

Abstract

In our previous study, we developed a genome-wide DNA methylation model for the diagnosis of prostate cancer, and we pointed out that a considerable average error is associated with the current method for the diagnosis of prostate cancer, which is predicated on pathological assessment of biopsied tissue. In this study, we utilized whole exome and transcriptome RNA-sequencing (RNA-seq) data that were derived from 468 tumor samples and 51 normal samples of prostatic tissue, and we analyzed over 20,000 genes per sample. We were able to develop a mathematical model that classified tumor tissue versus normal tissue with a high accuracy. The overall sensitivity was 97.01%, and the overall specificity was 94.12%. The input variables to the model were the mRNA expression values of the following nine genes: ANGPT1, MED21, AOX1, PLP2, HPN, HPN-AS1, EPHA10, NKX2-3, and LRFN1. The model was validated with unknown samples, with a 10-fold cross-validation, and a leave-one-out cross-validation. We present here a genomic model, based on a whole exome and transcriptome RNA-seq analysis of biopsied prostatic tissue, that could be utilized in the diagnosis of prostate cancer.
1. Introduction
1.1. Prostate Cancer
In our previous study,1 we pointed out that the current diagnosis of prostate cancer, based on pathological analysis of biopsied tissue, is associated with an average error of 25–30% in connection with underdetection and an average error of 1.3–7.1% in connection with overdetection.2,3 In some cases, the underdetection error is substantially higher.4 In addition, the error of a Gleason score—a pathological assessment scale used for grading biopsied prostate tumors—is estimated to be 39%.5 Those sources of error associated with the current method of prostate cancer diagnosis most likely stem from the fact that pathological assessment of tissue does not have the necessary resolution to detect—much less assess and monitor—those molecular processes, such as gene expression alterations, which initiate tumorigenesis and drive its early evolution and which occur at the molecular level. Furthermore, those molecular processes that initiate tumorigenesis and drive its early evolution begin to perturb a host of other molecular processes and mechanisms, such as DNA methylation and other gene expression alterations, years before cancer diagnosis,6 that is to say, years before cancer manifests itself at the tissue level.
1.2. Gene Expression and Cancer
Gene expression, the most fundamental cellular process, is driven by the nature and the state of cells and, equally importantly, by the environment in which they find themselves. That means that cell function and survival are the most dominant determinants of gene expression. Transcriptomics, the study of gene expression that relies on measurement of RNA molecules, is the most powerful and the most well-established method of assessing and monitoring the biology of cells.7 Moreover, transcriptomics affords the most expeditious means of observing changes imposed onto cells by their state or their environment. In the case of tumor cells, gene expression, like many other cellular processes, is appropriated, and it is used to activate or overexpress genes that are vital or beneficial to them and to suppress or silence genes that are detrimental to their survival. Following the advent of next-generation sequencing technologies, RNA sequencing (RNA-seq), with its unprecedented accuracy and sensitivity, quickly became the dominant technology for gene expression measurements.8 In addition to accuracy, sensitivity, and high resolution at the molecular level, RNA-seq revolutionized the entire field of transcriptomics by vastly expanding it so that whole exome and transcriptome sequencing, alternative splicing, RNA editing, fusion transcripts, allele-specific expression, post-transcriptional modifications, and many noncoding RNAs became essential components of it.7,9 As a result, cancer transcriptome profiling has started being introduced into the clinic to improve diagnosis, prognosis, and treatment.7
1.3. Hypothesis
The hypothesis of this study is that tumor cells have induced significant alterations in the gene expression of a number of genes and that, as a result of this, the transcriptomic profile of the tumor cells is significantly different from that of the normal cells.
1.4. Overview of the Model
We developed a multivariable function, whose nine input variables were the mRNA expression values of the following nine genes: ANGPT1, MED21, AOX1, PLP2, HPN, HPN-AS1, EPHA10, NKX2-3, and LRFN1. The model, this multivariable function, was developed during the training phase using approximately 70% of all available samples (328 tumor samples and 36 normal samples), and it was based on the transcriptomic analysis of over 20,000 genes per sample. After its development, the model was first validated with unknown samples [approximately 30% of all available samples (140 tumor samples and 15 normal samples)], which had been randomly preallocated at the beginning of the study and used only for testing purposes. Following its validation with unknown samples, the model was further tested with a 10-fold cross-validation and a leave-one-out cross-validation. The overall performance of the model, including the training and validation phases, was sensitivity = (454/468) = 97.01% and specificity = (48/51) = 94.12%.
2. Methods
2.1. Data
The data used in this study were the normalized data obtained from 468 prostatic tumor tissue samples and 51 normal prostatic tissue samples, generated from whole exome and transcriptome RNA-seq analysis of tissue using the Illumina HiSeq 2000 sequencer and downloaded from The Cancer Genome Atlas of the National Cancer Institute under the category PRAD.
2.2. Clinical Methods
All clinical methods used in this study were the same as the ones we employed in our previous study.1 Briefly, all tumor samples used in this study were primary tumors and had been obtained from subjects that did not have a history of prior and/or other malignancy. The Gleason scores of all 468 tumor samples ranged from 6 to 10 (Table S1).
2.3. Statistical Methods
The statistical methods used in this study were the same as the ones we used in our previous study and are presented in great detail there.1 Briefly, using only the data from the training set, a transcriptomic analysis was performed. Taking into account that the total number of variables (transcripts) per sample was 20,531 and utilizing the Bonferroni correction for multiple tests, the statistical significance level for the transcriptomic analysis was set at α = (0.05/20,531) = 2.43 × 10–6 (two-tailed). Therefore, in order for any variable to be deemed statistically significant, it must have a P value of < 2.43 × 10–6.
2.4. Development of the Model
A random sample selection was performed to generate the training and validation sets so that the training set comprised approximately 70% of the total number of samples from each group and the validation set comprised the remaining approximately 30% of the samples. Furthermore, in accordance with the same methods as in our previous study,1 the random selection of the tumor samples was modified in order for the training set to comprise approximately 70% of the tumor samples with a particular Gleason score and in order for the validation set to comprise approximately 30% of the tumor samples with a particular Gleason score (Tables S2 and S3).
Of the 20,531 variables (transcripts) assessed during the transcriptomic analysis using the data from the training set, 4484 met the above-mentioned criterion of significance and provided the variable pool for the development of the model. Employing the same methodology as in our previous studies,1,10−13 we were able to develop the following multivariable function (eq 1). Equation 1 exhibited the best performance in terms of sensitivity and specificity in the training phase and was selected for validation.
 |
1 |
In eq 1, X1 represents the mRNA expression value of the gene ANGPT1, X2 the gene MED21, X3 the gene AOX1, X4 the gene PLP2, X5 the gene HPN, X6 the gene HPN-AS1, X7 the gene EPHA10, X8 the gene NKX2-3, and X9 the gene LRFN1.
Since X1 to X9 are the respective mRNA expression values of the aforementioned nine genes and since, theoretically, mRNA expression values belong in the continuous interval [0, +∞), the theoretical domain of F (eq 1) is the interval [0, +∞). Moreover, since X5, X6, X7, X8, and X9 are all ≥0, the function F is continuous and is defined throughout its domain [0, +∞). From eq 1, it can be seen that F attains its minimum value when X1 = X2 = X3 = X4 = 0 and when, concomitantly, X5, X6, X7, X8, and X9 are very large. In this case, F → 0. Conversely, when X1, X2, X3, and X4 are very large and when, concomitantly, X5 = X6 = X7 = X8 = X9 = 0, F attains its maximum value. In this case, F → +∞. Therefore, the theoretical range of F is the interval (0, +∞). In practice, however, a very large value of mRNA expression may be in the order of 106. In fact, the largest expression value observed in this study was 6.9 × 106. Assuming, therefore, a very large expression value to be 107, we can observe the behavior of F in the two aforementioned extreme cases. In the case where X1 = X2 = X3 = X4 = 0 and X5 = X6 = X7 = X8 = X9 = 107, Fmin = 0.047, which is close to zero, the theoretical absolute minimum value to which F approaches. In the case where X1 = X2 = X3 = X4 = 107 and X5 = X6 = X7 = X8 = X9 = 0, Fmax = 6259.289. The transcriptomic analysis revealed that in the case of tumor cells, X1 to X4 were significantly underexpressed, whereas X5 to X9 were significantly overexpressed as compared with normal cells (Table 1). In view of this finding, the Fmin represents theoretically the worst possible transcriptomic state in which tumor cells could be. From a biological perspective, that makes sense because in that case, X1 to X4 are completely suppressed, while X5 to X9 are completely upregulated, and this transcriptomic state is the farthest from the normal one. Conversely, the Fmax represents theoretically the most extreme transcriptomic state in which normal cells could be: X1 to X4 are completely upregulated, while X5 to X9 are completely suppressed, a state that is the farthest from that of the tumor cells. The observed range of F, based on all 519 samples of this study, was [4.251, 44.127] (Table S1).
Table 1. mRNA Expression Analysis Results of the Nine Input Variables of the Model Based on the Data of the Training Seta.
| var. | gene symbol | gene ID | MT | SDT | MN | SDN | FC | P |
|---|---|---|---|---|---|---|---|---|
| X1 | ANGPT1 | 284 | 237.857 | 214.325 | 953.763 | 487.196 | –4.010 | 2.68 × 10–19 |
| X2 | MED21 | 9412 | 428.052 | 115.507 | 702.998 | 167.748 | –1.642 | 4.97 × 10–19 |
| X3 | AOX1 | 316 | 407.088 | 436.165 | 2187.538 | 2056.420 | –5.374 | 2.90 × 10–17 |
| X4 | PLP2 | 5355 | 751.555 | 429.088 | 1648.100 | 510.636 | –2.193 | 5.34 × 10–17 |
| X5 | HPN | 3249 | 5840.868 | 3401.767 | 1087.377 | 1031.123 | 5.372 | 2.24 × 10–18 |
| X6 | HPN-AS1 | 100,128,675 | 15.413 | 14.192 | 1.533 | 1.506 | 10.057 | 7.68 × 10–18 |
| X7 | EPHA10 | 284,656 | 236.742 | 129.399 | 53.527 | 31.985 | 4.423 | 6.04 × 10–19 |
| X8 | NKX2-3 | 159,296 | 36.437 | 39.944 | 1.935 | 4.304 | 18.831 | 1.24 × 10–17 |
| X9 | LRFN1 | 57,622 | 199.837 | 92.912 | 71.144 | 37.270 | 2.809 | 9.02 × 10–18 |
| TBPb | 6908 | 246.777 | 44.032 | 250.043 | 37.409 | –1.013 | 0.950 |
The fold change was calculated as follows: R = MT/MN. If R ≥ 1, then FC = R; if R < 1, then FC = −1/R.
Control gene.
Using a receiver operating characteristic (ROC) curve analysis on the results of the F function in the training phase and taking into account the respective variances of the two groups (T vs N), the cut-off point (COP) that demarcates a score of a tumor sample from that of a normal sample was determined to be COP = 23.162. An F score less than the COP designates a tumor sample, whereas an F score greater than or equal to the COP designates a normal sample. Using an ROC curve analysis in a future clinical trial validation study, the COP can be adjusted to optimize the performance of the model.
3. Results
3.1. Transcriptomic Analysis
The transcriptomic results in connection with the nine input variables of the model based on the data of the training set appear in Table 1. The variable name, the gene symbol, the NCBI gene ID, the mean mRNA expression value (MT) of the T (tumor) group with its standard deviation (SDT), the mean mRNA expression value (MN) of the N (normal) group with its standard deviation (SDN), the fold change (FC) (representing the mRNA expression change in the tumor cells as compared with that in the normal cells), and the probability of significance (P) for all nine variables are listed.
3.2. Training
In the training phase, the model (eq 1) identified correctly 316/328 tumor samples and 34/36 normal samples (Table S2). Therefore, the sensitivity was 96.34%, and the specificity was 94.44%. The ROC area-under-the-curve (AUC) statistics were as follows: AUC = 0.98044, its standard error was AUC SE = 0.00718, z-value = 66.94, and the 95% confidence interval of the AUC value was [0.95998, 0.99049] (Figure 1A). The T group had a mean F score and standard deviation of FT = 13.0445 ± 4.9146, and its median value was 12.4581. Based on 100,000 Monte Carlo and bootstrap simulations, the 99% confidence interval of the FT was determined to be [12.3270, 13.7350]. The N group had a mean F score and standard deviation of FN = 28.7701 ± 5.1239, and its median value was 29.3081. Similarly, based on 100,000 Monte Carlo and bootstrap simulations, the 99% confidence interval of the FN was determined to be [26.5718, 30.9196]. The Mann–Whitney U test statistics were as follows: UT = 231, UN = 11,577, z-value = 9.4652, and the approximate probability of significance with correction was P = 2.93 × 10–21 (Figure 2A,B).
Figure 1.

(A) ROC AUC curve of the model F in the training phase. (B) ROC AUC curve of the model F (overall performance).
Figure 2.

(A) Combination graph (box plot, density plot, and dot plot) of the model F in the training phase. Red circles denote statistical outliers. (B) Comparative histogram of the model F in the training phase.
3.3. Validation with Unknown Samples
In the validation phase, using the 155 preallocated unknown samples (140 tumor and 15 normal samples), the model (eq 1) identified correctly 138/140 tumor samples and 14/15 normal samples (Table S3). Therefore, the sensitivity was 98.57%, and the specificity was 93.33%. The unknown T samples had a mean F score and standard deviation of FT = 13.4263 ± 4.2591, and their median value was 12.8706. The unknown N samples had a mean F score and standard deviation of FN = 30.2478 ± 7.2911, and their median value was 30.5702. The Welch unequal-variance t-test statistics for the two groups of unknown samples in connection with their F scores were as follows: t = 8.7764, degrees of freedom = 15.04, and the probability of significance was P = 2.65 × 10–07.
3.4. Overall Performance
When we pooled together the 364 samples (328 tumor and 36 normal) from the training phase with the 155 unknown samples (140 tumor and 15 normal) from the validation phase, the model’s overall performance was as follows: sensitivity = (454/468) = 97.01% and specificity = (48/51) = 94.12% (Figure 3 and Table S1). The ROC AUC statistics were as follows: AUC = 0.97796, its standard error was AUC SE = 0.00901, z-value = 53.06, and the 95% confidence interval of the AUC value was [0.95113, 0.99014] (Figure 1B). The T group had a mean F score and standard deviation of FT = 13.1587 ± 4.7268, and its median value was 12.5619. Based on 100,000 Monte Carlo and bootstrap simulations, the 99% confidence interval of the FT was calculated to be [12.5949, 13.7157]. The N group had a mean F score and standard deviation of FN = 29.2047 ± 5.8073, and its median value was 29.3748. Similarly, based on 100,000 Monte Carlo and bootstrap simulations, the 99% confidence interval of the FN was calculated to be [27.1356, 31.2805]. The Mann–Whitney U test statistics were as follows: UT = 526, UN = 23,342, z-value = 11.2169, and the approximate probability of significance with correction was P = 3.37 × 10–29. The range of all 519 F scores was [4.251, 44.217] (Figure 4A,B). Table 2 shows the mRNA expression results of the nine input variables of the model based on the combined data of the training and validation sets. The variable name, the gene symbol, the NCBI gene ID, the mean mRNA expression value (MT) of the T (tumor) group with its standard deviation (SDT), the mean mRNA expression value (MN) of the N (normal) group with its standard deviation (SDN), the FC, and the probability of significance (P) for all nine variables are listed.
Figure 3.
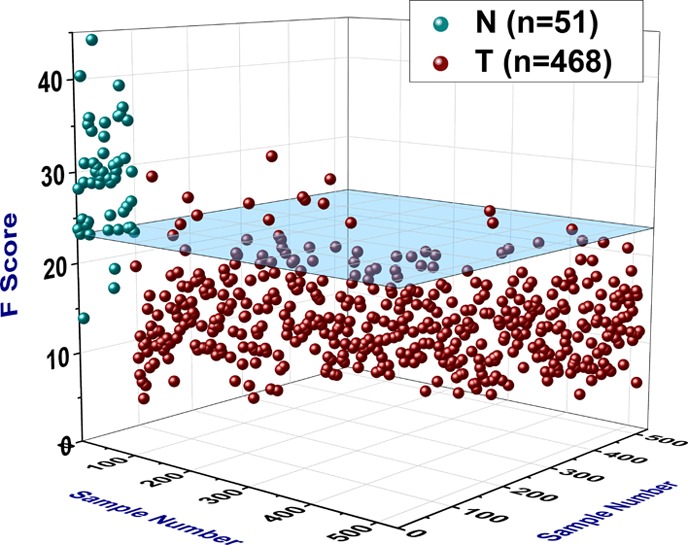
3D scatter plot illustrating the overall performance of the model F. The blue transparent plane is parallel to the x–y plane and intersects the z-axis (F score) at z = 23.162, which is the cut-off point (COP) used by the model such that an F score less than the COP signifies a tumor sample, whereas an F score greater than or equal to the COP signifies a normal sample. Therefore, the blue transparent plane represents the cut-off plane in the 3D space of all 519 samples.
Figure 4.

(A) Combination graph (box plot, density plot, and dot plot) of the model F (overall performance). Red circles denote statistical outliers. (B) Comparative histogram of the model F (overall performance).
Table 2. mRNA Expression Results of the Nine Input Variables of the Model Based on the Combined Data of the Training and Validation Setsa.
| var. | gene symbol | gene ID | MT | SDT | MN | SDN | FC | P |
|---|---|---|---|---|---|---|---|---|
| X1 | ANGPT1 | 284 | 243.843 | 208.138 | 994.899 | 542.859 | –4.080 | 5.38 × 10–25 |
| X2 | MED21 | 9412 | 429.729 | 111.912 | 709.904 | 164.239 | –1.652 | 2.59 × 10–26 |
| X3 | AOX1 | 316 | 414.447 | 428.663 | 2216.079 | 1938.192 | –5.347 | 4.18 × 10–24 |
| X4 | PLP2 | 5355 | 743.570 | 411.813 | 1673.877 | 548.394 | –2.251 | 2.19 × 10–23 |
| X5 | HPN | 3249 | 5721.015 | 3497.136 | 1066.070 | 1031.282 | 5.366 | 1.73 × 10–25 |
| X6 | HPN-AS1 | 100,128,675 | 15.033 | 13.945 | 1.555 | 1.710 | 9.670 | 2.19 × 10–24 |
| X7 | EPHA10 | 284,656 | 233.751 | 124.943 | 51.056 | 32.721 | 4.578 | 8.99 × 10–27 |
| X8 | NKX2-3 | 159,296 | 35.180 | 38.607 | 1.871 | 4.075 | 18.799 | 1.63 × 10–24 |
| X9 | LRFN1 | 57,622 | 197.464 | 93.427 | 69.171 | 36.344 | 2.855 | 5.60 × 10–25 |
| TBPb | 6908 | 246.959 | 44.381 | 251.222 | 38.559 | –1.017 | 0.732 |
The fold change was calculated as follows: R = MT/MN. If R ≥ 1, then FC = R; if R < 1, then FC = −1/R.
Control gene.
3.5. Cross-Validations
The 10-fold cross-validation yielded a misclassification rate of 5.59% (29/519) and a mean-squared error of 0.0559. The sensitivity was 94.44%, and the specificity was 94.12%. The Matthews correlation coefficient (MCC) was 0.7540. The training and testing sets for each of the 10 rounds, as well as the confusion matrix and other statistical information, appear in Table S4. Similarly, the leave-one-out cross-validation yielded a misclassification rate of 5.59% (29/519) and a mean-squared error of 0.0559. The sensitivity was 94.44%, the specificity was 94.12%, and the MCC was 0.7540. The results of all 519 rounds, as well as the confusion matrix and other statistical information, appear in Table S5.
4. Discussion
In this study, using whole exome and transcriptome RNA-seq, we developed and presented a genomic model that could be utilized in the diagnosis of prostate cancer. A subsequent clinical validation with a larger, multicenter retrospective study would constitute the next step for this model and would advance its application closer to the clinic. Such a multicenter study would be able to minimize the innate error of the current diagnostic method (reliance on morphology, that is, visual inspection of the tissue) to classify samples, and it would also provide more information about the few statistical outliers in this study. For example, in the T group, there were eight samples with the highest F scores for that group (from 26.323 to 31.580) (Figures 3 and 4A and Table S1), ranging from 2.79 to 3.90 standard deviations above the mean value of that group. All of those eight samples were determined to be statistical outliers, and they were misclassified by the model. If they were to be removed from the study as extreme outliers, then the performance of the model would increase considerably [sensitivity = (462/468) = 98.72%]. We should point out here that seven of those eight statistical outliers were also misclassified by the DNA methylation model in our previous study.1 This provides independent evidence and raises questions about the actual nature of those seven samples, all of which were determined to be tumor samples by the pathology report. From the different perspectives of two different molecular analyses (gene expression and DNA methylation), those seven samples should be classified in the heart of the N group. Furthermore, in the N group, there were three samples with the lowest F scores for that group (19.352, 17.211, and 13.886) (Figures 3 and 4A and Table S1), being 1.70, 2.07, and 2.64 standard deviations below the mean value of that group, respectively. Those samples constituted the only three misclassifications of the model with respect to the N group (Table S1). Interestingly, two out of those three samples, the ones with the two lowest F scores (17.211 and 13.886), were also misclassified by the DNA methylation model in our previous study.1 This suggests that those two samples, which were deemed normal by the pathology report, should be classified as tumor samples according to both the gene expression analysis and the DNA methylation analysis. The results of this study, as well as those of the previous one,1 demonstrate the capability of genomic methods and modeling to improve current cancer diagnosis. Furthermore, genomic methods and modeling, based on molecular analyses, can be used to improve cancer prognosis as well; genomic models can be developed and trained to detect a characteristic signature of the most aggressive tumor cells and to make accurate predictions about the survival of patients and about the future course of the disease.12,13
According to the results of our transcriptomic analysis (Tables 1 and 2), the genes ANGPT1, MED21, AOX1, and PLP2 (variables X1 to X4) were significantly suppressed in the tumor cells compared with normal cells. Suppression of ANGPT1 has been observed to correlate with progression and metastasis of prostate cancer, as well as with patients’ reduced relapse-free survival.14 Suppression of AOX1 has been observed in prostate cancer and has been associated with reduced patients’ survival and disease recurrence.15 Our transcriptomic analysis (Tables 1 and 2) further revealed that the genes HPN, HPN-AS1, EPHA10, NKX2-3, and LRFN1 (variables X5 to X9) were significantly upregulated in the tumor cells compared with normal cells. A number of studies have descried that HPN is an oncogene, the overexpression of which promotes tumorigenesis and invasiveness of tumor cells in various different types of cancer,16,17 particularly prostate cancer.18EPHA10 has been found to be overexpressed in breast, colon, and prostate cancers,19 and its overexpression has been associated with stage progression and metastasis of tumor cells in breast cancer.20 Overexpression of the homeobox gene NKX2-3 has been linked to tumorigenesis in marginal-zone B-cell lymphomas.21 Finally, LRFN1 has been reported to be overexpressed in prostate cancer,22 and its suppression has been observed to decrease cell viability and survival of pancreatic tumor cells.23
Acknowledgments
This study was supported by Genomix Inc. The funding agency had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b02995.
Overall performance of the model, training of the model, validation of the model with unknown samples, 10-fold cross-validation of the model, and leave-one-out cross-validation of the model (XLSX)
The authors declare no competing financial interest.
Supplementary Material
References
- Nikas J. B.; Nikas E. G. Genome-wide DNA methylation model for the diagnosis of prostate cancer. ACS Omega 2019, 4, 14895–14901. 10.1021/acsomega.9b01613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukimura O.; Coleman J. A.; de la Taille A.; Emberton M.; Epstein J. I.; Freedland S. J.; Giannarini G.; Kibel A. S.; Montironi R.; Ploussard G.; Roobol M. J.; Scattoni V.; Jones J. S. Contemporary role of systematic prostate biopsies: indications, techniques, and implications for patient care. Eur. Urol. 2013, 63, 214–230. 10.1016/j.eururo.2012.09.033. [DOI] [PubMed] [Google Scholar]
- Graif T.; Loeb S.; Roehl K. A.; Gashti S. N.; Griffin C.; Yu X.; Catalona W. J. Under diagnosis and over diagnosis of prostate cancer. J. Urol. 2007, 178, 88–92. 10.1016/j.juro.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Piert M.; Montgomery J.; Kunju L. P.; Siddiqui J.; Rogers V.; Rajendiran T.; Johnson T. D.; Shao X.; Davenport M. S. 18F-Choline PET/MRI: The Additional Value of PET for MRI-Guided Transrectal Prostate Biopsies. J. Nucl. Med. 2016, 57, 1065–1070. 10.2967/jnumed.115.170878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman M.; Ward K. C.; Osunkoya A. O.; Datta M. W.; Luthringer D.; Young A. N.; Marks K.; Cohen V.; Kennedy J. C.; Haber M. J.; Amin M. B. Frequency and determinants of disagreement and error in gleason scores: a population-based study of prostate cancer. Prostate 2012, 72, 1389–1398. 10.1002/pros.22484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Veldhoven K.; Polidoro S.; Baglietto L.; Severi G.; Sacerdote C.; Panico S.; Mattiello A.; Palli D.; Masala G.; Krogh V.; Agnoli C.; Tumino R.; Frasca G.; Flower K.; Curry E.; Orr N.; Tomczyk K.; Jones M. E.; Ashworth A.; Swerdlow A.; Chadeau-Hyam M.; Lund E.; Garcia-Closas M.; Sandanger T. M.; Flanagan J. M.; Vineis P. Epigenome-wide association study reveals decreased average methylation levels years before breast cancer diagnosis. Clin. Epigenet. 2015, 7, 67. 10.1186/s13148-015-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cieślik M.; Chinnaiyan A. M. Cancer transcriptome profiling at the juncture of clinical translation. Nat. Rev. Genet. 2018, 19, 93–109. 10.1038/nrg.2017.96. [DOI] [PubMed] [Google Scholar]
- Byron S. A.; Van Keuren-Jensen K. R.; Engelthaler D. M.; Carpten J. D.; Craig D. W. Translating RNA sequencing into clinical diagnostics: opportunities and challenges. Nat. Rev. Genet. 2016, 17, 257–271. 10.1038/nrg.2016.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa V.; Aprile M.; Esposito R.; Ciccodicola A. RNA-Seq and human complex diseases: recent accomplishments and future perspectives. Eur. J. Hum. Genet. 2013, 21, 134–142. 10.1038/ejhg.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikas J. B. Inflammation and immune system activation in aging: a mathematical approach. Sci. Rep. 2013, 3, 3254. 10.1038/srep03254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikas J. B.; Boylan K. L. M.; Skubitz A. P. N.; Low W. C. Mathematical prognostic biomarker models for treatment response and survival in epithelial ovarian cancer. Cancer Inform. 2011, 10, 233–247. 10.4137/CIN.S8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikas J. B. A mathematical model for short-term vs. long-term survival in patients with glioma. Am. J. Cancer Res. 2014, 4, 862–873. [PMC free article] [PubMed] [Google Scholar]
- Nikas J. B. Independent validation of a mathematical genomic model for survival of glioma patients. Am. J. Cancer Res. 2016, 6, 1408–1419. [PMC free article] [PubMed] [Google Scholar]
- Ayala G.; Morello M.; Frolov A.; You S.; Li R.; Rosati F.; Bartolucci G.; Danza G.; Adam R. M.; Thompson T. C.; Lisanti M. P.; Freeman M. R.; Di Vizio D. Loss of caveolin-1 in prostate cancer stroma correlates with reduced relapse-free survival and is functionally relevant to tumour progression. J. Pathol. 2013, 231, 77–87. 10.1002/path.4217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldrup C.; Mundbjerg K.; Vestergaard E. M.; Lamy P.; Wild P.; Schulz W. A.; Arsov C.; Visakorpi T.; Borre M.; Høyer S.; Ørntoft T. F.; Sørensen K. D. DNA methylation signatures for prediction of biochemical recurrence after radical prostatectomy of clinically localized prostate cancer. J. Clin. Oncol. 2013, 31, 3250–3258. 10.1200/JCO.2012.47.1847. [DOI] [PubMed] [Google Scholar]
- Xing P.; Li J. G.; Jin F.; Zhao T. T.; Liu Q.; Dong H. T.; Wei X. L. Clinical and biological significance of hepsin overexpression in breast cancer. J. Investig. Med. 2011, 59, 803–810. 10.2310/JIM.0b013e31821451a1. [DOI] [PubMed] [Google Scholar]
- Tervonen T. A.; Belitškin D.; Pant S. M.; Englund J. I.; Marques E.; Ala-Hongisto H.; Nevalaita L.; Sihto H.; Heikkilä P.; Leidenius M.; Hewitson K.; Ramachandra M.; Moilanen A.; Joensuu H.; Kovanen P. E.; Poso A.; Klefström J. Deregulated hepsin protease activity confers oncogenicity by concomitantly augmenting HGF/MET signalling and disrupting epithelial cohesion. Oncogene 2016, 35, 1832–1846. 10.1038/onc.2015.248. [DOI] [PubMed] [Google Scholar]
- Valkenburg K. C.; Hostetter G.; Williams B. O. Concurrent Hepsin overexpression and adenomatous polyposis coli deletion causes invasive prostate carcinoma in mice. Prostate 2015, 75, 1579–1585. 10.1002/pros.23032. [DOI] [PubMed] [Google Scholar]
- Nagano K.; Yamashita T.; Inoue M.; Higashisaka K.; Yoshioka Y.; Abe Y.; Mukai Y.; Kamada H.; Tsutsumi Y.; Tsunoda S. Eph receptor A10 has a potential as a target for a prostate cancer therapy. Biochem. Biophys. Res. Commun. 2014, 450, 545–549. 10.1016/j.bbrc.2014.06.007. [DOI] [PubMed] [Google Scholar]
- Nagano K.; Kanasaki S.; Yamashita T.; Maeda Y.; Inoue M.; Higashisaka K.; Yoshioka Y.; Abe Y.; Mukai Y.; Kamada H.; Tsutsumi Y.; Tsunoda S. Expression of Eph receptor A10 is correlated with lymph node metastasis and stage progression in breast cancer patients. Cancer Med. 2013, 2, 972–977. 10.1002/cam4.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles E. F.; Mena-Varas M.; Barrio L.; Merino-Cortes S. V.; Balogh P.; Du M.-Q.; Akasaka T.; Parker A.; Roa S.; Panizo C.; Martin-Guerrero I.; Siebert R.; Segura V.; Agirre X.; Macri-Pellizeri L.; Aldaz B.; Vilas-Zornoza A.; Zhang S.; Moody S.; Calasanz M. J.; Tousseyn T.; Broccardo C.; Brousset P.; Campos-Sanchez E.; Cobaleda C.; Sanchez-Garcia I.; Fernandez-Luna J. L.; Garcia-Muñoz R.; Pena E.; Bellosillo B.; Salar A.; Baptista M. J.; Hernandez-Rivas J. M.; Gonzalez M.; Terol M. J.; Climent J.; Ferrandez A.; Sagaert X.; Melnick A. M.; Prosper F.; Oscier D. G.; Carrasco Y. R.; Dyer M. J. S.; Martinez-Climent J. A. Homeobox NKX2-3 promotes marginal-zone lymphomagenesis by activating B-cell receptor signalling and shaping lymphocyte dynamics. Nat. Commun. 2016, 7, 11889. 10.1038/ncomms11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J. H.; Han Z. P.; Zou M. X.; Wang L.; Lv Y. B.; Zhou J. B.; Cao M. R.; Li Y. G. Analyzing the LncRNA, miRNA, and mRNA Regulatory Network in Prostate Cancer with Bioinformatics Software. J. Comput. Biol. 2018, 25, 146–157. 10.1089/cmb.2016.0093. [DOI] [PubMed] [Google Scholar]
- Kuuselo R.; Savinainen K.; Azorsa D. O.; Basu G. D.; Karhu R.; Tuzmen S.; Mousses S.; Kallioniemi A. Intersex-like (IXL) is a cell survival regulator in pancreatic cancer with 19q13 amplification. Cancer Res. 2007, 67, 1943–1949. 10.1158/0008-5472.CAN-06-3387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.