

Nature is nowhere accustomed more openly to display her secret mysteries than in cases where she shows tracings of her workings apart from the beaten paths; nor is there any better way to advance the proper practice of medicine than to give our minds to the discovery of the usual law of nature, by careful investigation of cases of rarer forms of disease.1
It is a special honour to stand before you to deliver the Harveian Oration as a physician who has devoted his professional life to one of the most recent of medicine's many branches: the field of clinical genetics. When it became a recognised UK specialty in 1976, I was completing my final examinations in Newcastle, but had already declared my intention to become our city's first appointee in this new field – an ambition I fulfilled 8 years later. It is noteworthy that when I became president of the European Society of Human Genetics 30 years after qualification, our specialty was still unrecognised at European level and I was able to help, along with my successor Milan Macek, with the completion of that process of adoption.2 With the emergence of genomic medicine, it might be assumed that the specialty is now more secure, yet the appropriate pressure to democratise the use of the new more powerful diagnostic tools has also challenged the distinct position of clinical geneticists. I believe they still have a role, as I hope this lecture will demonstrate.
My opening quotation from William Harvey, used by Sir Archibald Garrod in his Harveian Oration, illustrates a guiding principle of geneticists whose focus traditionally has been on the investigation of hereditary traits; Garrod's colleague William Bateson coined the term ‘genetics’ on this basis. Clinical genetics emphasises the observation of specific phenotypes in families and addresses the issues they raise, whereas genomics looks at the ‘big picture’.
Terminology and technology
Genetics and genomics
For many, the term ‘genomic era’ dates from an editorial by Guttmacher and Collins in 2003,3 but the concept is older; the words have their root in the Greek prefix gen-, to become, to create.4 The word ‘genome’ is originally German, attributed to Hans Winkler, adopted into English in the 1920s to describe all genes, while the term ‘genomics’ was born in Cold Spring Harbor when Thomas Roderick was sharing a beer,5,6 and was popularised by my mentor and teacher Victor McKusick, who chose it as the name for a new journal. As the ‘-omic’ revolution gained pace, we began to speak of transcriptomes, epigenomes, regulomes, metabolomes and proteomes, then giving rise to multiomics. The chair of the Global Alliance for Genomics and Health (GA4GH), Ewan Birney, has proposed that we use the term ‘genomics’ as a catch-all for the characterisation and quantification of genes, their interactions and influence on the organism.
In this context it might be argued that, far from being a 21st-century creation, genomics has a history as long as genetics, with efforts to link the chromosomes to disease extending back to the beginning of the 20th century when Boveri speculated correctly that chromosome abnormalities might underlie cancer.7 The clinical discipline of genetics can trace back efforts to understand familial patterns into ancient history, but its true emergence is better attributed to Archibald Garrod's description of the recessive trait alkaptonuria as the first description of an inborn error of metabolism.8 Genetics and genomics converged with Lejeune's recognition of trisomy 21 as the basis for Down's syndrome in 1959.9
Over the following three decades, aside from the delineation of the metabolic disorders, diagnostic advances relied predominantly on technological advances in chromosome analysis. In my personal experience, this reached its pinnacle when our group was able to harness the technique of fluorescence in situ hybridisation (FISH) to help solve the clinical phenotype surrounding those children with complex outflow tract cardiac defects and subtle facial features along with other features associated with disruption of branchial arch development, a condition best known as DiGeorge syndrome.10 Typically, children with this pattern had a high mortality due to the severity of the cardiac malformation, most often interruption of the ascending aorta.
On a visit to see a likely case in intensive care, staff mentioned that this was the third member of the family to be seen in the cardiac unit. A trip to west Cumbria with research fellow, now professor, David Wilson was soon arranged. The family displayed the much greater clinical spectrum, with two brothers having more minor heart problems and their mother a normal heart, but she had experienced psychiatric problems. The shared facial features reinforced the belief that they shared a common cause (Fig 1a) and we were able to use molecular techniques and FISH to show that the three boys had inherited a deletion in the long arm of chromosome 22. The connection to 22q11 was not novel, but the important message was that this was much more common and the phenotype was broader, such that it could be sufficiently mild to run as a dominant trait in families. A further lesson from the pursuit of understanding this common deletion, second only to trisomy 21 as a cause of heart malformations,11 and other similar microdeletions was that loss of a block of genes, a contiguous gene syndrome, meant that it was not simple to decide which of the lost genes was pivotal in causing the syndrome. The duplication underlying the high probability of this microdeletion illustrated the structural complexity of the genome which was emerging (Fig 1b). Around 4% of our genome is made up of duplicated segments, many very large, which predispose to additional structural errors.12 The advent of single nucleotide polymorphism (SNP) arrays has transformed clinical practice, replacing time-consuming karyotyping with arrays of millions of SNPs that ‘tag’ all segments of the genome. This allows areas of deletion or duplication to be identified, supplementing the information from sequencing.13
Fig 1.
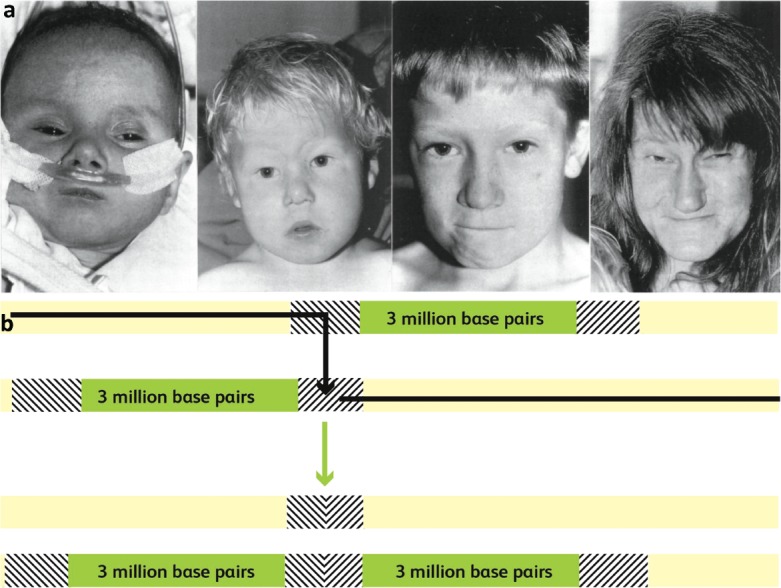
a) A baby with an interrupted aortic arch introduced us to older brothers with the milder problems of coarctation and a ventricular septal defect respectively. Their mother had a normal heart, but had experienced psychiatric problems. The three children had inherited a 22q11 deletion from their mother and the characteristic facial features of this ‘contiguous gene syndrome’. Adapted with permission from Wilson DI, Cross IE, Goodship JA et al. DiGeorge syndrome with isolated aortic coarctation and isolated ventricular septal defect in three sibs with a 22q11 deletion of maternal origin. Br Heart J 1991;66:308–12. b) A duplicated segment of the long arm of chromosome 22 can cause misalignment at gamete formation. If a chromosomal crossover hits this section, it results in one chromosome with a deletion and one with duplication. If the gamete which receives the deleted chromosome results in a pregnancy, the baby will have 22q11 deletion syndrome.
Gene hunting
The trickle of genes attached to well-recognised phenotypes became a flood in the last decade of the 20th century, as the tools provided by the Human Genome Project enabled ever more precise tracking of familial traits with the growing number of markers identified on chromosomes.
The most significant personal journey set out in Fig 2 involved Muriel, who arrived in a Carlisle clinic with her several daughters to discuss their risk of having inheriting from her the gene for Huntington's disease (HD). Despite her movement disorder, said to be of many years standing, Muriel rang alarm bells by being intellectually intact, providing the dates of birth of the whole family to assist in drawing up the family tree. This began a decade of intermittent progress in assembling a family tree, extending back to the 18th century, of a genetic disease that could cause a wide spectrum of neurological impairment, all pointing to a degenerative process in the basal ganglia, but with relative sparing of cognitive function. The unifying feature was that imaging revealed cavitation in the late stage quite unlike anything previously described, together with a wide collection of wrong neurological diagnoses. Finally, a coroner's post-mortem on a woman who had sadly drowned herself in the mistaken belief that she too had HD gave us another branch of the family, and enough meioses to narrow the location of the gene down to a relatively small segment of chromosome 19.
Fig 2.

A novel autosomal dominant neurodegeneration with brain iron accumulation, neuroferritinopathy. a) Mapped to chromosome 19 (19S596–19S866) by linkage analysis. b) Possible genes in the interval. c) Insertional mutation in ferritin light chain (FTL). d) Accumulation of iron–ferritin complex in basal ganglia neurones.
There was still some way to go, as this is a gene-rich chromosome. My scientific colleague Andy Curtis remembered mention of iron staining in the brain of people with Parkinson's disease, so decided to pick ferritin light chain from the list for first analysis as ‘it had something to do with iron’. It was an inspired choice. A single base insertion near the end of the gene causes a frame shift that changes the amino acids at the end of the ferritin protein and, crucially, moves the stop codon. The longer subunit disrupts the pore in the hollow dodecahedron, the 12-sided ferritin ‘container’ that is designed to isolate the toxic but essential iron molecules in the cell. The steady accumulation of this abnormal iron–ferritin complex disrupts the basal ganglia neurones, leading to their eventual destruction.14–16
This family shed light on the importance of iron accumulation in neurodegeneration. We subsequently showed that the iron deposition is demonstrable with magnetic resonance imaging (MRI) in children who carry the disease, making this a slow-motion version of the ‘inborn errors of metabolism’ that first caught Garrod's eye.17 This has become a potentially treatable disorder, although our initial efforts with iron chelation were not effective. The newer gene-editing techniques could cure this condition but its rarity makes such development difficult, even with the help of the ‘orphan disease – orphan drug’ legislation, which offers additional protection for such developments.
The long and the short of whole genome sequencing
The ‘next-generation sequencing’ revolution began in earnest in 2008, when new techniques emerged that were capable of generating and assembling millions of short-run sequences across the genome, replacing the reliable but relatively slow chain-termination technique for which Fred Sanger earned his second Nobel prize. The dominant technique developed by Shankar Balasubramanian and David Klenerman through their start-up, Solexa Ltd, in Cambridge and commercialised by US company Illumina Inc, uses a bridge polymerase chain reaction (PCR) to produce clusters of identical copies of each fragment in a flow cell which can then be ‘read’ by the fluorescent signal of the tagged bases as they bind – so-called ‘sequencing by synthesis’. Errors are reduced by sequencing each fragment in both directions before assembling the fragments against the reference genome to identify variants. The latest version, the Novaseq, can generate up to 48 whole genome sequences in a 48-hour run, equivalent to one per hour!
Bioinformatic techniques have been developed to try to deal with large-scale structural variation such as multiple copies of the same fragment side by side, but these are still far from perfect. Standard sequencing cannot cope with highly repetitive stretches, which are highly mutable and of major biological importance, particularly the telomeres on the chromosome ends that are essential to cell survival and a marker of age-related decline.
Short fragment sequencing does not detect DNA methylation, the process by which cytosines in the DNA strand are altered by addition of a methyl group. This and other ‘epigenetic’ alterations, such as modification of the histone bodies around which the DNA molecule is wound, are central to the control of gene expression.
Many clinically important DNA changes occur after conception, resulting in mosaicism. Even small areas of variation can cause major medical consequences. Most mosaicism is missed by sequencing, even if the correct tissue is sampled.
Clinically significant simple variants are also missed if there are gaps in the ‘jigsaw’ of assembled fragments. In general, the aim is to achieve an average of 30 ‘hits’ on each DNA base in order to ensure that the great majority of significant variants are detected, but for greater certainty much greater coverage is needed.
Finally, the massively parallel sequencing based on short fragments approach usually cannot determine which DNA strand is involved. This is particularly important where there are two pathogenic variants at the same locus. Relatives, usually parents, can be used to decide this, but this greatly increases cost and complexity. For recessive diseases, two changes to the same allele, in cis, result in an asymptomatic carrier, whereas faulty copies in trans will disrupt both copies or alleles and cause disease.
The end result of all these limitations is that ‘whole genome sequencing’ might be better described as ‘hole genome sequencing’.
This results in two conclusions of clinical relevance. The first is that the clinical case for storing sequence data is not straightforward. The general argument of recent years has been that it is worth generating sequence data and storing them as a resource for future healthcare of the individual. In reality, given the pressures on national health budgets, this will only happen at scale when the cost falls significantly below its present price. Having fallen so low, the cost of secure large-scale data storage and retrieval must be set against simply repeating the sequence analysis when needed, with almost certainly much improved quality. In practice, sequencing is still expensive and analysis continues to improve, so there is a case for storage as a translational research tool and to allow reanalysis with possible benefits for the individual, but this should not be oversold.
The second important issue relates to the core technique of sequencing and assembling millions of short sequences. There are two strong contenders for the crown of long-run sequencing: the American company Pacific Biosciences (PacBio for short) and the British company Oxford Nanopore. Both use techniques of reading very long stretches of DNA, which allow much simpler assembly of the genome and, crucially, make it possible to analyse separately the two copies of each gene and their surrounding regulatory segments. This will cause us to change basic teaching slides which state that there are 3 billion bases to be analysed when, in fact, there are 6 billion in the two haploid sets received from each parental gamete. This is not the place for a detailed analysis of the strengths and weaknesses of the two approaches and the health economics of these changes. Perhaps the clearest evidence of the near-future disruptive potential of long-range sequencing in clinical practice is the ongoing billion-dollar-plus attempt by Illumina to buy PacBio.
DNA testing at the bedside
Thomas J Watson of IBM apparently didn't say, in 1943, that the world would need five computers but an academic contemporary, Prof Douglas Hartree in Cambridge, did think in 1951 that all the calculations that would ever be needed in Britain could be done on the three digital computers which were then being built.18 Even in 1972, I remember vividly – during my programming course for the intercalated degree – waiting patiently for the mainframe to again pass by my terminal to enact, hopefully, my carefully typed Fortran instructions. Four-plus decades on, as we unify our storage needs in shared clouds, it can be imagined that indeed few such storage vehicles will be needed, but the number of computers in the world continues to escalate, having passed 2 billion in 2015.
As sequencing becomes more industrial, there is a similar logic at play; current English national genomic policy is built around provision of genomic laboratory services from a much reduced number of centres with only seven hubs for England, insisting that clinical diagnostics will be best served by centralised processing. In a Darwinian sense, this has short-term validity and the deal between Genomics England and Illumina Inc to develop a UK-based facility to provide sequences of clinical grade at minimum cost has much to commend it but, following a similar logic, this opens the large remote laboratories to competition from very low cost near-patient testing, targeted at specific questions of immediate relevance.
Q-POCTM and friends
Over more than a decade, I have had the privilege to work alongside a team led by Jonathan O'Halloran and Elaine Warburton focused on low cost high speed molecular diagnostics, originally as a medical adviser and now also as its chair. The company, QuantuMDx Group Ltd (www.quantumdx.com), has developed a point-of-care battery-powered device with accompanying sample handling and target amplification on low cost disposable cassettes, which can perform a multiplex amplification and diagnosis in less than 20 minutes in almost any setting and without technical expertise. Just as self-testing for pregnancy is now routine using lateral flow techniques, so it will soon become equally simple to diagnose common infections in village clinics, check for drug sensitivities in pharmacies or emergency rooms, do tumour markers in a district pathology lab or test for the common thalassaemia mutations in a rural clinic in east Asia.
As with any evolutionary niche, our small company is not alone in trying to fill this space, but the number of credible contenders able to deliver complex molecular analysis remains small (Table 1).
Table 1.
Leading contenders in the production of point-of-care DNA testing devices (in alphabetical order)
| Point-of-care diagnostic devices | |
|---|---|
| Abbott mPIMA | www.alere.com |
| BioMérieux BioFire | www.biofiredx.com |
| Bosch Vivalytic | https://bosch-vivalytic.com |
| Cepheid GeneXpert | www.cepheid.com |
| Curetis Unyvero | https://curetis.com |
| Meridian (GenePOC) RevoGene | www.genepoc-diagnostics.com |
| QIAGEN QiaStat-Dx | www.qiagen.com |
| QuantuMDx Q-POC | https://quantumdx.com/q-poc |
| Roche Liat | https://diagnostics.roche.com |
The field suffered a setback with the exposure of the fraudulent assertions of Elizabeth Holmes, CEO of the US company Theranos, which claimed to be able to replicate routine biochemistry tests on a finger-prick of blood.19 This claim always looked far-fetched, but diagnosis based on nucleic acid sequences, which are amenable to amplification, is more achievable. There is now a race to achieve a commercially viable system. A critical dimension is the need to address the huge market in low- and middle-income countries (LMICs), where reliable central laboratories are often absent or too remote given the challenges of sample transport.
Our attention has focused successfully on sample acquisition, chemistries and amplification, as well as developing a cheap biosensor. A simple sample caddy loads the single-use cassette, which in turn is loaded into a compact portable battery-operable device. Once the caddy lid is closed on the swab or sample, the entire system is safely contained and, after reading, the cassette is discarded along with its biological hazard.
With support from global charities, we will be able to mass-produce the cassettes and they can be commercially viable at the US$5–8 mark, judged by experts to be the threshold for viability in LMIC markets. DNA can be extracted if necessary using a proprietary filter which fails to bind the charged nucleic acid molecules, allowing the DNA to flow into the amplification chamber. The amplification uses a shuttle flow across heat zones generated within the device, the Q-POCTM. Amplified fragments reach the fluorescent reader chip in less than 12 minutes. This biosensor carries matching DNA fragments which signal binding of the amplified segments from the original specimen.
In this way dozens of ‘questions’ can be asked: does the sample contain any of the 14 oncogenic forms of human papillomavirus, are the variants in the CYP2C9 and VKORC1 genes that confer sensitivity to warfarin detected, is the drug-resistant sequence from Mycobacterium tuberculosis in the sample, and so on. Each biosensor is designed to answer a clinically important question quickly and cheaply at the point of need.
By happy coincidence, the almost 12-year odyssey behind this invention reached a critical juncture in 2019 when the locked down Q-POCTM system began the process of achieving accreditation in Europe, so-called CE-IVD marking. When this is completed in 2020, large-scale trials can commence.
Prediction
Prediction lies at the heart of the physician's role. In general medicine, the three essential questions are what's wrong, what's going to happen and can you do anything to improve things, or, more succinctly, diagnosis, prognosis and therapy. The clinical geneticist is faced with a bewildering array of rare and potentially familial diseases, but the additional questions are usually a logical extension of those three standards.
They are:
Why did it happen?
Will it happen again?
Will it be as bad?
Are there any tests?
Understanding cause is central to estimating risk of recurrence, while understanding the phenotypic spectrum helps with the question of severity. The issue of testing has been addressed from a technological perspective above, but not from a clinical perspective.
In the face of widespread enthusiasm for solving all by sequencing, there are a host of qualifications. Slight variations in the spelling changes in a gene can result in vastly differing outcomes: missense variants in the FGFR3 gene cause the classic dominant trait achondroplasia, while the same change nearby in the same gene might cause a modest height reduction in hypochondroplasia or life-limiting rib shortening in the extreme thanatophoric dysplasia.20 Conversely, the well-known pattern Noonan syndrome has been shown to be the result, in subtly differing forms, of errors in a several genes involved in the RasMAPK cell signalling pathway, resulting in widespread use of the term ‘RASopathies’.21
Two clinical stories illustrate the diversity and scale of the challenge. The first involves two families from near my family home in south Durham. Two brothers had a striking facial appearance (Fig 3a) and bilateral choanal atresia; the openings at the back of the nasal cavity had failed to form, causing obstructed breathing at birth. At around the same time a little girl, Sian, was born a few miles away and presented with the same malformation and a similar facial appearance, together with an unusual ring 18 chromosome, involving loss of a small piece at both ends which then joined, causing some loss of genetic material. A couple of years later, a colleague working in Birmingham presented a family with two brothers that looked very like the families I had seen and I suggested that this was a new syndrome. We wrote a paper and it became known as Burn–McKeown syndrome.22
Fig 3.
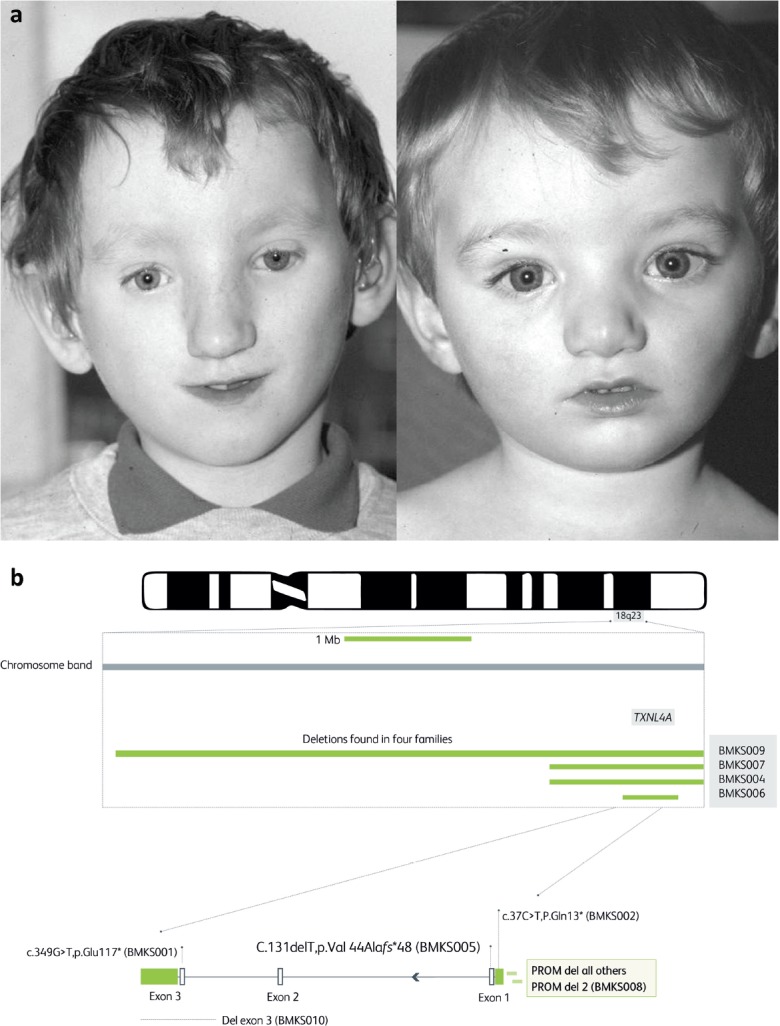
a) Two brothers who presented with facial malformations including the rare problem of choanal atresia, a failure to develop a nasal airway. A third local child, a little girl, was born soon after with the same problem. Two brothers in Birmingham were seen with the same features, allowing identification of the new disorder which became known as Burn–McKeown syndrome.22 b) Whole genome sequencing of DNA from 10 families with a person showing features of Burn–McKeown syndrome revealed, in nine, deletions or point mutations in the TXNL4A gene, responsible for part of the spliceosome. In all cases there was a second deletion involving the gene promoter. One case (BMK008) was homozygous for a slightly different promoter deletion. Adapted from Wieczorek D, Newman WG, Wieland T et al. Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn–McKeown syndrome. Am J Hum Genet 2014;95:698–707.
We speculated that this was a recessive disorder and that the gene might be on the tip of chromosome 18. A handful of other cases were reported around the world over the next decade, but repeated efforts including exome sequencing, which isolates and sequences all the exons or fragments of the coding genes in the genome, failed to find the underlying gene. Only when an international consortium was established and whole genome sequencing deployed was the cause found. The original hunch was correct. The gene was TXNL4A, a coding gene for a key component of the spliceosome. This structure is responsible for assembling the exons of each gene to create the messenger RNA from which the polypeptide chain is assembled. Not surprisingly, complete loss of both copies of this gene is a lethal trait, while loss of a single allele is tolerated.
In the rare families with the syndrome, the other copy of the gene had been downregulated by a deletion involving the upstream transcription factor, which acts as the ‘on/off’ switch for the gene, or they had two ‘downregulated’ copies (Fig 3b).23
This was the second time that such an obligatory compound heterozygote had been described, the first being the genetic basis for thrombocytopenia absent radius (TAR) syndrome.24 There have now been other examples where partial loss of function in the remaining allele of a critically important gene results in a major clinical problem. It's noteworthy that the original family agreed to be part of the pilot group for the 100,000 Genomes Project, but the gene defect has not been detected by Genomics England, and probably never would be without the specific knowledge obtained from pooling our clinical cases in the international consortium of geneticists.
This is because the standard method of finding gene defects that can cause recessive disease is to identify two coding variants in the same locus inherited from each parent. To narrow the focus, areas of homozygosity in the affected child are examined for a genetic change present in single copy in each of the parents. This approach is particularly useful in families with high consanguinity. It comes as a surprise to most that the average person carries at least two major recessive diseases. This is the primary reason why, across all societies, it is illegal for first-degree relatives to have children together. Around half are severely malformed or impaired. This clear disadvantage is likely to underlie the global barriers to incest in major religions.
Most societies allow first cousins to marry. First cousins share one-eighth of their genes in common, which means that if one has a recessive disease allele, there is a one in eight chance that their cousin will have it too, so they have a 1 in 32 or 3% chance of any child having that disease. In practice, the detailed observation of the Born in Bradford study showed a 6% birth defect rate in children of consanguineous parents, in keeping with the deduction from sequencing studies that we each carry at least two such diseases.25
Even where consanguinity is not recognised, there is a significant chance of the same allele turning up. Many of us grew up in the same geographical area as our partners. Families in the same rural area for more than three generations have a strong chance of being biologically linked. In future, there is likely to be a major debate about preconception care to check for shared recessive alleles, just as is already done for Tay–Sachs disease in orthodox Jewish families of Ashkenazi origin and for thalassaemia in Cyprus, where evolutionary selection by the malaria parasite drove up the frequency of a few beta-globin mutations and imposed a major burden on the island's healthcare system.26
Preconception care does not address the more common problem in most populations of de novo genetic defects. I was able to deploy the resources of the Collaborative Group on Genetics in Healthcare, which I led for the Department of Health, to encourage all genetic centres to support the Deciphering Developmental Disorders (DDD) project at the Sanger Centre. This excellent study used exome trios to identify a host of new syndromes, resulting in many cases from genetic errors unique to the child.27 This work is of major value to their parents, who can be reassured that recurrence of a de novo mutation is highly unlikely, barring rare gonadal mosaicism.
While the risk of numerical chromosomal abnormalities associated with the steadily rising average age of women having babies is well recognised and is addressed by the offer of prenatal testing, it is less well recognised that advancing paternal age also carries risks, but of point mutations not detected by the current screening methods. There can be little doubt that the ability to isolate fetal DNA from the circulating fragments in a mother's blood will lead to a growing demand for widespread molecular profiling of early pregnancies, especially with the more advanced age of many couples.
Clinical geneticists have wrestled with the ethical and technical challenges for professionals and families alike and will continue to be needed to help couples to make the right decision for them. A particular challenge of the genomic era is the range of information we are likely to have to deal with in that pressured environment. I had the pleasure to work with Prof Tom Shakespeare on these issues, and our ‘head to head’ video entitled ‘you should have been terminated’ found its way into a Science Museum display (www.youtube.com/watch?v=09VvOq83yMY). Tom's work on eugenics reminded us of the dangers of state-sponsored efforts to rid society of those with imperfections,28 while violence against clinicians in the USA draws attention to the threat against those who seek to offer families a choice.
The planned expansion of prenatal and postnatal sequencing will bring another challenge for society, that of the Inverse Care Law. First coined by Tudor Hart in 1971,29 it refers to the tendency to devote most resource to the care of those who least need it. Tudor Hart was a GP in the Welsh valleys and his words are equally valid today. I have the privilege of now chairing Newcastle Hospitals, where I have worked all my adult life. On a range of metrics this is arguably the best hospital in the NHS, yet the health gap between rich and poor in the population it serves continues to widen. The well-known social and environmental drivers are likely to be reinforced by genomics. There is a clear case for what has become known as anticipatory care, which lies at the heart of the prevention agenda, yet there is already evidence that it is the educated and wealthy who are most likely to avail themselves of genomic testing, which must inevitably draw ever more resources into investigation of those considered to be at genetic risk. As we move away from monogenic disorders, which already represent a major economic challenge, towards more intense attempts to predict common disease, the impact on healthcare will become more acute.
The problem with variants
The next clinical story involves two sisters shown to carry a spelling change in a cancer predisposition gene and the difficulty of deciding its significance; first it is necessary to illuminate the thorny issue of DNA variants and especially the dreaded VUS, or variant of uncertain significance.
I was inspired to become a geneticist in 1969 when my class was taken by our biology teachers to hear a lecture on the genetic code. The year before Marshall Nirenberg, Har Gobind Khorana and Robert W Holley had shared the Nobel Prize for Medicine for their discovery of the triplet code. They had solved the puzzle to explain how, using the four letters of the DNA alphabet, a complete instruction manual for a human, or any other organism, could be constructed. In order to have enough ‘words’ for each essential amino acid, there needed to be a code using three letters for each, but this meant that there was redundancy as the amino acids and grammar combined needed fewer than half of the 64 ways that four letters could be combined in sets of three. The result was described as a degenerate code where, in many cases, the third of the three letters made no difference to the amino acid selected for inclusion in the growing polypeptide chain. Thus, even before sequencing became possible, it was apparent that there would be a huge number of differences between people which would be synonymous, having no impact on the output.
Many amino acids are morphologically and chemically similar and/or a placed in a relatively unimportant position in the final protein structure, so mutations that swap them, known as missense mutations, are likely to have no clinical impact. Even if there is major disruption of the sequence, this might be compensated for by splice isoforms, where different combinations of exons are used to produce a range of physiologically viable proteins from the same gene. Conversely, a point mutation in a seemingly harmless location in an intervening sequence might create a false splice site, leading to incorporation of material that disrupts function.
The end result is that the typical human genome contains over 4 million variants of potential clinical significance that need to be filtered and sorted to decide which, if any, have clinical importance. What began as a preoccupation of a few geneticists and genomicists two decades ago has now become a critical issue in daily clinical practice, occupying countless hours for clinicians. Deciding whether a variant is pathogenic is a probability statement that uses a five-point classification first developed at a meeting hosted by the International Cancer Centre in Lyon, IARC, and later simplified for practical use by the American College of Medical Genetics and Genomics whereby grades 1 and 2 refer to definitely (>99%) or probably (>90%) benign and 4 or 5 as having equivalent probability of being pathogenic. Grade 3 is a VUS.
An initiative that emerged from the Human Genome Organisation became known as the Human Variome Project,30 led by the late Prof Dick Cotton and now managed from our centre under the charity Global Variome (www.humanvariomeproject.org). Based on free software developed in the Netherlands, the Leiden Open Variation Database, curated gene-specific databases have been developed for over 700 clinically important genes (www.lovd.nl/3.0/home). Despite being acknowledged by the World Health Organization and being a recognised non-governmental organisation by UNESCO, this international effort has struggled to secure long-term infrastructural investment. The LOVD output contributes to a major clinically led resource, DECIPHER,31 supported by the Wellcome Trust at the Sanger Centre, which pools structural and single nucleotide variants. The US National Institutes of Health have developed an equivalent free database called ClinVar (www.ncbi.nlm.nih.gov/clinvar), which is now a focus for a growing body of data, although it is limited by the quality of access to underpinning clinical data, while ClinGen is helping to formalise curation groups.
In 2013, the major genomic centres proposed a new initiative called the Global Alliance for Genomics and Health (www.ga4gh.org), and at its launch meeting in 2014, I proposed that we converge our genetic and genomic efforts on arguably the two most famous genes, BRCA1 and 2; the scale and clinical profile of breast cancer, the long-running battles over the question of patents and ownership, and the decision by world-famous Angelina Jolie to undergo mastectomy based on genetic testing have all combined to make these the two most famous sequences among the 20,000-plus protein-coding sequences and an ideal focus for a demonstration project.
To return to the two sisters: a colleague had investigated them because their Canadian aunt had suffered breast cancer in her 30s while one of the sisters had had a breast cancer in her 40s, so they had asked for the BRCA genes to be sequenced and found a missense change: replacement of a valine with alanine at position 1736. This had been judged to be a VUS, in part because the Canadian aunt did not carry it, but subsequently their cousin in Scotland had been shown to also carry the variant when she presented with ovarian cancer and had been similarly advised. I was asked to review it by one of the genetic counsellors and found that the variant lay in a biologically important part of the gene and an analysis by Sean Tavtigian at the Huntsman Institute in Utah had shown that the equivalent DNA repair gene to the human BRCA1 gene across a range of species always had a valine in this position.
Moreover, in addition to this evolutionary conservation, the amino acid valine is different from alanine in its physicochemical properties, giving this change a significant Grantham score.32 In short, it looked important but had been downgraded in 2005 based on a case of co-inheritance. A single patient with ovarian cancer had been found to have inherited this variant from one parent and a known pathogenic BRCA1 variant from the other. As it was thought that survival was impossible without a working copy, the Bayes probability calculation allocated this 10,000:1 odds, which cancelled out the other genetic and in silico data. In 2013, a further paper33 based on that single patient reported that she had syndromic features of a rare condition called Fanconi anaemia, which could be caused by loss of both copies of a range of DNA repair genes including BRCA2, so this patient was the first known survivor of two germline variants in BRCA1. The authors concluded that V1736A should be considered pathogenic, albeit with the possibility that its clinical severity might be reduced given this rare survival.
A ‘secret shopper’ enquiry in 2014 to clinical teams around the UK revealed a range of opinions; some regarded it to be a grade 3 VUS, others a likely pathogenic grade 4 and others as grade 5 pathogenic, including one team which included an author on the 2013 paper. A final team described it as a ‘high class 3’ after extensive searching of local and published databases. Within one integrated healthcare system, the same variant had elicited a range of interpretations, and involved collectively over 8 hours work by the different teams. I sought forgiveness by sponsoring the scientists involved to attend the upcoming European conference in Glasgow that year.
The message was clear. We were wasting precious resources and giving conflicting advice, which could lead to inappropriate surgery or avoidable death.
The BRCA Challenge was agreed by GA4GH leadership to be a driver project, which I co-chaired with Stephen Chanock of the US National Cancer Institute, and has brought together an international consortium, now coordinated via the University of California Santa Cruz. The resulting database has brought together variant data from around the world and the clinical significance of each is being assessed by an established international curation group, ENIGMA (https://enigmaconsortium.org), which assembles the evidence to determine the probability of pathogenicity and records this on the now public database, BRCA Exchange (www.BRCAexchange.org). By the time that our paper appeared in 2018,34 there were over 25,000 variants in the database; four times more than were in the public domain when the project began. Three other exciting innovations are a BRCA app, development of a functional assay for BRCA1 and integrated data from the National Cancer Registry with our English genetic centres.
The BRCA app developed by research partners in Zurich provides access to the BRCA Exchange database and has a push notification if a variant has changed its designation. It's not difficult to see a future where individuals share responsibility for keeping tabs on their family VUS and ensuring that their care team are alerted when knowledge changes.
Tests which examine gene function in vitro are a major help in resolving genomic questions; a recent report has exploited the new gene-editing technology to introduce every possible variant at every position across the important functional domains of BRCA1 and then introduce these modified sequences into a haploid cell, which is dependent for survival on its single working copy of BRCA1. If the cell dies, it suggests that the genetic change was of functional importance. In collaboration with Jay Shendure and Lea Starita, we have incorporated these new functional data into BRCA Exchange.35
I had the pleasure of forming a collaboration with Dr Jem Rashbass of the National Cancer Registration and Analysis Service (NCRAS) as part of the Department of Health committee developing a response the European guidance on rare diseases, and we quickly realised that there was an opportunity to link activity of the national genetics laboratories and the cancer registry. It is not widely recognised that the registry covering England and linked to Wales has one of, if not the, largest bodies of medical digital information on the planet, collecting as they do comprehensive patient data on some 300,000 cancers each year under Section 251 of the Health and Social Care Act. As part of the BRCA Challenge, we sought to demonstrate the power of linking genomic data to this record using a pseudonymisation approach, whereby software scrambles the NHS number and date of birth of an individual with 256 letters/numbers in order to produce an effectively uncrackable code. By using the same device at the genetics laboratory and the registry, it is possible to link genomic data with cancer outcomes without actually identifying the individual, allowing detailed prospective data to be collected across thousands of variants.
This approach has quickly led to the identification of pathogenic variants previously considered to be of uncertain significance simply because they had occurred across multiple centres in cancer patients and yet did not feature in the GnomAD database (http://gnomad.broadinstitute.org), which pools information from exome and genome sequencing across more than 140,000 individuals. In future, we can envisage these sorts of anonymised data linkages yielding immensely valuable data across the genomic medicine landscape.
The polygenic problem
Physicians approach genomics from the perspective of clinical presentation and for genomics to impact significantly on most practice, it must address common diseases where multiple genes, past and present environment and chance factors increase uncertainty. Much of the work of clinical geneticists involves helping to decide whether a range of clinical problems in a family are related, and if so, how. In this space, the latest manifestation of genomic prediction is the polygenic risk score,36,37 which seeks to pool genomic data and stratify on the overall pattern rather than search for individual variants. It is clear that this broad-brush approach can effectively stratify populations to inform more effective health policy, although the dangers are easily seen. Our current health secretary was impressed that he had been given a 15% risk of prostate cancer by this approach and would be seeing his GP about it, although it was pointed out that his lifetime risk was 12% just based on his sex and age. We deal badly with concepts of risk and probability, seeking to convert them into black and white choices.
The first appearance of the concept of polygenic risk emerged at the beginning of the 20th century from RA Fisher's convergence of Mendel's gene theory with the 19th-century work of Galton, who had deduced a blending of characters shown as a mathematical relationship between parental and adult offspring height. Fisher reasoned that Mendel's genes could act additively to produce a phenotype, an idea which became the multifactorial model wherein a biological trait with a Gaussian distribution crosses a threshold, leading to disease.
Having suffered frequent injuries since adolescence due to my stature, I constructed the teaching model shown in Fig 4 to illustrate the dilemma; imagine our height was determined not by hundreds of genes but just two and that each had only two alleles, heads or tails. There would be five categories of height – 4H, 3H1T, 2H2T, 3T1H AND 4T. It's easier to have two of each as the diagram shows, there being six combinations compared to only one each for four heads or four tails, so the proportions of the five categories are 1:4:6:4:1 and this simple model recreates, in crude form, the Gaussian curve. Imagine that those with three or four Ts were so tall that they often, or always, bumped their head when passing through a low door. The clinical phenotype becomes the head injury, not the height; what if genomic studies based on people presenting with frontal haematomas were undertaken? This might lead to one of the two genes being identified and a commercial test being offered, correctly asserting that those who are homozygous for the T allele of the ‘bump your head’ gene, which I've named BYH1, are at very high risk.
Fig 4.
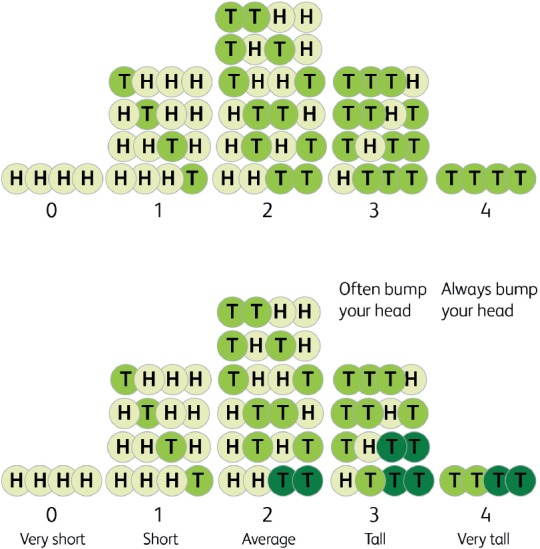
If height were determined by the combination of only two possible alleles at two loci, where the number of T alleles determined each of the five categories, a crude Gaussian distribution would result. If genomic research focused on head injury cases and identified one of the two loci, TT cases would be at high risk, yet two out of five at risk would be missed and one out of four said to be at risk would not be.
The figure shows the problem. While correctly spotting the 4T group, this test misses half of the 3T group, offering false reassurance. Among those of average height there is one who is homozygous T at BYH1, but is protected by the two H alleles at the other locus. This highly predictive test in a very simple ‘complex trait’ misses 2/5 (40%) of those at risk, while only three of the four identified are actually at risk. Factoring in the more than 700 genes now associated with height,38 the variable height of doors and the impact of maternal diet and love on our growth potential illustrates the danger of poor sensitivity and specificity and the often inappropriate use of resources that awaits us.
This trivial example has a serious backdrop; pressure on hospital laboratories, imaging services and endoscopy already strain capacity. Wald and Old have recently pointed out that an impressive odds ratio, while of great value to the discovery of causation, does not offer an effective screening device.39 Khera et al noted an odds ratio of 3.34 using the polygenic risk approach for the 5% in the highest risk category for coronary artery disease. Unfortunately, this represents only a 15% detection rate of affected people, with a 5% false positive rate. Thus the effect of genomic testing, especially if dispersed in an uncontrolled way, could be crippling.37
Prevention is better than cure
He is a better physician that keeps disease off us, than he that cures them being on us; prevention is so much better than healing because it saves the labour of being sick.40
Low-hanging fruit
The new 10-year NHS plan places great emphasis on prevention and its importance in squaring our future health needs with available resources. A theme of what might be called passive prevention runs through all the work of the clinical geneticist. While the purpose is to help families understand and manage debilitating diseases, an inevitable benefit is that many choose to take steps to avoid recurrence of the disease in themselves and future generations. When we take a genomic approach to preventive intervention, there is always the danger of slipping into a eugenic mode. This is least likely to be a problem when focusing on identification of adults for whom a simple low-cost therapy can prevent costly future disease.
Two obvious examples are familial hypercholesterolaemia (FH) which affects over 100,000 Britons, is easily identified by lipid measurement or genomic tests for the common variants and is easily prevented with very early introduction of statins, preferably in the teens.41
An even more dramatic example is haemochromatosis. While neuroferritinopathy is a vanishingly rare disease of accumulation of iron in the brain, its stablemate is extremely common. A remarkable 10–15% of the north European population carry one of two common mutations in the HFE gene, which results in them having a propensity to retain iron. No doubt a selective advantage in our colonisation of temperate climes, this genetic variation has a significant clinical burden attached. The major variant is the C282Y missense variant which, because of its common frequency, is found in the homozygous state in up to 0.5% of the population.42 I have a particular memory of counselling the family of Harry from my home town, who always looked healthy and seemed to have a tan despite never sunbathing. He was one of the estimated 9% of these homozygotes whose genetic difference coupled with a slightly above average iron intake led to severe deposition in his liver and pancreas, both of which failed in his mid-50s leading to his death.
A recent study based on the UK biobank has demonstrated that behind the cases of dramatic medical complications is a long tail of non-specific ill health with musculoskeletal pain, sarcopenia and frailty being significantly more common in the sixth and seventh decades.43 The burden is heavier in males, who are not protected by menstruation. Herein lies the rub: this disease could be eradicated by routine identification of homozygous gene carriers and a recommendation that they become enthusiastic blood donors. I have in the past discussed with colleagues connected to the Blood Transfusion Service that they might profitably campaign for a screening programme to identify these potential ‘super-donors’. A more pedestrian but equally effective approach is to avoid iron-rich food and drinks from an early age and, if needed, to combine venesection with regular iron chelation.
Cancer
Statins for FH and iron depletion for haemochromatosis are the lowest-hanging prevention fruit that widespread genomic medicine can gather, significantly improving the health of 1% of the whole population at modest cost. Approaching another 1% of the population carry a monogenic form of cancer which results in a high risk of developing one or more tumours at an early age. In some the health gains are self-evident, such as offering prophylactic colectomy to the 1 in 8,000 people who carry familial adenomatous polyposis (FAP) and would otherwise inevitably develop a colon cancer and probably die before their mid-40s. Searching for those at hereditary risk of cancer is made easier by the general investment in cancer prevention and treatment at population level, although there is the hidden cost of lifelong surveillance for those deemed to be at high risk. As genomic analysis becomes more routine for all cancers, those at risk due to germline defects will be more easily identified.
The urge to identify those with a monogenic underlying cause has been boosted by the development of specific therapies. In the case of BRCA1 and 2, their contribution to a repair pathway called homologous recombination means that cancers which have lost both copies of either gene are made of cells which are dependent for survival on DNA strand repair based on the enzyme poly(ADP-ribose) polymerase or PARP. In the 1990s, Newcastle's medicinal chemists and cancer research team developed the new drug class known as PARP inhibitors which have proved to be specifically effective in BRCA-deficient cancers, making genomic analysis more critical.44
More recently, a similar breakthrough has transformed interest in those with a germline defect in the mismatch repair system resulting in a hereditary predisposition to colorectal, endometrial and other cancers, especially of the upper genitourinary tract and uroepithelium. For a time called hereditary non-polyposis colon cancer (HNPCC), this form of cancer predisposition is now known as Lynch syndrome (LS) after the Utah-based gastroenterologist Henry T Lynch, who worked tirelessly until his recent death to ensure recognition of this important disorder, now known to be carried by around 1 in 400 people. Those with defects involving the two main genes, MSH2 and MLH1, have a 75% probability of developing one or more of the cancers before they reach old age.45
In 2017, the National Institute for Health and Care Excellence (NICE) announced that all colorectal cancers should be checked for evidence of mismatch repair deficiency in order to help identify families with LS. They were impressed by the clear evidence of benefit. Once identified, cancer deaths are greatly reduced by regular surveillance and prompt surgical response to symptoms. The case is further reinforced by the emergence of a new class of PD (programmed cell death) ligand inhibitors, which unleash lymphocytes on the highly immunogenic mismatch repair-deficient cancers. The first PD-L1 blocker was licensed for use in 2017, the first cancer drug to be directed at the molecular phenotype of the cancer rather than its anatomical location.46
Our research group has developed a new low-cost, high-volume assay suitable for next generation sequencing, which can sensitively and reliably identify LS cancers, and the much more common sporadic MSI (microsatellite instability)-high cancers based on a panel of markers linked to an informative polymorphism which allows a bioinformatic technique of ‘noise cancellation’ by focusing on those where instability is associated with allelic imbalance, suggesting a biological rather than a technical basis for the slippage in sequencing.47,48 This should help to greatly increase the identified proportion of the estimated 175,000 gene carriers in the UK.
The Cancer Prevention Programme (CaPP)
Emerging from our interest in finding the APC gene for FAP, our joint research team between Newcastle and Leeds had begun in the early 90s to pin down a gene for the more elusive hereditary non-polyposis colon cancer (HNPCC) when we were contacted by yeast geneticists in Boston who had an idea. The recent success of the Vogelstein group in mapping a gene for HNPCC in a Newfoundland family had resulted in an unusual observation. Instead of loss of heterozygosity in the tumours, which was being sought to further localise the causative gene, they had found a massive increase in DNA ‘spelling mistakes’. Kolodner and Fischel, as yeast geneticists, had seen this before as a result of disruption of mismatch repair, which detects and corrects DNA copying errors. We sent DNA from families we were working on, and they detected spelling changes in the human equivalent of the MSH2 repair gene.49 We were able, in early 1994, to rapidly deploy this knowledge and begin identifying, for the first time, family members at risk of this cancer syndrome.
This was an opportune moment as we had just succeeded in launching the first CaPP trial. Now known as the Cancer Prevention Programme, CaPP originally referred to the European Concerted Action funding programme. Inspired by a family I had visited where Jonathan, the 12-year-old son of an FAP gene carrier, had extra colonic features of the syndrome and osteomas on his forehead, but had yet to develop the adenomatous polyps in his bowel. It occurred to me that here was a perfect opportunity to develop what we would now call a genomically targeted prevention trial; FAP patients had a germline defect in the APC gene which predisposed, through a second hit, to thousands of polyps. The primary progression of the common sporadic colon cancer had been shown to be driven by loss of both copies of the APC gene in a colonic stem cell. FAP was, in other words, a high-risk model system for a common cancer. The high disease burden, routine surveillance and family support for research made this an ideal focus for a prevention trial.
Based on the MRC folic acid prevention trial for spina bifida, which I had helped to lead and had just reported,50 we used a factorial design in order to try out two possible low-cost interventions with experimental and epidemiological support for being effective in cancer prevention. The first was resistant starch, also known as fermentable fibre, which is fermented by gut bacteria to form short-chain fatty acids, particularly butyrate, that have beneficial anti-cancer effects. The second contender was aspirin.
Aspirin again
The young physician starts life with 20 drugs for each disease, and the old physician ends life with one drug for 20 diseases.51
From 1988 a series of epidemiological papers appeared, beginning with the report from Melbourne by Gabriel Kune that regular use of aspirin and other non-steroidal anti-inflammatories was associated with a reduced risk of cancer.52 Despite being 90 years old, aspirin remained a favourite of its inventors, what is now Bayer Pharma, and they agreed to supply the agent and placebo for all our trials in cancer prevention.
What became known as CaPP1 had launched across Europe and after a long gestation did produce interesting information,53 but we had underestimated the challenge of knowing whether our intervention had had an effect; despite recording endoscopists' assessments backed up with independent video recordings, it's difficult to know whether polyps sometimes numbering in the thousands have been significantly reduced, not to mention the challenge of giving a diet supplement and daily tablets to teenagers. Nevertheless, the idea of long-term chemoprevention, or therapeutic cancer prevention, had been launched.
The arrival of a new, and we now know much more numerous, group of people with a similar monogenic predisposition to colon cancer but without the polyps and developing later in adulthood made this a far more attractive target for intervention. Supported by an international research community, we set about developing a trial using the same interventions, aspirin and resistant starch versus placebos under the title CaPP2. After 5 years of fundraising and grant writing, the first recruit signed up in 1999 and we completed recruitment of 1,009 LS carriers across 16 countries and 43 research centres in the following 7 years.
LS has become the primary focus of my research in subsequent years. In 2008, our first report on CaPP254 revealed no apparent effect on neoplasia, mainly based on adenomas as the average of 2.5 years of treatment was, as expected, too short to impact on cancers and the epidemiology had demonstrated a several-year time lag between taking aspirin and the fall in cancer rates. Despite expert scepticism, we continued to follow the participants and in 2011 reported that, after an average of just under 5 years, those who had taken aspirin for 2 years developed fewer than half of the colorectal cancers seen in the placebo group.55 We are in the process of publishing the 10-year data, which reinforce the earlier report with a significant reduction of colorectal cancers now apparent on an ‘intention to treat’ analysis rather than when confined, as in the 2011 paper, to those who took aspirin ‘per protocol’ for 2 years. These data sit alongside a wealth of other information, mainly from the other large-scale aspirin cancer prevention trial, the Women's Health Study, which revealed an effect after the 10-year treatment programme ended,56 and the extensive work of Peter Rothwell and his team, who have re-examined cancer outcomes in people who took part in the original cardiovascular trials of aspirin over more than two decades.57,58
In 2017 we submitted a request to NICE that aspirin be recognised as a means of cancer prevention in LS, and in August 2019 they launched the recommendation for consultation based on the CaPP2 trial data. NICE notes that there is still uncertainty over the best dose to achieve cancer prevention while avoiding the well-recognised adverse effects of aspirin. Our follow-on dose non-inferiority trial, CaPP3, has just completed recruiting 1,882 gene carriers in the UK and in four other countries and will follow them over the next 5 years to see whether cancer and adverse event rates are equivalent on 100 mg, 300 mg or 600 mg daily aspirin (www.capp3.org).
A recent publication of the ASPREE trial results using aspirin in the over 70s suggests that adverse effects outweigh any benefit in older people,59 as was suspected based on early observational studies,60 but use more widely in late middle age could have a significant beneficial effect on population cancer incidence and overall mortality.61
We continue to explore the underlying mechanisms; while modulation of the response to inflammation appears to be at the centre of the anti-cancer effect, it is also necessary to explain the long time lag between commencement of aspirin and the fall in cancer incidence. Given the role of salicylates as a trigger for apoptosis in green plants to defend against infection and the loss of salicylates from the human diet due to modern farming methods, it is tempting to speculate that aspirin is replacing a lost nutrient and enhancing programmed cell death of cells that might one day become a cancer.62
This 30-year journey to establish aspirin as a cancer preventive is not yet over, but looks to have been successful. Nevertheless, the challenge of prevention should not be underestimated. Even with increasingly sophisticated genomic targeting to achieve maximum impact using minimal numbers of recruits, the bureaucratic oversight of randomised trials presents major challenges in a field where very long-term trials are needed and it is necessary to focus on agents with a long safety record that are usually beyond their patent life, making industry wary of involvement. This is added to the underlying anxiety that treating people who are well in order to avoid future disease makes any adverse effects less acceptable. You never meet the people who didn't get the disease you prevented, but you do meet those who develop side effects. Targeting those with a genetic predisposition remains an appealing approach, combining statistical power with the modern approach of co-production, gaining knowledge of benefit to those at high risk which can then benefit the general population, the human equivalent of the canaries who protected the miners of old.
A final thought
In 2007 I argued that the discovery of the structure of DNA might one day be seen as the single most important event in medical history, eclipsing even the huge public health benefits of clean water and vaccination, which pushed my case into bronze medal position in the contest run by the BMJ63 and I have endorsed the deployment of whole genome sequencing for all as a laudable aim, notwithstanding the continuing need for intelligent clinical interpretation and the avoidance of hyperbole.64 In that commentary I recalled our son planting a horse chestnut in our yard when he was 4 years old because he wanted a tree house and thus reasoned he should grow a tree. I took his picture when he left university in 2002 next to his now-substantial tree, which was still not big enough to support his abode, and argued that genomic medicine was indeed coming but then so was Jamie's tree house. In 2014, in consultation with grandchildren and with help from a carpenter neighbour, the tree house was built.
We stand on the edge of an era when genomic medicine will transform prediction and prevention to such an extent that we may lose the adjective, and simply call it ‘medicine’.
Acknowledgements
Many hundreds of colleagues and thousands of patients have made this presentation possible. I stand on their shoulders. The opinions are my own.
References
- 1.Garrod AE. The Harveian Oration: on the debt of science to medicine. Delivered before the Royal College of Physicians of London on St Luke's Day, October 18th. Br Med J 1924;2:747–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burn J. Recognition of clinical genetics in Europe. Eur J Hum Genet 2017;25(s2):S50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guttmacher AE, Collins FS. Welcome to the genomic era. N Engl J Med 2003;349:996–8. [DOI] [PubMed] [Google Scholar]
- 4.Liddell HG, Scott R. An intermediate Greek–English lexicon, founded upon the seventh edition of Liddell and Scott's Greek–English lexicon. New York: Harper & Brothers, 1889. [Google Scholar]
- 5.Oxford University Press Oxford English dictionary. Oxford: Oxford University Press, 2002. [Google Scholar]
- 6.Yadav SP. The wholeness in suffix -omics, -omes, and the word om. J Biomol Tech 2007;18:277. [PMC free article] [PubMed] [Google Scholar]
- 7.Boveri T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J Cell Sci 2008;121(Suppl 1):1–84. [DOI] [PubMed] [Google Scholar]
- 8.Garrod AE. The incidence of alkaptonuria: a study in chemical individuality. 1902 [classical article]. Yale J Biol Med 2002;75:221–31. [PMC free article] [PubMed] [Google Scholar]
- 9.Lejeune J, Turpin R, Gautier M. [Mongolism; a chromosomal disease (trisomy)]. Bull Acad Natl Med 1959;143:256–65. [PubMed] [Google Scholar]
- 10.Wilson DI, Cross IE, Goodship JA, et al. DiGeorge syndrome with isolated aortic coarctation and isolated ventricular septal defect in three sibs with a 22q11 deletion of maternal origin. Br Heart J 1991;66:308–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burn J, Goodship J. Developmental genetics of the heart. Curr Opin Genet Dev 1996;6:322–5. [DOI] [PubMed] [Google Scholar]
- 12.She XW, Jiang ZX, Clark RA, et al. Shotgun sequence assembly and recent segmental duplications within the human genome. Nature 2004;431:927–30. [DOI] [PubMed] [Google Scholar]
- 13.Manolio TA, Rowley R, Williams MS, et al. Opportunities, resources, and techniques for implementing genomics in clinical care. Lancet 2019;394:511–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curtis AR, Fey C, Morris CM, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet 2001;28:350–4. [DOI] [PubMed] [Google Scholar]
- 15.Chinnery PF, Curtis AR, Fey C, et al. Neuroferritinopathy. J Neurol Neurosur Ps 2002;73:213. [Google Scholar]
- 16.Burn J, Chinnery PF. Neuroferritinopathy. Semin Pediatr Neurol 2006;13:176–81. [DOI] [PubMed] [Google Scholar]
- 17.Keogh MJ, Jonas P, Coulthard A, Chinnery PF, Burn J. Neuroferritinopathy: a new inborn error of iron metabolism. Neurogenetics 2012;13:93–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bowden BV. The language of computers. Am Sci 1970;58:43–53. [Google Scholar]
- 19.Carreyrou J. Bad blood: Secrets and lies in a Silicon Valley startup. New York: Alfred A Knopf, 2018. [Google Scholar]
- 20.Foldynova-Trantirkova S, Wilcox WR, Krejci P. Sixteen years and counting: the current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum Mutat 2012;33:29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tajan M, Paccoud R, Branka S, Edouard T, Yart A. The RASopathy family: consequences of germline activation of the RAS/MAPK pathway. Endocr Rev 2018;39:676–700. [DOI] [PubMed] [Google Scholar]
- 22.Burn J, McKeown C, Wagget J, Bray R, Goodship J. New dysmorphic syndrome with choanal atresia in siblings. Clin Dysmorphol 1992;1:137–44. [PubMed] [Google Scholar]
- 23.Wieczorek D, Newman WG, Wieland T, et al. Compound heterozygosity of low-frequency promoter deletions and rare loss-of-function mutations in TXNL4A causes Burn–McKeown syndrome. Am J Hum Genet 2014;95:698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albers CA, Paul DS, Schulze H, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 2012;44:435–9, S1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheridan E, Wright J, Small N, et al. Risk factors for congenital anomaly in a multiethnic birth cohort: an analysis of the Born in Bradford study. Lancet 2013;382:1350–9. [DOI] [PubMed] [Google Scholar]
- 26.Angastiniotis MA, Hadjiminas MG. Prevention of thalassaemia in Cyprus. Lancet 1981;1:369–71. [DOI] [PubMed] [Google Scholar]
- 27.Deciphering Developmental Disorders Study Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017;542:433–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shakespeare T. Choices and rights: eugenics, genetics and disability equality. Disabil Soc 1998;13:665–81. [DOI] [PubMed] [Google Scholar]
- 29.Hart JT. The inverse care law. Lancet 1971;1:405–12. [DOI] [PubMed] [Google Scholar]
- 30.Cotton RG, et al. Recommendations of the 2006 Human Variome Project meeting. Nat Genet 2007;39:433–6. [DOI] [PubMed] [Google Scholar]
- 31.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 2009;84:524–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grantham R. Amino acid difference formula to help explain protein evolution. Science 1974;185:862–4. [DOI] [PubMed] [Google Scholar]
- 33.Domchek SM, Tang J, Stopfer J, et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov 2013;3:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cline MS, Liao RG, Parsons MT, et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet 2018;14:e1007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Findlay GM, Daza RM, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018;562:217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lambert SA, Abraham G, Inouye M. Towards clinical utility of polygenic risk scores. Hum Mol Genet 2019:ddz187 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 37.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet 2018;50:1219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marouli E, Graff M, Medina-Gomez C, et al. Rare and low-frequency coding variants alter human adult height. Nature 2017;542:186–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wald NJ, Old R. The illusion of polygenic disease risk prediction. Genet Med 2019;21:1705–7. [DOI] [PubMed] [Google Scholar]
- 40.Thomas Adams, 1618, quoted in. Burn J, Sheth H. Colon cancer prevention. In: Eeles RA, Berg CD, Tobias JS. (eds), Cancer prevention and screening: concepts, principles and controversies. Hoboken: Wiley, 2018:183–203. [Google Scholar]
- 41.Defesche JC, Gidding SS, Harada-Shiba M, Hegele RA, Santos RD, Wierzbicki AS. Familial hypercholesterolaemia. Nat Rev Dis Primers 2017;3:17093. [DOI] [PubMed] [Google Scholar]
- 42.Barton JC, Edwards CQ. HFE hemochromatosis. In: Adam MP, Ardinger HH, Pagon RA, et al. (eds), GeneReviews®. Seattle: University of Washington, 1993. [Google Scholar]
- 43.Tamosauskaite J, Atkins JL, Pilling LC, et al. Hereditary hemochromatosis associations with frailty, sarcopenia and chronic pain: evidence from 200,975 older UK Biobank participants. J Gerontol A Biol Sci Med Sci 2019;74:337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yap TA, Plummer R, Azad NS, Helleday T. The DNA damaging revolution: PARP inhibitors and beyond. Am Soc Clin Oncol Educ Book 2019;39:185–95. [DOI] [PubMed] [Google Scholar]
- 45.Dominguez-Valentin M, Sampson JR, Seppala TT, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med 2019. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Redford L, Alhilal G, Needham S, et al. A novel panel of short mononucleotide repeats linked to informative polymorphisms enabling effective high volume low cost discrimination between mismatch repair deficient and proficient tumours. PloS One 2018;13:e0203052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gallon R, Mühlegger B, Wenzel SS, et al. A sensitive and scalable microsatellite instability assay to diagnose constitutional mismatch repair deficiency by sequencing of peripheral blood leukocytes. Hum Mutat 2019;40:649–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027–38. [DOI] [PubMed] [Google Scholar]
- 50.MRC Vitamin Study Research Group Prevention of neural tube defects: results of the Medical Research Council Vitamin Study. Lancet 1991;338:131–7. [PubMed] [Google Scholar]
- 51.Osler W. The principles and practice of medicine: designed for the use of practitioners and students of medicine. New York: D Appleton and company, 1892. [Google Scholar]
- 52.Kune GA, Kune S, Watson LF. Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res 1988;48:4399–404. [PubMed] [Google Scholar]
- 53.Burn J, Bishop DT, Chapman PD, et al. A randomized placebo-controlled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev Res (Phila) 2011;4:655–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burn J, Bishop DT, Mecklin JP, et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N Engl J Med 2008;359:2567–78. [DOI] [PubMed] [Google Scholar]
- 55.Burn J, Gerdes AM, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011;378:2081–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cook NR, Lee IM, Zhang SM, Moorthy MV, Buring JE. Alternate-day, low-dose aspirin and cancer risk: long-term observational follow-up of a randomized trial. Ann Intern Med 2013;159:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rothwell PM, Wilson M, Elwin CE, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010;376:1741–50. [DOI] [PubMed] [Google Scholar]
- 58.Rothwell PM, Cook NR, Gaziano JM, et al. Effects of aspirin on risks of vascular events and cancer according to bodyweight and dose: analysis of individual patient data from randomised trials. Lancet 2018;392:387–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McNeil JJ, Nelson MR, Woods RL, et al. Effect of aspirin on all-cause mortality in the healthy elderly. N Engl J Med 2018;379:1519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paganini-Hill A. Aspirin and colorectal cancer: the Leisure World cohort revisited. Prev Med 1995;24:113–5. [DOI] [PubMed] [Google Scholar]
- 61.Cuzick J, Thorat MA, Bosetti C, et al. Estimates of benefits and harms of prophylactic use of aspirin in the general population. Ann Oncol 2015;26:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dhanoya T, Burn J. Colon cancer and salicylates. Evol Med Public Health 2016;2016:146–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Burn J. Discovery of structure of DNA: the best is yet to come. BMJ 2007;334(Suppl 1):s9. [DOI] [PubMed] [Google Scholar]
- 64.Burn J. Should we sequence everyone's genome? Yes. BMJ 2013;346:f3133. [DOI] [PubMed] [Google Scholar]