

Abstract
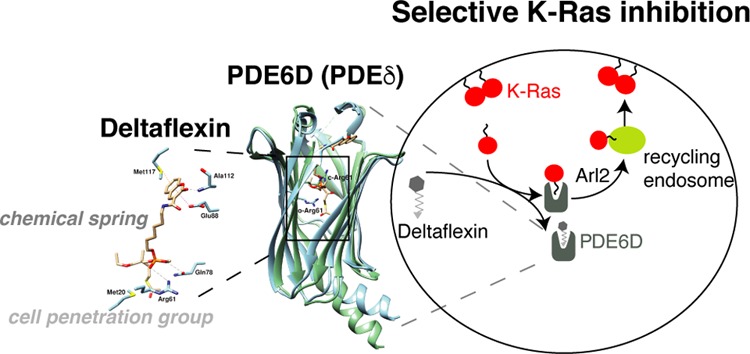
The trafficking chaperone PDE6D (also referred to as PDEδ) has been nominated as a surrogate target for K-Ras4B (hereafter K-Ras). Arl2-assisted unloading of K-Ras from PDE6D in the perinuclear area is significant for correct K-Ras localization and therefore activity. However, the unloading mechanism also leads to the undesired ejection of PDE6D inhibitors. To counteract ejection, others have recently optimized inhibitors for picomolar affinities; however, cell penetration generally seems to remain an issue. To increase resilience against ejection, we engineered a “chemical spring” into prenyl-binding pocket inhibitors of PDE6D. Furthermore, cell penetration was improved by attaching a cell-penetration group, allowing us to arrive at micromolar in cellulo potencies in the first generation. Our model compounds, Deltaflexin-1 and -2, selectively disrupt K-Ras, but not H-Ras membrane organization. This selectivity profile is reflected in the antiproliferative activity on colorectal and breast cancer cells, as well as the ability to block stemness traits of lung and breast cancer cells. While our current model compounds still have a low in vitro potency, we expect that our modular and simple inhibitor redesign could significantly advance the development of pharmacologically more potent compounds against PDE6D and related targets, such as UNC119 in the future.
Introduction
The oncogene Ras is one of the best-established cancer targets without an approved inhibitor. Ras drug development efforts in the 1990s were thwarted by the failure of farnesyltransferase inhibitors (FTI) in clinical trials.1 At that time, it was underappreciated that the highly mutated K-Ras and N-Ras can be alternatively prenylated by geranylgeranyltransferase I, reinstating Ras plasma membrane localization and thus activity, even in the presence of FTIs.2
Recently, there has been a resurgence in Ras drug development, with both direct and indirect targeting approaches.3 The Shokat group has pioneered direct, covalent inhibitors that target a switch II pocket in GDP-bound K-RasG12C.4 Efficacy of these compounds relies on the intrinsic GTPase activity of that mutant and trapping of an inhibitor-bound inactive state.5 Therefore, inhibitor efficacy can be increased by enriching the cellular pool of GDP-bound K-RasG12C, such as through inhibition of upstream receptor tyrosine kinases (e.g., EGFR, FGFR1, AXL) or Ras/Sos1 inhibitors, such as BAY-293.6
Switch II pocket inhibitors have rapidly evolved7 and are now in clinical trials.8 Thus, it is for the first time possible to assess the effect of selectively targeting a driver oncogene. While lung cancer patient data are encouraging, the durable responses and adaptive immunity that is seen in animal models are striking and may indicate the significance of K-Ras inhibition for curing cancer.
Given that other mutant K-Ras alleles are not targeted by these covalent inhibitors, different K-Ras inhibition approaches are still needed. Therefore, other noncovalent Ras binders with inhibitory potential remain interesting. They were typically discovered by computational approaches, such as the Kobe-compounds or pyrazolopyrimidine allosteric compounds, which block Ras-effector interaction and have micromolar cellular potencies.9,10
The indirect target PDE6D is a trafficking chaperone of farnesylated proteins, which may suggest that its inhibition affects the same clients as inhibition of farnesyltransferase. However, PDE6D has a different client profile than farnesyltransferase, as it cannot facilitate intracellular diffusion of proteins that are also palmitoylated.11,12 Moreover, PDE6D has the potential to accommodate geranyl-geranylated clients.12 Thus, PDE6D inhibition selectively affects K-Ras4B (hereafter K-Ras) trafficking but has much less effect on trafficking of dual-palmitoylated H-Ras.11,13
In order to relay signaling, K-Ras needs to be localized predominantly to the plasma membrane. This requires energy-dependent vesicular transport of K-Ras to the plasma membrane from the recycling endosome, where it is collected after PDE6D-assisted diffusion from internal cellular membranes.11,13 Unloading of K-Ras from PDE6D in the perinuclear compartment requires the binding of GTP-Arl2 to PDE6D, which results in an allosteric conformational change in PDE6D that effectively releases its cargo.14 Unfortunately, this ejection mechanism also applies to the first two generations of PDE6D inhibitors Deltarasin15 and Deltazinone,16 developed by the Waldmann group. Only their third generation of PDE6D inhibitors, the Deltasonamides, can largely withstand Arl2-mediated ejection, as they were highly optimized for sub-nanomolar affinity. However, these compounds have a low partitioning coefficient, suggesting low cell penetration.17 Unfortunately, this theme of micromolar cellular potency, despite nanomolar affinity to the target in vitro, also continues with more recent PDE6D inhibitors that emerged through substantial fragment-based discovery and structure-based virtual screening efforts by the Xiong and Sheng groups.18
In order to advance PDE6D inhibitor development, it may therefore be necessary to change the overall concept, as the discovery of various chemotypes with high in vitro affinity has been well demonstrated. We here present Deltaflexins, which have only micromolar in vitro, yet also micromolar in cellulo potencies. We have overcome the approximately 1000-fold in vitro to in cellulo potency gap, by employing an innovative compound engineering approach.
Results and Discussion
Generic Design of PDE6D Inhibitors with a Flexible Hexyl-Linker and Cleavable Cell Penetration Group
In order to improve the resilience of inhibitors against Arl2-mediated ejection from the PDE6D prenyl-binding pocket, a flexible hexyl linker that was envisaged to act like a buffering spring, was attached to a moiety that would provide major contacts to the “top” of the pocket. The design was completed by a simple phosphodiester “bottom” moiety, which was protected with a previously developed cell penetration group that was tunable for pharmacological stability.19,20 In our first-generation model compound, Deltaflexin-1 (15), the generic, heterocylic coumarin was chosen as “top”-moiety, as it would allow for H-bonding and π-stacking (Figure 1A). In addition, a panel of seven control compounds was synthesized that allowed us to assess the contribution of each of the design elements (Figures 1A, S1A).
Figure 1.

Newly designed inhibitors compete with K-Ras for PDE6D in vitro. (A) Design of Deltaflexin compounds, where in the first generation a generic heterocycle “top” was linked via a flexible hexyl “spring” to the protected phosphodiester “bottom”. (B) SPR data of Deltarasin and compounds 15 (Deltaflexin-1), 14 and 17; n = 3. N-terminal avi-tagged K-RasFMe was captured on a neutravidin sensor chip and the inhibitory activity of compounds for the PDE6D/avi-K-RasFMe interaction was measured. (C) Dose–response curve of Deltarasin and compounds 15 (Deltaflexin-1), 14 and 17 from the SPR data. (D) Docked structure of Deltaflexin-1 into the crystal structure of PDE6D in the open (cyan, PDB code 4JV8) and Arl2-GTP-bound, closed state (green, PDB code 1KSJ). Right shows details of PDE6D (open) residues in proximity of docked Deltaflexin-1.
In order to validate the inhibitory activity of the compounds on the PDE6D/K-Ras interaction, we established a surface plasmon resonance (SPR) assay. Farnesylated and carboxymethylated K-Ras (K-RasFMe) were captured on the sensor chip surface via an avi-tag, and the binding of PDE6D was measured (Kd PDE6D/avi-K-RasFMe = 3.6 ± 0.1 μM) (Figure S1B, Table 1). Deltaflexin-1 (15) bound with a surprisingly good, low micromolar Kd that was only 2.6-fold higher than what we determined for Deltarasin (Figure 1B,C, Table 1). This was encouraging given that this was the first generation of compounds. Importantly, the control compound 17 with a “stiff”, methyl cyclohexylmethyl linker had essentially the same affinity to PDE6D as compound 15 and therefore represented an excellent control for testing the significance of the flexible hexyl linker later on in cells (Figure 1C, Table 1). The relatively hydrophobic 14, which lacked the cell-penetration bottom-moiety exhibited a reduced binding activity (Figure 1C, Table 1). Surprisingly, compound 19 the phosphorylated (charged) derivative of 14 showed an increase in the SPR response, suggesting stabilization of the PDE6D/avi-K-RasFMe complex. Direct target binding data were corroborated by a previously published fluorescence anisotropy (FA) assay, which detects displacement of a fluorescently labeled, farnesylated peptide derived from the PDE6D client Rheb (F-Rheb)14 (Figure S1C, Table S1).
Table 1. Inhibitory Activities of Deltaflexins and Control Compounds on Binding of PDE6D to avi-K-RasFMe as Determined by SPR.
| inhibition
of PDE6D/avi-K-RasFMe |
||
|---|---|---|
| compound | IC50 ± SEM/μM | Kd ± SEM/μM |
| Deltarasin | 1.9 ± 0.1 | 1.4 ± 0.1 |
| 15 (Deltaflexin-1) | 4.87 ± 0.03 | 3.61 ± 0.02 |
| 19 | inconclusive | |
| 14 | 18.9 ± 0.2 | 14.1 ± 0.2 |
| 17 | 6.7 ± 0.1 | 4.97 ± 0.07 |
| 22 | 10.27 ± 0.03 | 7.62 ± 0.02 |
| 23 (Deltaflexin-2) | 3.94 ± 0.03 | 2.92 ± 0.02 |
Computational docking of Deltaflexin-1 into the PDE6D crystal structure suggested that similar to the Deltasonamide derivatives, the phosphotriester bottom moiety of Deltaflexin-1 forms hydrogen bonds with Arg61 and Gln78, at the base of the PDE6D pocket (Figures 1D, S1D,E).16,17 In addition, the top-moiety contacted Glu88, Ala112, and Met117. Therefore, three out of the seven contacts that were also engaged by the most optimized deltasonamide derivative (compound 8(17)) were utilized by Deltaflexin-1, consistent with the overall similar positioning of the compound (Figure S1E). A comparison of Deltaflexin-1 docked into PDE6D in the open and closed (Arl2-bound) state illustrates that the potential steric clash between the protected phosphodiester at the bottom of the pocket and Arg61 would likewise persist (Figure 1D). However, the presence of the adjacent flexible hexyl linker may enable the protected phosphodiester bottom moiety of Deltaflexin-1 to evade this clash.
Deltaflexin-1 Disrupts K-Ras/PDE6D Interaction and K-Ras Membrane Organization in Cells
Next, we tested for on-target activity in cells, using a FRET-assay, which directly detects the interaction between K-RasG12V and PDE6D. Deltaflexin-1 significantly reduced the FRET to a level comparable to that of Deltarasin (Figure 2A). This activity was also seen with three derivatives that employed different chemical coupling between top and linker moieties, suggesting a notable degree of flexibility for compound synthesis (Figure S2A). By contrast, the control compound, 17, with a “stiff” methyl-cyclohexylmethyl linker moiety, lacked in cellulo activity (Figure 2A), despite identical in vitro activity (Table 1). Likewise, 14 and 19 without the cell-penetration bottom-moiety did not block the interaction (Figure 2A). In support of the middle moiety requiring a specific length, shortening of the methylen-linker to four carbons abrogated the inhibitory activity in 20 (Figure 2A). Similar results were obtained using the previously described FRET-pair of PDE6D and another client, the farnesylated Ras-like GTPase Rheb (Figure S2B).11
Figure 2.

Deltaflexin-1 inhibits K-Ras/PDE6D interaction and selectively disrupts K-Ras membrane organization (A) PDE6D/K-Ras interaction by FLIM-FRET. HEK cells were co-transfected with mGFP tagged K-RasG12V and mCherry tagged PDE6D. Transfected cells were treated with 0.1% dimethyl sulfoxide (DMSO) control or 5 μM Deltaflexin-1, 17, 14, Deltarasin, or 500 nM FTI-277 for 24 h; n = 3. (B,C) Ras membrane organization measured with nanoclustering-FRET in HEK cells coexpressing mGFP or mCherry tagged K-RasG12V (B) or mGFP or mCherry tagged H-RasG12V (C). Cells were cotransfected with siRNA-PDE6D for 48 h or treated with 0.1% DMSO control, 5 μM Deltaflexin-1, 17, 14, Deltarasin, or 0.2 μM FTI-277 for 24 h; n = 3. (D) K-Ras nanoclustering-FRET dose response curve of Deltaflexin-1 or Deltarasin. Cells were treated for 24 h; n = 3. The numbers on the bars indicate numbers of analyzed cells. Graphs show mean values ± SEM. Statistical significance levels are annotated as ****p < 0.0001; ns, not significant.
PDE6D inhibition is expected to suppress K-Ras localization and activity more than H-Ras.13 We therefore employed an additional cellular FRET-assay that detects FRET due to nanoscale clustering (nanoclustering-FRET) of Ras oligomers, including dimers, on the membrane.21 We previously demonstrated that nanoclustering-FRET can quantify subcellular distribution changes that may be difficult to establish by confocal imaging (Figure S2C).22,23 Thus, we confirmed that FTI selectively affects H-Ras membrane anchorage more than that of K-Ras, as the latter can be alternatively prenylated by geranylgeranyltransferase I2,24 (Figure 2B,C). Given that rate-limiting steps of the PDE6D-mediated trafficking cycle selectively facilitate plasma membrane anchorage of K-Ras, but not H-Ras, knockdown of PDE6D selectively reduced the K-Ras but not H-Ras nanoclustering-FRET (Figures 2B,C and S2D). By contrast, the reference PDE6D-inhibitor, Deltarasin, reduced FRET of both Ras isoforms about equally. This indicates pan-Ras-directed, off-target effects of Deltarasin that may underlie its relatively high general toxicity.16 Deltaflexin-1 on the other hand selectively reduced K-RasG12V but not H-RasG12V FRET as expected (Figure 2B,C). This was confirmed with the three linkage derivatives, further corroborating the flexibility at this position in our generic design (Figure S2E,F). Importantly, Deltaflexin-1 demonstrated an in cellulo potency in this FRET-assay [IC50(15) = 1.65 ± 0.95 μM (mean ± standard error of the mean (SEM))] that was comparable to the in vitro affinity (Kd = 3.61 ± 0.02 μM), although it was not as potent as Deltarasin in cellulo [Figure 2D, IC50(Deltarasin) = 0.7 ± 0.4 μM (mean ± SEM)].
Previous work suggested that the pharmacological stability of the cell penetration group can be tuned within the esterase cleavable S-acyl moiety by the electronegativity of the 2-substituent, such as ethoxycarbonyl (relatively labile) or methyl (more stable).20 Indeed, heating of Deltaflexin-1 before application to cells decreased its cellular activity to a level similar to that of 14 or 19, which both do not have the cell penetration group (Figure S2G).
In conclusion, these results demonstrate that the hexyl-spring and cell penetration group enable a similar in cellulo potency of Deltaflexin-1 as observed in vitro. Importantly, Deltaflexin-1 acts on-target in cells allowing for a selective inhibition of K-Ras activity, whereas the reference compound Deltarasin affects equally K-Ras and H-Ras.
Deltaflexin-1 Selectively Inhibits Oncogenic K-Ras Driven Cell Proliferation and Tumorosphere Formation
In order to further validate the K-Ras selectivity in cells, we examined the effect of Deltaflexin-1 and derivatives on 2D proliferation of colorectal cancer cell lines. Deltaflexin-1 inhibited the K-RasG13D mutated HCT116 (IC50 = 11 μM, CI 95% 8.1–16 μM) significantly more than the Ras wt HT-29 cell line (IC50 = 40 μM, CI 95% CI 25–72 μM, p < 0.0001) (Figure 3A,B). Similar micromolar activities and oncogenic K-Ras selectivity (p < 0.0001) were also seen in the K-RasG13D-mutated and -dependent MDA-MB-231 (IC50 = 7.2 μM, CI 95% 5.1–9.7 μM) and H-RasG12D-mutated and -dependent Hs 578T (IC50 = 21 μM, CI 95% 13–35 μM) breast cancer cell lines (Figure S3A,B,G). Both activity and K-Ras selectivity in colon and breast cancer cells were corroborated by Deltaflexin-1 linkage derivatives (Figure S3C–F).
Figure 3.

Deltaflexin-1 selectively inhibits oncogenic K-Ras-driven cell proliferation and mammosphere formation. (A,B) Dose-dependent cell viability in response to 72 h treatment with Deltaflexin-1 at concentrations ranging from 0 to 20 μM in HCT116 cells (A) and HT-29 cells (B). Graphs show mean values ± SEM, n = 4. (C,D) Sphere formation efficiency (SFE) of MDA-MB-231 cells (C), n = 11, and Hs 578T cells (D), n = 7, cultured in suspension culture for 6 days, followed by a 72 h incubation with 5 μM Deltaflexin-1 or Deltarasin, 0.5 μM FTI or 0.1% DMSO control. Graphs show mean values ± SEM. (E) Deltaflexin-1 dose-dependent effect on SFE with the same protocol as in (C,D). Graphs show mean values ± SEM, n = 4. The SFE is significantly different between MDA-MB-231 and Hs 578T above 0.63 μM. (C–E) Statistical significance levels are annotated as ns, not significant; *p < 0.05; **p < 0.01; ****p < 0.0001; ns, not significant. (F) Representative images of MDA-MB-231 and Hs 578T spheroids at day 10 of suspension culture, after indicated treatments as in (C,D). Scale bar of 200 μm is representative for all images.
Thus, half-maximal inhibition of K-Ras-mutant cancer cell viability by Deltaflexin-1 occurred at approximately 7- to 11-fold higher concentrations than in the cellular K-Ras nanoclustering-FRET assay, which also means that the in vitro and cancer cell inhibitory activities were very similar. By contrast, the difference between cellular inhibition and in cellulo on-target activity of the bis-sulfonamide inhibitor Deltasonamide 2 was 12- to 25-fold, while an even greater difference is apparent for this compound when compared to the in vitro affinity (650- to 1300-fold).17 It is plausible to assume that the better in vitro/cell inhibition activity relation of Deltaflexin-1 compared to Deltasonamide 2 is due to its spring moiety.
While these results demonstrate selectivity for cancer cells expressing mutant K-Ras, the comparison of proliferation in 2D is not as discriminatory as the nanoclustering-FRET assay. However, in the past, we established that compounds which selectively decrease K-Ras-, but not H-Ras-nanoclustering-FRET, have a high potential as inhibitors of stemness properties of cancer cells, as measured by the formation of tumorospheres.22,25 Accordingly, the selective effect of PDE6D on K-Ras but not H-Ras (Figure 2B,C) makes PDE6D a good target to enable such anticancer stemness activity. Indeed, tumorosphere formation of K-RasG12V, but not H-RasG12V transfected or control HEK cells,26 was selectively decreased after knockdown of PDE6D (Figure S3H–J). Similarly, mammospheres derived from K-Ras mutant MDA-MB-231 were inhibited by Deltaflexin-1 more significantly than those of H-Ras mutant Hs 578T (Figure 3C–F). Control compounds 17 and 20 with a stiff or shortened linker, respectively, had no significant effect (Figure 3C,D). This K-Ras-mutant mammosphere selectivity was largely confirmed with the Deltaflexin-1 linkage derivatives (Figure S3K,L), nevertheless suggesting some off-target effects on tumorosphere growth by the different linkages.
Second Generation Deltaflexins with Improved Chemotype
Encouraged by these results, a second generation of inhibitors was developed based on Deltaflexin-1. We replaced the potentially toxic coumarin moiety with a substituted terephthalic acid moiety, thus generating a partial hybrid with a bis-sulfonamide Deltasonamide inhibitor (compound 8(17)) (Figure 4A). In addition, the bottom moiety was varied to characterize the activity of the pharmacologically more stable 2-methyl-substituent on the S-acyl cell penetration group. While compound 23 with the more stable bottom-moiety had a comparable affinity to 15, its analogue 22 bound with a 2.6-fold lower affinity (Figure 4B, Table 1, Figure S4A, Table S1).
Figure 4.

Partial scaffold hybridization creates second-generation inhibitors. (A) Second-generation compounds have an altered “top” moiety taken from a previously published PDE6D inhibitor. Note the variation of the protecting group between 22 and 23, as well as 24 and 25. Linker length is varied between the 22, 23 vs 24, 25. (B) SPR data of avi-K-RasFMe/PDE6D treated with 22 and 23 (Deltaflexin-2) and dose response curves constructed from SPR kinetic data; n = 3. (C) PDE6D/K-Ras interaction by FLIM-FRET. HEK cells were cotransfected with mGFP-tagged K-RasG12V and mCherry-tagged PDE6D. Transfected cells were treated with 0.1% DMSO control or 5 μM (n = 3) and 10 μM (n = 1) compounds 22, 23, 24, 25, or 5 μM Deltaflexin-1, Deltarasin for 24 h;. (D) K-Ras membrane organization measured with nanoclustering-FRET in HEK cells coexpressing mGFP or mCherry tagged K-RasG12V for 48 h or treated with 0.1% DMSO control, and various concentrations of compounds 22, 23, 24, 25, or 5 μM Deltaflexin-1, Deltarasin for 24 h; n = 3. For all FRET-data, the numbers on the bars indicate the number of analyzed cells. (E) Dose-dependent cell viability of lung cancer cell lines, NCI-H358 and A375 grown as spheroids under low attachment and serum free conditions in response to 72 h treatment with ARS-1620, Vemurafenib, FTI-277, Deltarasin, Deltaflexin-1, and Deltaflexin-2 at concentrations ranging from 0 to 20 μM; n ≥ 3. Higher efficacy results in a higher drug sensitivity score (DSS), an area under the curve (AUC) metric. Graphs show mean values ± SEM. Statistical significance levels are annotated as **p < 0.01; ****p < 0.0001; ns, not significant.
Computational docking supported these data, as overall similar positioning and similar contacts as for Deltaflexin-1 were observed for the second-generation compounds in the PDE6D pocket (Figure S4B). It appeared though that the substitution of the top moiety from an aromatic two-ring system to one aromatic ring allowed for more flexibility of the top moiety orientation. Moreover, the potential for a new hydrogen bond with Ser115 was identified for both 22 and 23. In addition, the bending of 23 could contribute to the formation of an additional hydrogen bond with Glu88, which is not observed in 22 (Figure S4B). The relative strength of these interactions may contribute to the observed differences in the binding affinities.
In agreement with the in vitro data, second-generation compound 23 performed as well as 15 and somewhat better than 22 in disrupting the K-RasG12V/PDE6D interaction as assessed by FRET-experiments in cells (Figure 4C). Similarly, amongst the 4n-methylen-linker counterparts of 22 and 23, 25 performed better than 24 (Figure 4A,C). These data further support that the replacement of the relatively labile 2-ethoxycarbonyl with the more stable 2-methyl on the S-acyl cell penetration group is beneficial. Finally, 22 and 23 (hereafter named Deltaflexin-2) led to a significant dose-dependent reduction of the K-RasG12V nanoclustering-FRET compared with 24 and 25 (Figure 4D). No effect on H-RasG12V FRET was observed (Figure S4C).
Recently, exquisite K-RasG12C selectivity was demonstrated for the covalent inhibitor ARS-1620.7 In agreement with a significant overall activity, quantitative scoring of the chemical inhibition of PDE6D with Deltaflexin-2 revealed that it had 70% activity of ARS-1620 in KRAS-G12C mutant NCI-H358 lung cancer cells grown as tumorospheres. Moreover, Deltaflexin-2 was inactive in the B-RafV600E mutant A375 lung metastasis melanoma cell line, as was ARS-1620 (Figure 4E). These data confirm that Deltaflexins significantly inhibit cell spheroid growth in a K-Ras selective manner. By contrast, no such selectivity was found for Deltarasin, which showed equally high activity in both cell lines, again suggesting a general off-target toxicity.
Similarly, K-RasG13D-mutant MDA-MB-231- but not H-RasG12V-mutant Hs 578T-mammosphere formation was selectively decreased by Deltaflxin-2, while the counterpart with the shorter flexible linker 25 was inactive (Figure S4D,E). In line with the Ras-mutation status, the mammosphere formation of MDA-MB-231 and Hs 578T was selectively abrogated by the knockdown only of the mutated Ras isoform (Figure S4F–I), confirming that this assay can report on the selectivity for oncogenic K-Ras (MDA-MB-231) or H-Ras (Hs 578T). However, knockdown of PDE6D strongly affected sphere formation of both cell lines (Figure S4F,G).
We therefore conclude that Deltaflexins can block stemness traits of cancer cells and have a clear in cellulo selectivity for K-Ras as compared to H-Ras. However, our data also suggest that the stemness inhibiting activity of PDE6D inhibitors depends at least in breast cancer cell lines on other clients or activities of PDE6D.
In this work, we have demonstrated that the attachment of a “chemical spring” and cell penetration group to a generic heterocycle enables the de novo design of low micromolar PDE6D inhibitors in the first generation. This redesign approach is strikingly simple and together with the flexible coupling chemistry should allow for improvements of existing PDE6D inhibitors. As validated by the second-generation compounds, our approach is compatible with compound-scaffold hybridization of existing PDE6D compounds. Thus, we expect to quickly advance the development of more potent derivatives in the near future. Moreover, our strategy may apply to inhibitors of closely related targets, such as UNC119,27 in addition to PDE6D inhibitors.
Past generations of PDE6D inhibitors underwent substantial structure-based optimization to arrive at sub and low nanomolar in vitro activities, but surprisingly they exhibited ∼1000-fold less antiproliferative potential in cancer cells. Thus, the relation of Deltaflexin-2 in vitro and in cellulo potencies is ∼60- to 180-times better than that of Deltasonamide-2. Moreover, as compared to Deltarasin, Deltaflexins demonstrated the expected selectivity for K-Ras over H-Ras in cells. Therefore, the current Deltaflexins can serve as valuable tool compounds to investigate the PDE6D associated anticancer mechanisms. While we demonstrate the K-Ras selectivity of Deltaflexins, the comparison of PDE6D- and KRAS-knockdown data suggests K-Ras-independent effects of PDE6D inactivation on stemness traits of cancer cells. Other activities of PDE6D or clients other than K-Ras could be important in this context. For example, the PDE6D client INPP5E (inositol polyphosphate-5-phosphatase E) localizes to the primary cilium in a PDE6D-dependent manner.28−30 In line with a potential contribution of INPP5E inhibition to the antistemness effect, primary ciliogenesis and associated Hedgehog signaling have recently been linked to epithelial–mesenchymal transition and stemness promotion in triple negative breast cancer.31
Nonetheless, K-Ras overactivity appears to be an important biomarker for the efficacy of PDE6D inhibitors.32 In order to establish the exact therapeutic spectrum of the single-agents, a broader panel of cancer cells should be tested with PDE6D inhibitors in the future. Given that carboxymethylation of the prenylated C-termini of small GTPase clients provides additional affinity, it is foreseeable that concurrent inhibition of isoprenylcysteine carboxyl methyltransferase, by inhibitors such as cysmethynil, will be synergistic with PDE6D inhibitors.12,32
While targeting of signaling or trafficking hubs, such as PDE6D, could intuitively lead to more side effects, this approach may in the end be advantageous from the drug development point of view. Given that different cancer types and subtypes have diverse sets of drivers,33,34 drugs that target a hub may affect several cancer drivers at once. Such hub inhibitors may therefore be more easily applicable to several types of cancer. This strategy is, for example, pursued for inhibitors of Hsp90, which is a major hub of kinases.35,36 Irrespective of their exact mechanism of action, PDE6D inhibitors therefore remain attractive as potential cancer drugs.
Methods
DNA Constructs and siRNA
Plasmids pmGFP/mCherry-H-RasG12V and pmGFP/mCherry-K-RasG12V were previously described.22,23 Plasmids for mCit-Rheb and mCherry-PDE6D were previously described.11 The human gene-directed siRNAs were purchased from Dharmacon as ON-TARGET plus SMART siRNA pools. The catalogue numbers are as follows: scrambled siRNA (cat. no. D-001810-10-05), human KRAS (cat. no. L-005069-00, lot 170710), human HRAS (cat. no. L-004142-00, lot 160414), and human PDE6D (cat. no. L-004310-00). Opti-MEM and transfection reagents Lipofectamine 3000 (cat. no. L3000001) and RNAiMAX (cat. no. 13778030) were purchased from Thermo Fisher Scientific. JetPrime (cat. no. 114-75) was obtained from Polyplus, Illkrich-Graffenstaden, France. FuGENE HD (cat. no. E2311) was purchased from Promega Biotech AB, Nacka, Sweden.
Cell Culture
HEK293 EBNA cells were kind gift of Prof. Florian M Wurm, EPFL, Lausanne, Switzerland. MDA-MB-231, Hs 578T, HCT116, HT-29, NCI-H358, and A-375 cell lines were obtained from ATCC. HEK293 EBNA, Hs 578T, HCT116, and HT-29 cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM, cat. no. D6171, Sigma-Aldrich, Helsinki, Finland), supplemented with 10% fetal bovine serum (FBS) (cat. no. S1810, Biowest, Nuaille, France) and 2 mM l-glutamine (cat. no. G7513, Sigma-Aldrich). MDA-MB-231, NCI-H358, and A375 cells were cultured in Roswell Park Memorial Institute medium (RPMI, cat. no. R5886, Sigma-Aldrich), containing 10% FBS and 2 mM l-glutamine. All cells were incubated at 37 °C, with 5% CO2, in a humidified cell incubator. Cells were subcultured twice a week.
Fluorescence Lifetime Imaging Microscopy-FRET
HEK293 EBNA cells were seeded in 12-well plates onto sterile coverslips. The next day, cells were transfected using FuGENE HD or jetPRIME transfection reagent with a total of 800 ng of plasmids. For donor samples, cells were transfected with mGFP or mCitrine-tagged plasmid. In FRET pairs, donors and acceptors were transfected at a 1:3 ratio. Acceptors were mCherry-tagged Ras plasmids in the nanoclustering-FRET experiments for membrane organization studies. For interaction studies, pmGFP-K-RasG12V and pmCherry-PDE6D, or pmCitrine-Rheb and pmCherry-PDE6D, were used all at the same 1:3 ratios with a total of 800 ng plasmids being used in each experiment. After 24 h of transfection, cells were treated with either 0.1% DMSO control or various concentrations of test compound or Deltarasin (cat. no. 9001536, Cayman Chemical, Tallinn, Estonia), or FTI-277 (cat. no. Sc-215058, Santa Cruz Biotechnology, Dallas, USA) or FTI-2628 (cat. no. sc-221635, Santa Cruz Biotechnology) for 24 h and fixed in 4% paraformaldehyde (PFA) before mounting with Mowiol 4-88 (cat. no. 81381, Sigma-Aldrich). The donor fluorescence lifetime was measured using a fluorescence microscope (Zeiss AXIO Observer D1) with a fluorescence lifetime imaging attachment (Lambert Instruments, Groningen, The Netherlands), as previously described in refs (37, 38). The fluorescence lifetime of at least 40 cells per treatment was measured in each experiment. The percentage of the apparent FRET efficiency (Eapp) was determined using the measured lifetimes of donor–acceptor pairs (τDA) of samples and the average donor lifetime (τD), based on the equation: Eapp= (1 – τDA/τD) × 100%.
Tumorosphere Assays
Mammosphere formation assays were performed in F-bottom 96-well suspension culture plates (cat. no. 655185, Cellstar, Greiner Bio-One, Frickenhausen, Germany). 1500 cells per well were seeded in 50 μL DMEM or RPMI medium containing 1× B27 (cat. no. 17504044, Gibco, Thermo Fisher Scientific), 25 ng/mL EGF (cat. no. E9644, Sigma-Aldrich), and 25 ng/mL FGF (cat. no. RP-8628, Thermo Fisher Scientific). Cells were cultured for 6 days and then treated with test compounds or DMSO control (0.1% v/v) for additional 3 days. The cells were resupplemented with fresh growth medium every 2 days. For knockdown experiments and for K-Ras and H-Ras transfections, cells were seeded in 6-well plates and treated with either 50 nM scrambled siRNA or siRNA targeting PDE6D KRAS or HRAS or with mGFP-K-RasG12V or mGFP-H-RasG12V plasmids, using jetPRIME transfection reagent or Lipofectamine 3000. On the next day, cells were replated into 96-well plates for suspension cell culture. Mammosphere formation was analyzed under an EVOS FL microscope (Thermo Fisher Scientific) and spheres exceeding 50 μm in diameter were counted. The SFE was expressed as percentage normalized to vehicle-treated control. In protein overexpression or knockdown experiments, SFE was normalized to the empty vector or scrambled siRNA-transfected controls, respectively.
Tumorospheres of lung cancer cells were generated by seeding NCI-H358 or A-375 cells (1500 cells per well) as a suspension culture in F-bottom 96-well suspension culture plates (cat. no. 655185, Cellstar, Greiner Bio-One) in 50 μL RPMI medium supplemented with 0.5% methylcellulose, 25 ng/mL EGF, 25 ng/mL FGF, 1× B-27 supplement, and 2 mM l-glutamine. Cells were initially grown for 72 h and treated with freshly thawed compounds. After another 72 h of incubation, the cell viability was assessed using the alamarBlue assay (cat. no. DAL1100, Invitrogen, Carlsbad, CA, USA). A 10% final volume of alamarBlue reagent was added to each well of the plate and incubated for 4 h at 37 °C. Then, the fluorescence intensity was measured using a FLUOstar OPTIMA plate reader (BMG Labtech, Germany) with an excitation wavelength of 560 ± 5 nm and emission wavelength of 590 ± 5 nm. The obtained fluorescence intensity data were normalized to DMSO control. To quantitatively profile the drug sensitivity from the dose–response data, the DSS algorithm was employed.39 The DSS score was calculated according to the formula
The curve fitting parameters were used to calculate the AUC relative to the total area (TA) between the 10% threshold (DMSO at 0.1%) and 100% inhibition (benzethonium chloride at 100 μM). The integrated response is divided by the logarithm of the top asymptote (log a).
2D Cell Viability Assay
Hs 578T, HCT-116, and HT29 cells were plated onto 96-well cell culture plates at a density of 500 cells/well and allowed to attach for 24 h. Freshly thawed aliquots of test compounds were then added ice-cold at indicated concentrations. DMSO (0.1% v/v) was used as control. Plates were further incubated for 72 h. The cell viability was assessed using the alamarBlue reagent, according to manufacturer’s instructions. The fluorescence intensity was read with the excitation wavelength of 530 ± 10 nm and emission wavelength of 590 ± 10 nm using a Synergy H1 plate reader (BioTek, Winooski, VT, USA).
Western Blotting
Cells were lysed in RIPA buffer (cat. no. 89900, Thermo Scientific) containing protease (cat. no. 88666, Thermo Scientific) and phosphatase inhibitors (cat. no. 0496845001, Roche, Mannheim, Germany) and then sonicated for 5 min in ice-cold water. Proteins were separated on 10% sodium dodecyl sulfate polyAcrylamide gel electrophoresis (SDS-PAGE) gels and transferred to nitrocellulose membranes (cat. no. NBA 083C001EA, Protran, Waltham, USA) using a Transferblot Turbo Transfer System (Bio-Rad, Hercules, CA, USA). Briefly, the membranes were blocked in 1xTBS containing 0.1% Tween-20 and 5% w/v nonfat dry milk for 30 min and incubated with primary antibodies PDE6D (cat. no. sc-166854, Santa Cruz Biotechnology, Paso Robles, CA, USA.) or β-actin (cat. no. A1978, Sigma-Aldrich) overnight, at 4 °C. The membranes were then incubated with secondary antibodies antimouse (cat. no. sc-2954, Santa Cruz Biotechnology) for 1 h at room temperature. Protein bands were detected using a ChemiDoc MP instrument (Bio-Rad) after treatment with ECL reagent (cat. no. 170-5061, BioRad). The protein-band intensities were measured using Image Lab software (Bio-Rad).
RT-qPCR Analysis for Gene Knockdowns
MDA-MB-231 and Hs 578T cells were seeded in 6-well plates and transfected with 50 nM of siRNA KRAS or HRAS or scrambled, negative control siRNA (QIAGEN, cat. no. 1022076, QIAGEN, Hilden, Germany) using Lipofectamine. After 24 h of transfection, total RNA was isolated using NucleoZol according to the manufacturer’s protocol (cat. no.: 7040404, Macherey-Nagel, Hoerdt, France). Reverse transcription was performed with 1 μg of total RNA using SuperScript III Reverse Transcriptase (cat. no.: 18080093, Invitrogen). The knockdowns of KRAS and HRAS gene transcripts were analyzed by real-time qPCR using SsoAdvanced Universal SYBR Green Supermix (Bio-Rad), on the CFX-connect real-time PCR instrument (Bio-Rad). The transcripts were selectively amplified using specific primers producing amplicons for KRAS (total) and HRAS. The gene β-actin was used as reference. The following primers were used: for KRAS (total), forward 5′-tacagtgcaatgagggacca-3′, reverse 5′-tcctgagcctgttttgtgtct-3′ (amplicon 206 bp); for HRAS, forward 5′-ctgaccatccagctgatcca-3′, reverse 5′-tggcaaacacacacaggaag-3′ (amplicon 196 bp); for ACTB (β-actin), forward 5′-ggggtgttgaaggtctcaaa-3’; reverse 5′-ggcatcctcaccctgaagta-3′.40
Confocal Imaging
MDCK cells, stably overexpressing mGFP-KRasG12V, were seeded onto coverslips in 12-well cell culture plates. The cells were treated with 0.1% DMSO control or 5 μM Deltarasin or Deltaflexin-1. After 24 h cells were fixed in 4% PFA and mounted with Mowiol, cells were imaged using a laser scanning microscope (LSM 780) Carl Zeiss Microscopy, Jena, Germany.
Compound Syntheses
Purchased compounds were used without further purifications. Chemicals and solvents were purchased from the companies Sigma-Aldrich, VWR, and Thermo Fisher Scientific. Solvents were dried over 3 or 4 Å molecular sieves. Commercially available solvents for semipreparative high-performance liquid chromatography (HPLC) (acetonitrile HPLC grade) were used. Solvent mixtures are understood as volume/volume. Extended information on the complete compound syntheses and analytics are described in Supporting Information S1.
Molecular Modeling
Docking studies were carried out by the GOLD module41 of Discovery Studio 4.5 (Accelrys Inc. San Diego, USA, http://www.accelrys.com/) using the crystal structure of PDE6D determined at 1.45 Å resolution (PDB code 4JV8)15 as the receptor molecule. Water molecules and the bound ligand were removed from the structure prior to the docking. Binding sites were identified by using the binding site identification tool in Discovery Studio. The binding site that matched also the binding site identified by the crystal structure was selected for docking. The ligands were generated with ChemDraw Professional 15.1 (PerkinElmer Informatics, www.cambridgesoft.com) and exported as *.mol files to the Discovery Studio. Hydrogen atoms were added to the protein and the ligand. The Goldscore fitness function was used to score the poses.
SPR Binding and Inhibition Assays
SPR binding experiments were performed on a Biacore S200 instrument (GE Healthcare Life Sciences, Marlborough, MA, USA) as described previously.42 Farnesylated and methylated K-Ras-GDP (K-RasFMe), with an N-terminal avi-tag, was captured on neutravidin-coupled CM5 sensor chips (capture density 500–1500 RU). PDE6D (2-fold serial dilution, 20–0.039 μM) was injected over the K-RasFMe surface in 20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES) (pH 7.5), 150 mM NaCl, 0.01%, Tween 20, 5 mM MgCl2, and 5% DMSO at 30 μL/min and with a contact time of 60 s. The inhibitory activity of compounds for PDE6D/K-RasFMe interaction was measured using 1.25 μM PDE6D and a serial dilution of compound (2-fold, 100–0.39 μM) using the same buffer and flow rate. PDE6D (1.25 μM) was diluted with Deltarasin (3-fold, 50 μM to 68 nM) was used as a positive control. All titrations were run over three flow cells with variable surface densities of K-RasFMe. The data were processed by subtracting binding responses on the reference flow cells as well as the buffer subtraction. The samples were also corrected for DMSO mismatches using a DMSO standard curve. The equilibrium binding constant and IC50 values were calculated using the Biacore S200 Evaluation software. The IC50 value obtained from SPR and FA assay was converted into Kd using a modified Cheng–Prusoff equation
where KD is the dissociation constant between PDE6D/K-RasFMe and [L] is the concentration of PDE6D used in SPR experiments.44
FA Assay
The binding of compounds to PDE6D was assessed in an FA assay using fluorescein-tagged farnesylated Rheb (F-Rheb) peptide as the probe.14,43 PDE6D was produced as described below. FA assays were performed on a black low volume, round-bottom, non-binding surface 384-well plate (Corning, #4514) in an assay buffer composed of 30 mM Tris, 150 mM NaCl, and 3 mM dithiothreitol. To a three-fold dilution of compounds in assay buffer (300 μM to 1.7 nM), a complex of 0.25 μM F-Rheb peptide and 2 μM PDE6D was added, and the reaction mix was incubated for 15 min at RT. Then, the FP was recorded on a Synergy H1 hybrid plate reader (BioTek) equipped with a polarization cube with an excitation wavelength of 485 ± 10 nm and emission wavelength of 528 ± 10 nm. The FP data were plotted against the logarithmic concentration of compounds and fit to log inhibitor vs response–variable slope (four parameters) equation in Prism (GraphPad), and the IC50 was determined.
Cloning, Expression, and Purification of Recombinant Proteins
PDE6D was recombinantly produced in Escherichia coli BL21 (DE3) transformed with pDest-His6-MBP-PDE6D grown at 37 °C until OD of 0.6–0.8 and then induced with 0.5 mM of β-d-1-thiogalactopyranoside overnight at 16 °C.12 The bacterial lysate was prepared in a binding buffer (20 mM HEPES, pH 7.6, 300 mM NaCl, 5 mM MgCl2, 1 mM TCEP (Tris(2-carboxyethyl)phosphine hydrochloride), and protease inhibitor tablet from Roche) containing 0.5 mg/mL of lysozyme. Soluble proteins were isolated after sonication and centrifugation steps. The protein His6-MBP-TEV-PDE6D was purified on HisTrap HP column (GE Healthcare) in binding buffer containing imidazole and eluted with a 20 column-volume gradient from 35 to 500 mM imidazole. The eluted fractions were analyzed on SDS-PAGE. The fractions containing His6-MBP-TEV-PDE6D were treated with tobacco etch virus (TEV) protease (cat. no.: T4455, Sigma-Aldrich) (1:25 w/w, TEV/fusion protein) for 1 h at room temperature and then dialyzed overnight in binding buffer without protease inhibitor. The cleavage product containing His6-MBP and PDE6D was loaded a second time on a HisTrap HP column in binding buffer. The peak fractions (OD280) corresponding to the nonbound protein PDE6D were collected, pooled, concentrated using Amicon Ultra Centrifugal Filters (Merck Millipore, Molsheim, France), and analyzed on a SDS-PAGE gel with Coomassie blue. The presence of purified PDE6D was confirmed by Western blotting using a monoclonal anti-PDE6D antibody (cat. no.: sc-376724, Santa Cruz Biotechnology; dilution ratio 1:200).
The baculovirus expression vector for production of processed K-Ras4B has been previously described.44 An avi-tagged full-length K-Ras4B clone was generated by introducing an avi sequence (LNDIFEAQKIEWG) between the TEV protease cleavage site (ENLYFQG) sequence and amino acids 2–188 of K-Ras4B. The baculovirus expression vector for the production of processed avi-K-Ras4B was produced using the methods described previously.44 Expression of farnesylated and carboxymethylated K-Ras4B (K-RasFMe) and avi-K-Ras-FMe followed the methods as described.42 Purification of processed avi-tagged full-length K-Ras4B was as described44 with modifications. Protein isolated after the removal of the N-terminal His6-MBP tag (21.5 μM avi-K-Ras-FMe) was biotinylated by incubating for 2 h at room temperature with 2.8 μM His6-BirA, 3.6 mM ATP, 215 μM biotin in a buffer of 20 mM HEPES, pH 7.3, 300 mM NaCl, 5 mM MgCl2, 1 mM TCEP (Tris(2-carboxyethyl)phosphine hydrochloride), and 1:1000 v/v protease inhibitor (P8849 Sigma-Aldrich). The reaction was then dialyzed at 4 °C for 16 h against 20 mM HEPES, pH 7.3, 300 mM NaCl, 5 mM MgCl2, and 1 mM TCEP. The dialyzed reaction was passed over a HisTrap FF column (GE Healthcare), equilibrated with dialysis buffer, to remove His6-BirA. The biotinylated avi-K-Ras-FMe has a slight affinity for the column and elutes from the column at low (<50 mM) imidazole concentrations. Fractions were analyzed by coomassie-stained SDS-PAGE, and positive fractions were pooled and dialyzed against 20 mM HEPES, pH 7.3, 300 mM NaCl, 1 mM MgCl2, and 1 mM TCEP for 3 h at room temperature. The protein concentration was determined by absorbance at 280 nm, and aliquots were snap frozen in liquid nitrogen and stored at −80 °C.
Statistical Analysis
GraphPad Prism software was used for the statistical analysis. The sample size n for each data set is provided in the relevant figure legends and represents independent experiments. Unless otherwise stated, statistical significance was evaluated with the Student’s t-test or One-way ANOVA complemented by multiple comparison post hoc tests, as appropriate. A p-value of <0.05 is considered statistically significant, and the statistical significance levels are annotated as * = p < 0.05; ** = p < 0.01; *** = p < 0.001; **** = p < 0.0001, or ns = not significant.
Acknowledgments
The authors are grateful for the support from the cell imaging core (CIC, Turku Bioscience Centre, Turku, Finland). The infrastructure support from Biocenter Finland is acknowledged. We thank Prof. Philippe I. H. Bastiaens (Max-Planck Institute Dortmund, Germany) for providing the mCitrine-Rheb and mCherry-PDEδ constructs. We thank Lange Yakubu Saleh for the synthesis of compound 20 and Anuer Al-Rammahi for the synthesis of compound 17. We thank Eyad K. Fansa (Max-Planck Institute Dortmund, Germany) for providing the F-Rheb peptide. We acknowledge the protein production support for avi-KRAS-FMe and KRasFMe from Vanessa Wall, Matt Drew, and Jennifer Mehalko and Dominic Esposito for providing the pDest-His6-MBP-PDE6D plasmid (NCI-RAS Initiative, Frederick National Laboratory for Cancer Research, USA). This project was funded in whole or in part with federal funds from National Cancer Institute, NIH Contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, and the mention of trade names, commercial products, or organizations does not imply endorsement by the US Government.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03639.
Compound affinities by anisotropy; linkage derivatives and binding data; linkage derivative cell activity and heat passivation data; linkage derivative antiproliferation effect and genetic dependencies; additional second generation Deltaflexin data and knockdown controls; and compound synthesis and characterization (PDF)
Author Contributions
F.A.S. designed, performed, and analyzed all FLIM-FRET and WB experiments. C.A. and S.O. designed, performed, and analyzed sphere formation and cell proliferation experiments. A.S., O.M.A., H.L., M.O., and P.V. collaboratively designed the compounds 5, 9, 13, and 15. M.O., P.V., P.R., and D.A. collaboratively designed the compounds 17, 19, 20, 22, 23, 24, and 25. A.S. and P.R. prepared the compounds under supervision of M.O. M.C. prepared PDE6D protein. G.b.M. performed and analyzed FA experiments. L.B. and A.G.S. designed, performed, and analyzed SPR experiments. TT prepared avi-KRasFMe. A.C.P. performed computational docking and prepared graphs of structures. M.O. and P.V. wrote the chemical synthesis description. D.A. conceived the study, designed experiments, interpreted results, and wrote the manuscript together with F.A.S. All authors commented on the manuscript.
This work was supported by a CIMO fellowship to A.S. and F.A.S. P.V. received the support from the Academy of Finland Project (#308931). D.A. received the support from the Academy of Finland Key Project (#304638) grant, the Sigrid Juselius Foundation, and the Cancer Society of Finland.
The authors declare the following competing financial interest(s): We have filed a patent around second generation inhibitor Deltaflexin-2.
Supplementary Material
References
- Papke B.; Der C. J. Drugging RAS: Know the enemy. Science 2017, 355, 1158–1163. 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- Lerner E. C.; Zhang T.-T.; Knowles D. B.; Qian Y.; Hamilton A. D.; Sebti S. M. Inhibition of the Prenylation of K-Ras, but Not H- or N-Ras, Is Highly Resistant to CAAX Peptidomimetics and Requires Both a Farnesyltransferase and a Geranylgeranyltransferase I Inhibitor in Human Tumor Cell Lines. Oncogene 1997, 15, 1283–1288. 10.1038/sj.onc.1201296. [DOI] [PubMed] [Google Scholar]
- Spiegel J.; Cromm P. M.; Zimmermann G.; Grossmann T. N.; Waldmann H. Small-Molecule Modulation of Ras Signaling. Nat. Chem. Biol. 2014, 10, 613–622. 10.1038/nchembio.1560. [DOI] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P.; Solomon M.; Li L.-S.; Hansen R.; Rosen N. Allele-Specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science 2016, 351, 604–608. 10.1126/science.aad6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillig R. C.; Sautier B.; Schroeder J.; Moosmayer D.; Hilpmann A.; Stegmann C. M.; Werbeck N. D.; Briem H.; Boemer U.; Weiske J.; Badock V.; Mastouri J.; Petersen K.; Siemeister G.; Kahmann J. D.; Wegener D.; Böhnke N.; Eis K.; Graham K.; Wortmann L.; Nussbaum von F.; Bader B. Discovery of Potent SOS1 Inhibitors That Block RAS Activation via Disruption of the RAS-SOS1 Interaction. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 2551. 10.1073/pnas.1812963116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes M. R.; Zhang J.; Li L.-S.; Hansen R.; Peters U.; Guo X.; Chen Y.; Babbar A.; Firdaus S. J.; Darjania L.; Feng J.; Chen J. H.; Li S.; Li S.; Long Y. O.; Thach C.; Liu Y.; Zarieh A.; Ely T.; Kucharski J. M.; Kessler L. V.; Wu T.; Yu K.; Wang Y.; Yao Y.; Deng X.; Zarrinkar P. P.; Brehmer D.; Dhanak D.; Lorenzi M. V.; Hu-Lowe D.; Patricelli M. P.; Ren P.; Liu Y. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. 10.1016/j.cell.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Canon J.; Rex K.; Saiki A. Y.; Mohr C.; Cooke K.; Bagal D.; Gaida K.; Holt T.; Knutson C. G.; Koppada N.; Lanman B. A.; Werner J.; Rapaport A. S.; Miguel T. S.; Ortiz R.; Osgood T.; Sun J.-R.; Zhu X.; McCarter J. D.; Volak L. P.; Houk B. E.; Fakih M. G.; O’Neil B. H.; Price T. J.; Falchook G. S.; Desai J.; Kuo J.; Govindan R.; Hong D. S.; Ouyang W.; Henary H.; Arvedson T.; Cee V. J.; Lipford J. R. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Shima F.; Yoshikawa Y.; Ye M.; Araki M.; Matsumoto S.; Liao J.; Hu L.; Sugimoto T.; Ijiri Y.; Takeda A.; Nishiyama Y.; Sato C.; Muraoka S.; Tamura A.; Osoda T.; Tsuda K.-I.; Miyakawa T.; Fukunishi H.; Shimada J.; Kumasaka T.; Yamamoto M.; Kataoka T. In Silico Discovery of Small-Molecule Ras Inhibitors That Display Antitumor Activity by Blocking the Ras-Effector Interaction. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 8182. 10.1073/pnas.1217730110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M. J.; Pagba C. V.; Prakash P.; Naji A. K.; van der Hoeven D.; Liang H.; Gupta A. K.; Zhou Y.; Cho K.-J.; Hancock J. F.; Gorfe A. A. Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega 2019, 4, 2921–2930. 10.1021/acsomega.8b03308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra A.; Grecco H. E.; Pisupati V.; Perera D.; Cassidy L.; Skoulidis F.; Ismail S. A.; Hedberg C.; Hanzal-Bayer M.; Venkitaraman A. R.; Wittinghofer A.; Bastiaens P. I. H. The GDI-like solubilizing factor PDEδ sustains the spatial organization and signalling of Ras family proteins. Nat. Cell Biol. 2011, 14, 148–158. 10.1038/ncb2394. [DOI] [PubMed] [Google Scholar]
- Dharmaiah S.; Bindu L.; Tran T. H.; Gillette W. K.; Frank P. H.; Ghirlando R.; Nissley D. V.; Esposito D.; McCormick F.; Stephen A. G.; Simanshu D. K. Structural basis of recognition of farnesylated and methylated KRAS4b by PDEδ. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E6766–E6775. 10.1073/pnas.1615316113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmick M.; Vartak N.; Papke B.; Kovacevic M.; Truxius D. C.; Rossmannek L.; Bastiaens P. I. H. KRas Localizes to the Plasma Membrane by Spatial Cycles of Solubilization, Trapping and Vesicular Transport. Cell 2014, 157, 459–471. 10.1016/j.cell.2014.02.051. [DOI] [PubMed] [Google Scholar]
- Ismail S. A.; Chen Y.-X.; Rusinova A.; Chandra A.; Bierbaum M.; Gremer L.; Triola G.; Waldmann H.; Bastiaens P. I. H.; Wittinghofer A. Arl2-GTP and Arl3-GTP Regulate a GDI-Like Transport System for Farnesylated Cargo. Nat. Chem. Biol. 2011, 7, 942–949. 10.1038/nchembio.686. [DOI] [PubMed] [Google Scholar]
- Zimmermann G.; Papke B.; Ismail S.; Vartak N.; Chandra A.; Hoffmann M.; Hahn S. A.; Triola G.; Wittinghofer A.; Bastiaens P. I. H.; Waldmann H. Small molecule inhibition of the KRAS-PDEδ interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- Papke B.; Murarka S.; Vogel H. A.; Martín-Gago P.; Kovacevic M.; Truxius D. C.; Fansa E. K.; Ismail S.; Zimmermann G.; Heinelt K.; Schultz-Fademrecht C.; Saabi Al; Baumann M.; Nussbaumer P.; Wittinghofer A.; Waldmann H.; Bastiaens P. I. H. Identification of Pyrazolopyridazinones as PDEδ Inhibitors. Nat. Commun. 2016, 7, 11360. 10.1038/ncomms11360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín-Gago P.; Fansa E. K.; Klein C. H.; Murarka S.; Janning P.; Schürmann M.; Metz M.; Ismail S.; Schultz-Fademrecht C.; Baumann M.; Bastiaens P. I. H.; Wittinghofer A.; Waldmann H. A PDE6δ-KRas Inhibitor Chemotype with up to Seven H-Bonds and Picomolar Affinity that Prevents Efficient Inhibitor Release by Arl2. Angew. Chem., Int. Ed. 2017, 56, 2423–2428. 10.1002/anie.201610957. [DOI] [PubMed] [Google Scholar]
- Chen D.; Chen Y.; Lian F.; Chen L.; Li Y.; Cao D.; Wang X.; Chen L.; Li J.; Meng T.; Huang M.; Geng M.; Shen J.; Zhang N.; Xiong B. Fragment-based drug discovery of triazole inhibitors to block PDEδ-RAS protein-protein interaction. Eur. J. Med. Chem. 2019, 163, 597–609. 10.1016/j.ejmech.2018.12.018. [DOI] [PubMed] [Google Scholar]
- Hecker S. J.; Erion M. D. Prodrugs of Phosphates and Phosphonates. J. Med. Chem. 2008, 51, 2328–2345. 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- Kiuru E.; Ahmed Z.; Lönnberg H.; Beigelman L.; Ora M. 2,2-Disubstituted 4-Acylthio-3-Oxobutyl Groups as Esterase- and Thermolabile Protecting Groups of Phosphodiesters. J. Org. Chem. 2013, 78, 950–959. 10.1021/jo302421u. [DOI] [PubMed] [Google Scholar]
- Prakash P.; Sayyed-Ahmad A.; Cho K.-J.; Dolino D. M.; Chen W.; Li H.; Grant B. J.; Hancock J. F.; Gorfe A. A. Computational and Biochemical Characterization of Two Partially Overlapping Interfaces and Multiple Weak-Affinity K-Ras Dimers. Sci. Rep. 2017, 7, 40109. 10.1038/srep40109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada I. M. D.; Lectez B.; Siddiqui F. A.; Oetken-Lindholm C.; Sharma M.; Abankwa D. Opposite Feedback From mTORC1 to H-Ras and K-ras4B Downstream of SREBP1. Sci. Rep. 2017, 7, 8944. 10.1038/s41598-017-09387-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posada I. M. D.; Lectez B.; Sharma M.; Oetken-Lindholm C.; Yetukuri L.; Zhou Y.; Aittokallio T.; Abankwa D. Rapalogs Can Promote Cancer Cell Stemness in Vitro in a Galectin-1 and H-Ras-Dependent Manner. Oncotarget 2017, 8, 44550–44566. 10.18632/oncotarget.17819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Blaskovich M. A.; Knowles D.; Qian Y.; Ohkanda J.; Bailey R. D.; Hamilton A. D.; Sebti S. M. Antitumor Efficacy of a Novel Class of Non-Thiol-Containing Peptidomimetic Inhibitors of Farnesyltransferase and Geranylgeranyltransferase I. Cancer Res. 1999, 59, 4919. [PubMed] [Google Scholar]
- Najumudeen A. K.; Jaiswal A.; Lectez B.; Oetken-Lindholm C.; Guzmán C.; Siljamäki E.; Posada I. M. D.; Lacey E.; Aittokallio T.; Abankwa D. Cancer Stem Cell Drugs Target K-Ras Signaling in a Stemness Context. Oncogene 2016, 35, 5248–5262. 10.1038/onc.2016.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debeb B. G.; Zhang X.; Krishnamurthy S.; Gao H.; Cohen E.; Li L.; Rodriguez A. A.; Landis M. D.; Lucci A.; Ueno N. T.; Robertson F.; Xu W.; Lacerda L.; Buchholz T. A.; Cristofanilli M.; Reuben J. M.; Lewis M. T.; Woodward W. A. Characterizing Cancer Cells with Cancer Stem Cell-Like Features in 293T Human Embryonic Kidney Cells. Mol. Cancer 2010, 9, 180. 10.1186/1476-4598-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejuch T.; Garivet G.; Hofer W.; Kaiser N.; Fansa E. K.; Ehrt C.; Koch O.; Baumann M.; Ziegler S.; Wittinghofer A.; Waldmann H. Small-Molecule Inhibition of the UNC119-Cargo Interaction. Angew. Chem., Int. Ed. 2017, 129, 6277–6282. 10.1002/ange.201701905. [DOI] [PubMed] [Google Scholar]
- Humbert M. C.; Weihbrecht K.; Searby C. C.; Li Y.; Pope R. M.; Sheffield V. C.; Seo S. ARL13B, PDE6D, and CEP164 Form a Functional Network for INPP5E Ciliary Targeting. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 19691–19696. 10.1073/pnas.1210916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S.; Wright K. J.; Corre S. L.; Micalizzi A.; Romani M.; Abhyankar A.; Saada J.; Perrault I.; Amiel J.; Litzler J.; Filhol E.; Elkhartoufi N.; Kwong M.; Casanova J.-L.; Boddaert N.; Baehr W.; Lyonnet S.; Munnich A.; Burglen L.; Chassaing N.; Encha-Ravazi F.; Vekemans M.; Gleeson J. G.; Valente E. M.; Jackson P. K.; Drummond I. A.; Saunier S.; Attié-Bitach T. A HomozygousPDE6DMutation in Joubert Syndrome Impairs Targeting of Farnesylated INPP5E Protein to the Primary Cilium. Hum. Mutat. 2013, 35, 137–146. 10.1002/humu.22470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fansa E. K.; Kösling S. K.; Zent E.; Wittinghofer A.; Ismail S. PDE6δ-Mediated Sorting of INPP5E Into the Cilium Is Determined by Cargo-Carrier Affinity. Nat. Commun. 2016, 7, 11366. 10.1038/ncomms11366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guen V. J.; Chavarria T. E.; Kröger C.; Ye X.; Weinberg R. A.; Lees J. A. EMT Programs Promote Basal Mammary Stem Cell and Tumor-Initiating Cell Stemness by Inducing Primary Ciliogenesis and Hedgehog Signaling. Proc. Natl. Acad. Sci. U.S.A. 2017, 114, E10532–E10539. 10.1073/pnas.1711534114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter-Vann A. M.; Baron R. A.; Wong W.; dela Cruz J.; York J. D.; Gooden D. M.; Bergo M. O.; Young S. G.; Toone E. J.; Casey P. J. A Small-Molecule Inhibitor of Isoprenylcysteine Carboxyl Methyltransferase with Antitumor Activity in Cancer Cells. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 4336–4341. 10.1073/pnas.0408107102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D. R.; Wu Y.-M.; Lonigro R. J.; Vats P.; Cobain E.; Everett J.; Cao X.; Rabban E.; Kumar-Sinha C.; Raymond V.; Schuetze S.; Alva A.; Siddiqui J.; Chugh R.; Worden F.; Zalupski M. M.; Innis J.; Mody R. J.; Tomlins S. A.; Lucas D.; Baker L. H.; Ramnath N.; Schott A. F.; Hayes D. F.; Vijai J.; Offit K.; Stoffel E. M.; Roberts J. S.; Smith D. C.; Kunju L. P.; Talpaz M.; Cieślik M.; Chinnaiyan A. M. Integrative Clinical Genomics of Metastatic Cancer. Nature 2017, 548, 297–303. 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway L. A.; Lander E. S. Lessons From the Cancer Genome. Cell 2013, 153, 17–37. 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Butler L. M.; Ferraldeschi R.; Armstrong H. K.; Centenera M. M.; Workman P. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol. Cancer Res. 2015, 13, 1445–1451. 10.1158/1541-7786.mcr-15-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M.; Krykbaeva I.; Koeva M.; Kayatekin C.; Westover K. D.; Karras G. I.; Lindquist S. Quantitative Analysis of Hsp90-Client Interactions Reveals Principles of Substrate Recognition. Cell 2012, 150, 987–1001. 10.1016/j.cell.2012.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán C.; Oetken-Lindholm C.; Abankwa D. Automated High-Throughput Fluorescence Lifetime Imaging Microscopy to Detect Protein-Protein Interactions. J. Lab. Autom. 2016, 21, 238–245. 10.1177/2211068215606048. [DOI] [PubMed] [Google Scholar]
- Guzmán C.; Šolman M.; Ligabue A.; Blaževitš O.; Andrade D. M.; Reymond L.; Eggeling C.; Abankwa D. The Efficacy of Raf Kinase Recruitment to the GTPase H-Ras Depends on H-Ras Membrane Conformer-Specific Nanoclustering. J. Biol. Chem. 2014, 289, 9519–9533. 10.1074/jbc.m113.537001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav B.; Pemovska T.; Szwajda A.; Kulesskiy E.; Kontro M.; Karjalainen R.; Majumder M. M.; Malani D.; Murumägi A.; Knowles J.; Porkka K.; Heckman C.; Kallioniemi O.; Wennerberg K.; Aittokallio T. Quantitative Scoring of Differential Drug Sensitivity for Individually Optimized Anticancer Therapies. Sci. Rep. 2014, 4, 5193. 10.1038/srep05193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai F. D.; Lopes M. S.; Zhou M.; Court H.; Ponce O.; Fiordalisi J. J.; Gierut J. J.; Cox A. D.; Haigis K. M.; Philips M. R. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 779–784. 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking 1 1Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Agamasu C.; Ghirlando R.; Taylor T.; Messing S.; Tran T. H.; Bindu L.; Tonelli M.; Nissley D. V.; McCormick F.; Stephen A. G. KRAS Prenylation Is Required for Bivalent Binding with CaM in a Nucleotide Independent Manner. Biophys. J. 2019, 116, 1049–1063. 10.1016/j.bpj.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaževitš O.; Mideksa Y. G.; Solman M.; Ligabue A.; Ariotti N.; Nakhaeizadeh H.; Fansa E. K.; Papageorgiou A. C.; Wittinghofer A.; Ahmadian M. R.; Abankwa D. Galectin-1 Dimers Can Scaffold Raf-Effectors to Increase H-Ras Nanoclustering. Sci. Rep. 2016, 6, 24165. 10.1038/srep24165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillette W. K.; Esposito D.; Blanco M. A.; Alexander P.; Bindu L.; Bittner C.; Chertov O.; Frank P. H.; Grose C.; Jones J. E.; Meng Z.; Perkins S.; Van Q.; Ghirlando R.; Fivash M.; Nissley D. V.; McCormick F.; Holderfield M.; Stephen A. G. Farnesylated and Methylated KRAS4b: High Yield Production of Protein Suitable for Biophysical Studies of Prenylated Protein-Lipid Interactions. Sci. Rep. 2015, 5, 15916. 10.1038/srep15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.