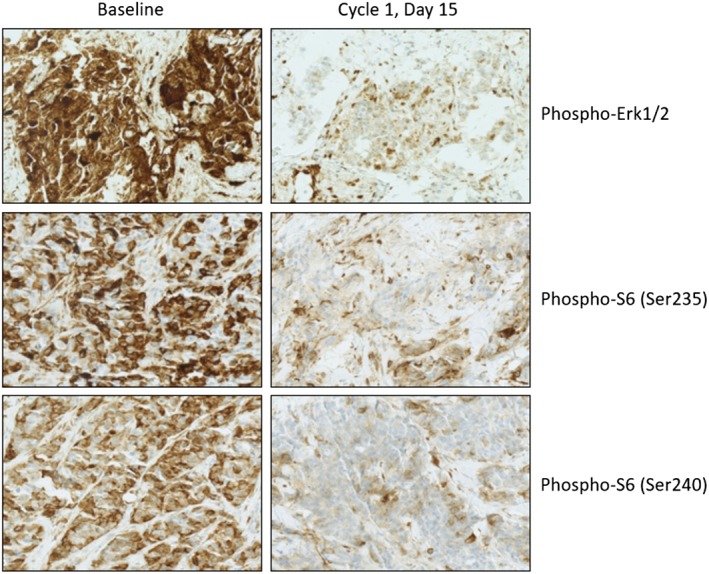
Immunohistochemical staining on pre‐ and postbaseline tumor biopsies (representative images shown at ×25 magnification).
Discussion
In this study, we show that buparlisib, an oral pan‐PI3K inhibitor, and binimetinib, an oral MEK1/2 inhibitor, have overlapping toxicities when given in combination; the RP2D of this combination is lower than the MTD for single‐agent buparlisib (100 mg/day) and binimetinib (60 mg b.i.d.) 17, 20, 22. Although the pharmacokinetics of binimetinib are comparable to those observed in a single‐agent phase I study, the peak concentration of buparlisib and exposure at steady state were lower than expected from single‐agent data 17. Despite the lower exposures of buparlisib, there was significant interpatient variability, and a significant proportion of patients required dose reductions and treatment discontinuation even at the RP2D.
In terms of efficacy, the most striking responses were seen in KRAS/NRAS/BRAF‐mutant ovarian cancer, where standard therapies are ineffective 26. In contrast, a phase II study of single‐agent selumetinib reported a 15% response rate irrespective of BRAF or KRAS status 26. The doubling of response rates seen in our study was similarly observed in a previous MEK‐PI3K trial 11, thereby lending support to a combination strategy. A phase III study (MILO; NCT01849874) of single‐agent binimetinib compared with standard chemotherapy in genomically unselected patients with low‐grade serous (LGS) ovarian cancer failed to meet its futility endpoint 27. Based on findings from our study, further exploration of combination therapy in this patient population with selected genomic alterations is warranted.
Although dual PI3K and MAPK pathway inhibition has strong preclinical rationale across a range of cancer types, the efficacy of the combination was not as robust in the clinic, perhaps with the exception of ovarian cancer (PR in 5/18 [27.8%] patients treated at RP2D). Several factors should be considered. First, toxicities from the combination resulted in frequent dose reductions, and the consequent suboptimal pathway modulation likely contributed to reduced efficacy. Second, although tumors may share common driver alterations, coexisting mutations and their genomic context—whether truncal, shared, or private—may explain the significant differences in responses between the cancers. In support, whole exome sequencing of LGS has shown a high prevalence of activating mutations in the MAPK pathway (~70%) with few “passenger” mutations likely explaining the sensitivity of this histology to combination strategy 28. Dual inhibition of PI3K and MEK in colon cancer cell lines demonstrated antitumor activity except in those harboring TP53 gene mutations 29. In targeting PI3K‐ and MAPK‐mutant pathways, the impact of “passenger” mutations needs further elucidation. Third, beyond genomic alterations, the dependency of a tumor on a particular pathway can be mediated by epigenetic factors that dictate therapeutic vulnerabilities, requiring different doses of targeted therapy to achieve sufficient pathway inhibition. This was previously illustrated in a histology‐independent, BRAF V600E–mutant basket study with vemurafenib that showed differential activities between histologies 30.
In this study, toxicity with the combination generally required dose reduction of both agents, and many patients discontinued because of intolerable toxicities. Data from other phase Ib studies have suggested that long‐term tolerability of the combination at RP2D is challenging 11, 31 and that overlapping toxicities of rash, diarrhea, and fatigue limits the ability to dose both agents continuously 22, 32. Pharmacodynamic studies in BRAF‐mutant melanoma indicate that profound (>80%) inhibition of the pathway is required for clinical responses 33. In this study, RP2D patient biopsies at baseline and after treatment (Day 15) demonstrated median pERK H‐score (cytoplasm) of 210 (range, 0–300) and 190 (range, 60–300), respectively; median pERK H‐score (nucleus) of 180 (range, 0–300) and 160 (range, 0–300); and median pS6 (Ser235 and Ser240) H‐score (cytoplasm) of 120 (range, 3–280) and 110 (range, 9–140). This shows that with continuous dosing, the downstream targets of MAPK and PI3K pathway are not adequately inhibited at the Day 15 time point, likely because of early and acquired compensatory resistance. Furthermore, we found elevated ERK signaling after treatment, which can paradoxically have antiproliferative effects 34.
A potential way of overcoming toxicities while achieving adequate PD inhibition is to evaluate alternate schedules and doses. MEK inhibitors are limited by a narrow therapeutic index and accumulative toxicities. Pulsatile dosing of buparlisib and/or binimetinib may overcome issues related to toxicities 35. This strategy was successfully used in the combination study of selumetinib, a MEK inhibitor, with MK‐2206, an AKT inhibitor, where pulsatile dosing was better tolerated than a continuous dosing schedule 36. The advantage of pulsatile dosing may not only mitigate toxicities but also delay resistance (acquired mutations or receptor upregulations) 37, 38, 39. To the best of our knowledge, clinical trials have not formally evaluated the PK/PD relationship of pulsatile dosing strategies, where the degree of PD inhibition at early time points (24–72 hours after dosing) would be critical. Based on our findings, we advocate clinical trial designs in which either one or both drugs are administered at or above the established MTD in a pulsatile fashion 40 to induce tumor regression and mitigate toxicities.
This study further expands the “basket” concept from selection based on a single genomic mutation (e.g., BRAF V600E) to selection based on multiple genomic alterations that converge on a common pathway. It should be noted that tissue type and quality are important for these genomic studies, as highlighted by the BELLE‐2 trial of buparlisib plus fulvestrant that showed that PIK3CA mutations in circulating tumor DNA, but not tissue specimens, may help select patients who could benefit from PI3K‐inhibitor therapy 23. Moving forward, comprehensive molecular profiling of fresh tissue or liquid biopsies might be important for matching patients with rational combinations of targeted drugs and may address some of the challenges associated with biomarker development.
Limitations
The study limitations include the relatively small number of patients in each tumor group of the expansion phase and the limited number of tumor samples available for PD analyses.
Conclusion
The toxicity profile of the combination regimen evaluated in this study resulted in a lower dose intensity than anticipated. However, an encouraging signal was observed in patients with ovarian cancer. Based on these data, further exploration of this combination may be warranted to define a better‐tolerated dose and/or schedule, such as alternative scheduling with noncontinuous/pulsatile dosing of either agent, which could be explored further in RAS/BRAF‐mutant tumors.
Author Contributions
Conception/design: Novartis Pharmaceuticals Corporation
Provision of study material or patients: Aditya Bardia, Mrinal Gounder, Jordi Rodon, Filip Janku, Martijn P. Lolkema, Joe J. Stephenson, Philippe L. Bedard, Martin Schuler, Cristiana Sessa, Patricia LoRusso, Michael Thomas, Heiko Maacke, Helen Evans, Yongjian Sun, Daniel S.W. Tan
Collection and/or assembly of data: Aditya Bardia, Mrinal Gounder, Jordi Rodon, Filip Janku, Martijn P. Lolkema, Joe J. Stephenson, Philippe L. Bedard, Martin Schuler, Cristiana Sessa, Patricia LoRusso, Michael Thomas, Heiko Maacke, Helen Evans, Yongjian Sun, Daniel S.W. Tan
Data analysis and interpretation: Aditya Bardia, Mrinal Gounder, Jordi Rodon, Filip Janku, Martijn P. Lolkema, Joe J. Stephenson, Philippe L. Bedard, Martin Schuler, Cristiana Sessa, Patricia LoRusso, Michael Thomas, Heiko Maacke, Helen Evans, Yongjian Sun, Daniel S.W. Tan
Manuscript writing: Aditya Bardia, Mrinal Gounder, Jordi Rodon, Filip Janku, Martijn P. Lolkema, Joe J. Stephenson, Philippe L. Bedard, Martin Schuler, Cristiana Sessa, Patricia LoRusso, Michael Thomas, Heiko Maacke, Helen Evans, Yongjian Sun, Daniel S.W. Tan
Final approval of manuscript: Aditya Bardia, Mrinal Gounder, Jordi Rodon, Filip Janku, Martijn P. Lolkema, Joe J. Stephenson, Philippe L. Bedard, Martin Schuler, Cristiana Sessa, Patricia LoRusso, Michael Thomas, Heiko Maacke, Helen Evans, Yongjian Sun, Daniel S.W. Tan
Disclosures
Aditya Bardia: Novartis, Pfizer, Genentech/Roche, Radius Health, Merck, Spectrum Pharmaceuticals, Immunomedics, Daiichi, Sanofi (C/A), Biotheranostics (RF); Mrinal Gounder: Amgen, Daiichi Sankyo, Karyopharm Therapeutics, TRACON Pharmaceuticals, Epizyme (H); Amgen, Daiichi Sankyo, Karyopharm Therapeutics; Epizyme (C/A); Amgen (speaker's bureau); Amgen, Daiichi Sankyo, Karyopharm Therapeutics, Epizyme (travel); Jordi Rodon: GlaxoSmithKline (H), Novartis, Eli Lilly, Orion Pharmaceuticals, Servier Pharmaceuticals, Peptomyc, Merck Sharp & Dohme, Kelun Pharmaceutical/Klus Pharma, Spectrum Pharmaceuticals Inc, Pfizer, Roche Pharmaceuticals, Ellipses Pharma (C/A, travel), Bayer, Novartis, Spectrum Pharmaceuticals, Tocagen, Symphogen, BioAtla, Pfizer, GenMab, CytomX, KELUN‐BIOTECH, Takeda‐Millenium, GlaxoSmithKline, Ipsen (RF), Ellipses Pharma (SAB); Filip Janku: Novartis, Genentech, BioMed Valley Discoveries, Astellas, Agios, Plexxikon, Deciphera, Piqur, Symphogen, Bristol‐Myers Squibb, Asana, Upsher‐Smith Laboratories (RF), Guardant Health, IFM Therapeutics, Synlogic, Deciphera (SAB), Trovagene, Immunomet (C/A), Trovagene (OI); Martijn P. Lolkema: Novartis Pharmaceuticals (C/A); Philippe L. Bedard: Bristol‐Myers Squibb, Roche/Genentech, Sanofi, Pfizer (C/A), Bristol‐Myers Squibb, Roche/Genentech, Novartis, Servier, Nektar, Oncothyreon, Seattle Genetics, Eli Lilly, Sanofi, Merck, AstraZeneca, GlaxoSmithKline, PTC Therapeutics, Mersana, Zymeworks (RF); Martin Schuler: Boehringer Ingelheim, Bristol‐Myers Squibb, Novartis, Roche (C/A); AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Novartis (RF); Patricia LoRusso: Agenus, Agios, Cybrexa, CytomX, FivePrime, Genentech, Halozyme (SAB), Roche‐Genentech, imCORE Alliance, Sotio (C/A); Heiko Maacke: Novartis (E, OI); Helen Evans: Novartis (E, OI); Daniel S.W. Tan: Merck, Pfizer, Novartis, Boehringer Ingelheim, Roche, Takeda (H), Novartis, Bayer, Boehringer Ingelheim, Celgene, Astra Zeneca, Eli‐lily, Loxo (C/A), Novartis, Astra Zeneca, GlaxoSmithKline, Bayer, Pfizer (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Tables
Acknowledgments
We thank Dr. Rona Yaeger at Memorial Sloan Kettering Cancer Center, New York, New York, for critical review of the manuscript. The valuable contributions of Drs. Analía Azaro and Cristina Cruz at Vall D'Hebron Institute of Oncology, VHIO, Barcelona, Spain, to the study and their dedication to the patients are gratefully acknowledged. We acknowledge the contributions of Dr. Jose Baselga in the initial development of this manuscript. Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation and Array BioPharma; we thank Philip Sjostedt and Julie O'Grady (The Medicine Group, LLC, New Hope, Pennsylvania) for medical editorial assistance with this manuscript. M. Gounder was supported by MSKCC Cancer Center Core grant (P30‐CA008748‐53). http://clinicaltrials.gov Identifier: NCT01363232. M.P.L. is currently affiliated with Erasmus Medical Center Cancer Center, Erasmus Medical Center, Rotterdam, The Netherlands.
Disclosures of potential conflicts of interest may be found at the end of this article.
References
- 1. Auner V, Kriegshauser G, Tong D et al. KRAS mutation analysis in ovarian samples using a high sensitivity biochip assay. BMC Cancer 2009;9:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barras D. BRAF mutation in colorectal cancer: An update. Biomark Cancer 2015;7:9–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Irahara N, Baba Y, Nosho K et al. NRAS mutations are rare in colorectal cancer. Diagn Mol Pathol 2010;19:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tan C, Du X. KRAS mutation testing in metastatic colorectal cancer. World J Gastroenterol 2012;18:5171–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol 2014;5:875–885. [DOI] [PubMed] [Google Scholar]
- 6. Allen LF, Sebolt‐Leopold J, Meyer MB. CI‐1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin Oncol 2003;30:105–116. [DOI] [PubMed] [Google Scholar]
- 7. Davies MA, Fox PS, Papadopoulos NE et al. Phase I study of the combination of sorafenib and temsirolimus in patients with metastatic melanoma. Clin Cancer Res 2012;18:1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Califano R, Landi L, Cappuzzo F. Prognostic and predictive value of K‐RAS mutations in non‐small cell lung cancer. Drugs 2012;72:28–36. [DOI] [PubMed] [Google Scholar]
- 9. Haagensen EJ, Kyle S, Beale GS et al. The synergistic interaction of MEK and PI3K inhibitors is modulated by mTOR inhibition. Br J Cancer 2012;106:1386–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Britten CD. PI3K and MEK inhibitor combinations: Examining the evidence in selected tumor types. Cancer Chemother Pharmacol 2013;71:1395–1409. [DOI] [PubMed] [Google Scholar]
- 11. Bedard PL, Tabernero J, Janku F et al. A phase Ib dose‐escalation study of the oral pan‐PI3K inhibitor buparlisib (BKM120) in combination with the oral MEK1/2 inhibitor trametinib (GSK1120212) in patients with selected advanced solid tumors. Clin Cancer Res 2015;21:730–738. [DOI] [PubMed] [Google Scholar]
- 12. Engelman JA, Chen L, Tan X et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med 2008;14:1351–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoeflich KP, Merchant M, Orr C et al. Intermittent administration of MEK inhibitor GDC‐0973 plus PI3K inhibitor GDC‐0941 triggers robust apoptosis and tumor growth inhibition. Cancer Res 2012;72:210–219. [DOI] [PubMed] [Google Scholar]
- 14. Ascierto PA, Schadendorf D, Berking C et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non‐randomised, open‐label phase 2 study. Lancet Oncol 2013;14:249–256. [DOI] [PubMed] [Google Scholar]
- 15. Winski S, Anderson D, Bouhana K et al. MEK162 (ARRY‐162), a novel MEK 1/2 inhibitor, inhibits tumor growth regardless of KRas/Raf pathway mutations. EJC Suppl 2010;8:56. [Google Scholar]
- 16. Lee PA, Wallace E, Marlow A et al. Preclinical development of ARRY‐162, a potent and selective MEK 1/2 inhibitor. Cancer Res 2010;70(suppl 8):2515a. [Google Scholar]
- 17. Bendell JC, Papadopoulos K, Jones SF et al. A phase I dose‐escalation study of MEK inhibitor MEK162 (ARRY‐438162) in patients with advanced solid tumors. Mol Cancer Ther 2011;10(suppl):B243a. [Google Scholar]
- 18. Finn RS, Javle MM, Tan BR et al. A phase I study of MEK inhibitor MEK162 (ARRY‐438162) in patients with biliary tract cancer. J Clin Oncol 2012;30(suppl 4):220a. [Google Scholar]
- 19. PR Newswire . Array BioPharma announces phase 3 binimetinib trial meets primary endpoint for NRAS‐mutant melanoma. Available at http://www.prnewswire.com/news-releases/array-biopharma-announces-phase-3-binimetinib-trial-meets-primary-endpoint-for-nras-mutant-melanoma-300193548.html. Accessed February 13, 2019.
- 20. Rodon J, Braña I, Siu LL et al. Phase I dose‐escalation and ‐expansion study of buparlisib (BKM120), an oral pan‐class I PI3K inhibitor, in patients with advanced solid tumors. Invest New Drugs 2014;32:670–681. [DOI] [PubMed] [Google Scholar]
- 21. Maira SM, Pecchi S, Huang A et al. Identification and characterization of NVP‐BKM120, an orally available pan‐class I PI3‐kinase inhibitor. Mol Cancer Ther 2012;11:317–328. [DOI] [PubMed] [Google Scholar]
- 22. Bendell JC, Rodon J, Burris HA et al. Phase I, dose‐escalation study of BKM120, an oral pan‐class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012;30:282–290. [DOI] [PubMed] [Google Scholar]
- 23. Baselga J, Im SA, Iwata H et al. PIK3CA status in circulating tumor DNA (ctDNA) predicts efficacy of buparlisib (BUP) plus fulvestrant (FULV) in postmenopausal women with endocrine‐resistant HR+/HER2– advanced breast cancer (BC): First results from the randomized, phase III BELLE‐2 trial. Cancer Res 2015;76(suppl):S6‐01a. [Google Scholar]
- 24. Hoeflich KP, O'Brien C, Boyd Z et al. In vivo antitumor activity of MEK and phosphatidylinositol 3‐kinase inhibitors in basal‐like breast cancer models. Clin Cancer Res 2009;15:4649–4664. [DOI] [PubMed] [Google Scholar]
- 25. Babb J, Rogatko A, Zacks S. Cancer phase I clinical trials: Efficient dose escalation with overdose control. Stat Med 1998;17:1103–1120. [DOI] [PubMed] [Google Scholar]
- 26. Farley J, Brady WE, Vathipadiekal V et al. Selumetinib in women with recurrent low‐grade serous carcinoma of the ovary or peritoneum: An open‐label, single‐arm, phase 2 study. Lancet Oncol 2013;14:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Array BioPharma. Array BioPharma announces decision to discontinue MILO study in ovarian cancer. Available at http://investor.arraybiopharma.com/phoenix.zhtml?c=123810&p=RssLanding&cat=news&id=2152468#sthash.N6Lw0zd7.dpufhttp://investor.arraybiopharma.com/phoenix.zhtml?c=123810&p=RssLanding&cat=news&id=2152468. Accessed February 13, 2019.
- 28. Grisham RN, Sylvester BE, Won H et al. Extreme outlier analysis identifies occult mitogen‐activated protein kinase pathway mutations in patients with low‐grade serous ovarian cancer. J Clin Oncol 2015;33:4099–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Garcia‐Garcia C, Rivas MA, Ibrahim YH et al. MEK plus PI3K/mTORC1/2 therapeutic efficacy is impacted by TP53 mutation in preclinical models of colorectal cancer. Clin Cancer Res 2015;21:5499–5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hyman DM, Puzanov I, Subbiah V et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shimizu T, Tolcher AW, Papadopoulos KP et al. The clinical effect of the dual‐targeting strategy involving PI3K/AKT/mTOR and RAS/MEK/ERK pathways in patients with advanced cancer. Clin Cancer Res 2012;18:2316–2325. [DOI] [PubMed] [Google Scholar]
- 32. Ascierto P. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non‐randomised, open‐label phase 2 study. Lancet Oncol 2013;14:249–256. [DOI] [PubMed] [Google Scholar]
- 33. Bollag G, Hirth P, Tsai J et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF‐mutant melanoma. Nature 2010;467:596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Woods D, Parry D, Cherwinski H et al. Raf‐induced proliferation or cell cycle arrest is determined by the level of Raf activity with arrest mediated by p21Cip1. Mol Cell Biol 1997;17:5598–5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yap TA, Omlin A, de Bono JS. Development of therapeutic combinations targeting major cancer signaling pathways. J Clin Oncol 2013;31:1592–1605. [DOI] [PubMed] [Google Scholar]
- 36. Tolcher A, Khan K, Ong M et al. Antitumor activity in RAS‐driven tumors by blocking AKT and MEK. Clin Cancer Res 2015;21:739–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Little AS, Balmanno K, Sale MJ et al. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci Signal 2011;4:er2. [DOI] [PubMed] [Google Scholar]
- 38. Manchado E, Weissmueller S, Morris JP 4th et al. A combinatorial strategy for treating KRAS‐mutant lung cancer. Nature 2016;534:647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das Thakur M, Salangsang F, Landman AS et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature 2013;494:251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yap T, Bjerke L, Clarke P et al. Drugging PI3K in cancer: Refining targets and therapeutic strategies. Curr Opin Pharmacol 2015;23:98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Tables