

Abstract
Photodynamic therapy (PDT) is a treatment in which photoactive compounds delivered to cancerous tissues are excited with light and then transfer the absorbed energy to adjacent tissue oxygen molecules to generate toxic singlet oxygen (1O2). As 1O2 is produced only where light and photosensitizers (PSs) are combined, PDT holds promise as a minimally invasive, highly selective treatment for certain cancers. The practical application of PDT requires easily synthesized, water-soluble PSs that have low dark toxicities, high 1O2 quantum yields, and efficient absorption of 650–850 nm near-infrared (NIR) light, which deeply penetrates tissue. We recently developed a linear tetrapyrrole metal complex, Pd[DMBil1]–PEG750, that meets most of these criteria. This complex is remarkably effective as a PS for PDT against triple-negative breast cancer (TNBC) cells but, critically, it does not absorb NIR light, which is necessary to treat deeper tumors. To enable NIR activation, we synthesized a new derivative, Pd[DMBil1]–PEG5000–SH, which bears a thiol functionality that facilitates conjugation to NIR-absorbing gold nanoshells (NSs). Upon excitation with pulsed 800 nm light, NSs emit two-photon-induced photoluminescence spanning 500–700 nm, which can sensitize the attached PSs to initiate PDT. Additionally, NSs produce heat upon 800 nm irradiation, endowing the NS–PS conjugates with an auxiliary photothermal therapeutic (PTT) capability. Here, we demonstrate that NS–PS conjugates are potent mediators of NIR-activated tandem PDT/PTT against TNBC cells in vitro. We show that Pd[DMBil1]–PEG5000–SH retains the photophysical properties of the parent Pd[DMBil1] complex, and that NS–PS generate 1O2 under pulsed 800 nm irradiation, confirming activation of the PSs by photoluminescence emitted from NSs. TNBC cells readily internalize NS PS conjugates, which generate reactive oxygen species in the cells upon pulsed NIR irradiation to damage DNA and induce apoptosis. Together, these findings demonstrate that exploiting photoluminescent NSs as carriers of efficient Pd[DMBil1] PSs is an effective strategy to enable NIR light-activated tandem PDT/PTT.
Introduction
Photodynamic therapy (PDT) is an attractive treatment for certain cancers and offers a number of advantages over more conventional treatment modalities. During PDT, a light-absorbing compound is applied directly to the area requiring treatment or administered systemically via injection into the bloodstream and allowed time to accumulate in the tumor prior to irradiation. Provided that the PDT agent supports a triplet photochemistry, light activation initiates energy transfer from the photosensitizer (PS) to nearby molecular oxygen, forming excited singlet oxygen (1O2) in situ, which induces cellular damage. The effects of 1O2 are constrained within an approximately 100 nm radius of the source,1 resulting in highly localized cell death.2,3 Aside from the potential to confine the effects of treatment to targeted tissues through careful control of the illuminated area and/or preferential accumulation of the PS in the tumor,4−8 PDT is also less invasive and gives better cosmetic outcomes than surgical excision9 and it does not cause the debilitating side effects encountered with radiotherapy or chemotherapy.10 Additionally, PDT can stimulate antitumor immunity in contrast to the immunosuppressant nature of many other treatment modalities.11,12
Despite its many potential advantages, PDT has yet to be adopted into the arsenal of commonly used cancer treatments because the development of a single PS endowed with optimal photophysical and pharmacological characteristics has remained elusive. The group of compounds that have been investigated for use in PDT is dominated by macrocyclic tetrapyrroles belonging to the porphyrinoid family,13−16 but has been expanding to include additional classes of molecules.17−19 These compounds generate 1O2 effectively, but possess varying unfavorable attributes such as challenging or low-yielding syntheses, a tendency to aggregate or precipitate in biorelevant, aqueous-based solutions, high inherent cellular dark toxicity, or poor absorption in the near infrared (NIR) spectral regions (650–850 nm) that are best suited to deeply penetrate biological tissues. Consequently, an active area of research centers on development of improved PSs for use in PDT. In support of this effort, we have introduced a family of stable and synthetically accessible linear tetrapyrrole metal complexes known as biladienes. These complexes absorb across a broad range of visible wavelengths and generate 1O2 with quantum yields that range from <0.2% to 80%, depending on the metal ion coordinated within the biladiene core.20,21 Recently, we reported a water-soluble derivative of the most promising complex, Pd[DMBil1]–PEG750, and demonstrated its ability to act as a highly effective PS for PDT of triple-negative breast cancer (TNBC) cells with extremely low toxicity in the dark and a remarkably high phototoxicity index (PI; ratio of LD50/ED50) of ∼5300 under excitation with λ > 500 nm light.22 Excitingly, the PI of this PS was ∼200 and ∼3000 times higher than those of hematoporphyrin dihydrochloride and isohematoporphoyrin, two commonly utilized photosensitizers. Despite these advances, Pd[DMBil1]–PEG750 only absorbs at wavelengths shorter than 600 nm, undercutting its potential as a viable PDT agent for treatment of most solid tumors because of limited tissue penetration attainable using those visible wavelengths of light. To enable PDT of deeper-seated tumors, strategies for NIR activation of Pd[DMBil1]-based photosensitizers must be developed.
One potential strategy to enable NIR activation of Pd[DMBil1]-based PS for use in PDT would be to chemically modify the complex to red-shift its absorption spectrum. The method commonly used to enhance absorption at longer wavelengths involves extending the conjugated π system of the chromophore. Such strategies generally require several additional synthetic steps, and may fail to produce a bathochromic shift of the magnitude needed to push the absorption envelope into the NIR. Additionally, changing the structure and electronic conjugation of the PS can dramatically impact the system’s photophysics. Reduction in the quantum yield, lifetime, and energy of the 3PS* state formed upon excitation can all decrease or shut down 1O2 production. An alternative strategy, which avoids the need for synthetic modification of the PS core, is to use upconverting materials to transform longer wavelength incident NIR light into shorter wavelengths that the PDT agent can absorb. This strategy for NIR-activated PDT has been explored for other visible light-activated 1O2 photosensitizers, which have been combined with upconverting nanomaterials such as lanthanide-doped nanoparticles, quantum dots, carbon nanomaterials, silica nanoparticles, polymer nanoparticles, or gold nanorods.23−27 In this work, we have combined the promising Pd[DMBil1]-based PS with NIR-activated silica core/gold shell nanoshells (NSs) to enable NIR-activated PDT. This system offers several advantages over prior conjugates, including the use of a designer PS that has 1O2 quantum yields much greater than commercial PSs, and the use of NSs that are already being evaluated in clinical trials.28 The combination of these two entities enables effective treatment to be achieved at much lower doses of the nanoconjugates and light than possible with previously reported systems.
NSs with diameters of ∼150 nm absorb broadly in the visible to NIR spectral region with λmax ≈ 800 nm.29,30 When irradiated with a femtosecond (fs)-pulsed 800 nm laser, NS photoluminescence is λ = 500–700 nm, which overlaps with the absorption spectrum of the Pd[DMBil1] chromophore.21 NSs have a significant two-photon absorption cross section comparable to values reported for gold nanorods and some quantum dots (∼2000 GM).31,32 In addition to upconverted emission, NSs also generate heat under NIR irradiation. As a sufficient increase in temperature induces cell death, NSs and other gold nanomaterials have received substantial attention for their ability to mediate photothermal therapy (PTT) in solid tumors.33−37 Accordingly, we anticipated that irradiation with 800 nm light would cause gold NS–Pd[DMBil1] photosensitizer conjugates (NS–PS) to generate 1O2 to enable NIR-activated PDT in complement with generation of heat to enable tandem PDT/PTT (Scheme 1). The oxygen-independent nature of PTT permits light-induced cellular toxicity to be initiated in tissues that may be too hypoxic for effective PDT,38 allowing for improved therapeutic effects deep within solid tumors where oxygen levels tend to be low. Moreover, as increased intratumoral blood flow has been observed during PTT, the tandem strategy outlined above has the potential to enhance the PDT effect in solid/hypoxic tumors by boosting cellular oxygen levels.39 The possible benefits of combining these two therapies have inspired increasing research into dual PDT/PTT approaches,40−42 and multimodal phototherapies that can be activated by a single wavelength of light are especially attractive.
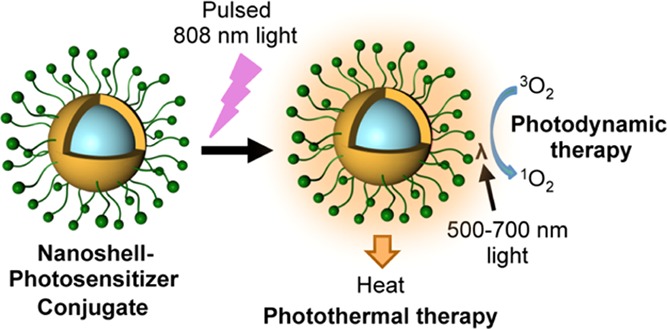
Scheme 1. Structure of NS–PS Conjugates and Mechanism of NIR Light-Activated Tandem PDT/PTT.

In response to 800 nm pulsed light, NSs emit photoluminescence that can activate PSs tethered to their surface via a poly(ethylene glycol) (PEG) chain, to trigger PDT. Simultaneously, the NSs can produce heat to enable PTT.
In this work, the synthetic strategy developed previously for Pd[DMBil1]–PEG75022 was adapted by replacing the methoxy-terminated 750 Da PEG with a longer thiol-terminated 5000 Da PEG to neatly obtain the biladiene complex Pd[DMBil1]–PEG5000–SH that can be readily conjugated to NSs. The photophysical properties of this new compound remain largely unchanged compared to those of Pd[DMBil1]–PEG750, and the thiol functionality facilitates attachment to the gold surfaces of the NSs. The NS–PS conjugates continue to produce 1O2 under 550 nm irradiation and heat under 800 nm irradiation, indicating that neither the photodynamic capability of Pd[DMBil1]–PEG5000–SH nor the photothermal capability of the NSs are lost after linking the two components together. Additionally, NS–PS show a clear ability to make 1O2 upon irradiation with 800 nm light, which confirms that the Pd[DMBil1] can be activated by the photoluminescence emitted from the NSs. Studies with MDA-MB-231 TNBC cells reveal that the NS–PS conjugates are well tolerated in the dark, but efficiently mediate apoptotic cell death under pulsed 800 nm light. Furthermore, TNBC cells incubated with NS–PS show enhanced levels of reactive oxygen species (ROS) and DNA damage following light treatment. Overall, this work demonstrates that linking photoluminescent gold NSs to 1O2 photosensitizers that absorb light in the visible region of the spectrum is an effective strategy to enable NIR-activated tandem PDT/PTT. As shown here, a single wavelength of tissue-penetrating NIR light can be utilized to activate NSs, resulting in the emission of photoluminescence that yields luminescence resonance energy transfer (LRET)-based sensitization of the PS for PDT, as well as simultaneous production of heat for complimentary PTT. As NSs have already entered clinical trials with promising results,28 and Pd[DMBil]-based PSs have a substantially higher phototoxicity index than other agents used for PDT, the NS–PS conjugates reported here are poised to substantially advance the field of photoactivated cancer therapy.
Results and Discussion
Synthesis of and 1O2 Sensitization by Pd[DMBil1]–PEG5000–SH
Pd[DMBil1]–PEG5000–SH was synthesized as shown in Scheme 2 using the same synthetic strategy employed previously for Pd[DMBil1]–PEG750.22 Briefly, the biladiene core was prepared in four steps using methodology previously developed for the synthesis of biladienes21 and related tetrapyrroles containing sp3-hybridized mesocarbons.43−45 Installation of the HS–PEG5000–NH2 chain was facilitated by the presence of the biladiene’s ancillary 5- and 15-pentafluorophenyl rings, which can be substituted with mercaptoacetic acid via nucleophilic aromatic substitution of the para-fluorine substituents46 to generate Pd[DMBil1]–SCH2CO2H. PEGylation of the carboxylic acid-appended biladiene was achieved using standard amide coupling chemistry. Treatment of Pd[DMBil]–SCH2CO2H with N-hydroxysuccinimide (NHS) and 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide hydrochloride (EDC) to form the corresponding NHS mercaptoacetate intermediate was followed by reaction with triethylamine and 5 kDa HS–PEG5000–NH2 chain to deliver the desired Pd[DMBil1]–PEG5000–SH derivative. The length of the HS–PEG5000–NH2 chain (5 kDa) was chosen to ensure stability and biocompatibility of the final NS–PS conjugate (Scheme 1) upon tethering of the Pd[DMBil1]–PEG5000–SH to NSs.29,47−49 Synthetic procedures and characterization data for Pd[DMBil1]–PEG5000–SH are detailed in the Experimental Methods and Supporting Information. The structure of this compound was verified by a combination of 1H, 13C, and 19F NMR spectroscopy (Figures S1–S3).
Scheme 2. Synthesis of Pd[DMBil1]–PEG5000–SH.

The steady-state photophysical properties of Pd[DMBil1]–PEG5000–SH are consistent with those previously observed for Pd[DMBil1]21 and Pd[DMBil1]–PEG750.22 When dissolved in methanol, Pd[DMBil1]–PEG5000–SH displays absorption maxima at 402 and 481 nm, with a shoulder around 543 nm. These features also appear in the absorption profiles of Pd[DMBil1] and Pd[DMBil1]–PEG750 at nearly identical wavelengths as shown in Figure 1a and Table 1. The maximum extinction coefficient of Pd[DMBil1]–PEG5000–SH (35 300 M–1 cm–1) is enhanced slightly compared to that of Pd[DMBil1] (29 200 M–1 cm–1), and is close to the maximum extinction coefficient observed for Pd[DMBil1]–PEG750 (34 600 M–1 cm–1). Functionalization with the longer thiol-terminated PEG chain appears to cause a modest increase in the extinction coefficient of the longer wavelength shoulder from the values of 7200 and 7500 M–1 cm–1 observed for Pd[DMBil1] and Pd[DMBil1]–PEG750, respectively, to 8700 M–1 cm–1 for Pd[DMBil1]–PEG5000–SH. Upon excitation at λex = 460 nm, a nitrogen-saturated methanol solution of Pd[DMBil1]–PEG5000–SH produces two broad emission features spanning the ranges of λem = 500–700 nm and λem = 725–850 nm, as shown in Figure 1b. Pd[DMBil1] and Pd[DMBil1]–PEG750 display two similar emission features in nitrogen-saturated methanol under the same conditions, and the second feature has been attributed to phosphorescence because of its large Stoke’s shift and complete disappearance upon exposure to air.21,22 As shown in Figure 1b, the second feature of Pd[DMBil1]–PEG5000–SH is also quenched under air, confirming its identity consistent with the long wavelength band corresponding to phosphorescence. These results demonstrate that the thiol-terminated PEG substituent does not significantly attenuate intersystem crossing or the triplet photochemical properties of the Pd[DMBil1] core to the triplet excited state. Table 1 compiles the emission characteristics of Pd[DMBil1], Pd[DMBil1]–PEG750, and Pd[DMBil1]–PEG5000–SH. The fluorescence quantum yield of Pd[DMBil1]–PEG5000–SH (3.1 × 10–4) is only marginally higher than Φfl of Pd[DMBil1] (1.3 × 10–4) and Pd[DMBil1]–PEG750 (1.4 × 10–4). Additionally, the phosphorescence signal measured for Pd[DMBil1]–PEG5000–SH has a maximum at 755 nm, which is essentially identical to the phosphorescence maxima of Pd[DMBil1] (753 nm) and Pd[DMBil1]–PEG750 (756 nm). Pd[DMBil1]–PEG5000–SH has a phosphorescence quantum yield of 6.1 × 10–5, which is slightly lower than Φphos of Pd[DMBil1] (1.3 × 10–4), but close in value to that measured for Pd[DMBil1]–PEG750 (7.8 × 10–5). Overall, the longer 5 kDa PEG chain and presence of a thiol functional group do not significantly perturb the electronic/photochemical properties of the palladium biladiene photosensitizer.
Figure 1.

(a) Room-temperature absorption spectra recorded for Pd[DMBil1] (gray), Pd[DMBil1]–PEG750 (blue), and Pd[DMBil1]–PEG5000–SH (black) in methanol. (b) Room-temperature emission spectra recorded for Pd[DMBil1]–PEG5000–SH dissolved in either nitrogen-saturated (red) or air-saturated (orange) methanol upon excitation (λex = 460 nm).
Table 1. Photophysical Properties of Pd[DMBil1], Pd[DMBil1]–PEG750 and Pd[DMBil1]–PEG5000–SH.
| compound (solvent) | λabs/nm (ε × 103 M–1 cm–1) | λfl/nm (Φfl) | λphos/nm (Φphos) | ΦΔ |
|---|---|---|---|---|
| Pd[DMBil1] (methanol) | 401 (16.5), 483 (29.2), 540 (7.2) | 557 (1.3 × 10–4) | 753 (1.3 × 10–4) | b0.80 |
| Pd[DMBil1]–PEG750 (methanol) | 402 (16.6), 483 (34.6), 540 (7.5) | 568 (1.4 × 10–4) | 756 (7.8 × 10–5) | b0.57 |
| Pd[DMBil1]–PEG5000–SH (methanol) | 402 (17.6), 481 (35.3), 543 (8.7) | 511 (3.1 × 10–4) | 755 (6.1 × 10–5) | b0.66 |
| Pd[DMBil1]–PEG750 (Milli-Q water) | 398 (15.6), 495 (35.8), 551 (11.8) | a580 (2.0 × 10–4) | c0.13 | |
| Pd[DMBil1]–PEG5000–SH (Milli-Q water) | 400 (13.7), 488 (29.5), 546 (10.1) | 585 (1.3 × 10–4) | c0.48 |
Measured in pH 7.4 phosphate buffered saline.
Measured after irradiation with λexc = 500 nm.
Measured after irradiation with λexc = 550 nm.
The steady-state photophysical properties of Pd[DMBil1]–PEG5000–SH were also examined in ultrapure water (Table 1 and Figure S4), to gain insight into how this new derivative might behave under biocompatible aqueous environments. The most prominent absorption feature of Pd[DMBil1]–PEG5000–SH and the longer wavelength shoulder shifted bathochromically from 481 and 543 nm in methanol to 488 and 546 nm in water. Additionally, the extinction coefficients of the two absorption maxima of Pd[DMBil1]–PEG5000–SH in water (13 700 and 29 500 M–1 cm–1) were attenuated slightly compared to their values in methanol (17 600 and 35 300 M–1 cm–1), whereas the intensity of the longer wavelength shoulder increased from 8700 to 10 100 M–1 cm–1. Pd[DMBil1]–PEG5000–SH was found to obey the Beer–Lambert Law in water (Figure S5), suggesting that it is not prone to aggregation within the range of concentrations tested (8.0–40.0 μM). For comparison, Pd[DMBil1]–PEG750 showed very similar absorption characteristics in ultrapure water, but exhibited a slightly more pronounced red shift and enhancement of the long wavelength shoulder (Figure S4 and Table 1). The aqueous emission profiles of the two PEGylated derivatives were also nearly identical. Figure S4 shows that fluorescence (but not phosphorescence) was observed from Pd[DMBil1]–PEG5000–SH in nitrogen-saturated ultrapure water. Previously, we noted that Pd[DMBil1]–PEG750 failed to phosphoresce in pH 7.4 phosphate buffered saline (PBS) under a nitrogen atmosphere, which was attributed to rapid deactivation of the triplet excited state through energy transfer to a water overtone.22 The fluorescence features of both complexes are red-shifted compared to their positions in methanol, and Pd[DMBil1]–PEG5000–SH has a fluorescence maximum at around 585 nm in ultrapure water, whereas that of Pd[DMBil1]–PEG750 occurs at about 580 nm in pH 7.4 PBS(Table 1). The Φfl of Pd[DMBil1]–PEG5000–SH in ultrapure water was calculated to be 1.3 × 10–4, which is a slight decrease from the value of 3.1 × 10–4 calculated in methanol. The Φfl calculated previously for Pd[DMBil1]–PEG750 in PBS(2.0 × 10–4) was only marginally higher.22 These results suggest that the photophysical characteristics of Pd[DMBil1]–PEG5000–SH in water are reminiscent of Pd[DMBil1]–PEG750, and it may behave similarly as an effective 1O2 photosensitizer in aqueous environments.
To verify that Pd[DMBil1]–PEG5000–SH retains the ability of the parent tetrapyrrole to sensitize 1O2 efficiently, its ability to generate this ROSs was ascertained in both methanol and ultrapure water. Using 1,3-diphenylisobenzofuran (DPBF) as a probe for 1O2 production50 and [Ru(bpy)3](PF6)2 as a reference photosensitizer with ΦΔ = 0.81 in methanol,51 the 1O2 quantum yield of Pd[DMBil1]–PEG5000–SH was determined to be 0.66 under irradiation with 500 nm light (in methanol). This value is only slightly lower than the quantum yield measured for the original Pd[DMBil1] photosensitizer (ΦΔ = 0.81), and is slightly higher than the quantum yield found for Pd[DMBil1]–PEG750 in methanol (ΦΔ = 0.57), as summarized in Table 1. The ability of Pd[DMBil1]–PEG5000–SH to photosensitize the formation of 1O2 was also determined in water using singlet oxygen sensor green (SOSG) as a probe52 and methylene blue as a reference photosensitizer (ΦΔ = 0.52).53,54 Pd[DMBil1]–PEG5000–SH exhibited an impressive quantum yield of 0.48 upon excitation with 550 nm light. This value is significantly higher than that measured for Pd[DMBil1]–PEG750 in ultrapure water (ΦΔ = 0.13), and is comparable to the 1O2 quantum yields of PSs that are used clinically as PDT agents.17 The results of these studies indicate that the 5 kDa thiol-terminated PEG substituent retains the palladium biladiene’s ability to efficiently photosensitize 1O2 production in both polar protic and aqueous environments.
Characterization of NS–PS and NS–PEG
NSs composed of silica cores (∼120 nm) and thin gold shells (∼15 nm) were synthesized according to previously published methods.30 For our studies, we developed two types of NS formulations: (1) NSs coated with Pd[DMBil1]–PEG5000–SH (NS–PS) and (2) NSs coated with only mPEG5000–SH (NS–PEG). UV–vis spectrophotometry revealed that the NS–PS had a peak plasmon resonance at 800 nm (Figure 2a), which was tuned to be consistent with the wavelength of the pulsed laser for optimal PTT and PDT effects. The photosensitizer peak is not noticeable after conjugation as its concentration (ca. 0.2 μM) in the solution of NSs at OD1 is below the limit of detection (Figure S5). However, a slight bathochromic-shift of NS–PS over bare NSs is a positive sign of successful functionalization with the Pd[DMBil1]–PEG5000–SH. The functionalization was further confirmed by dynamic light scattering (DLS), which showed an increased hydrodynamic diameter from bare NSs (151.9 ± 1.6 nm) to NS–PEG (172.6 ± 2.8 nm) and NS–PS (201.9 ± 1.6 nm) (Figure 2b). Slight increases in zeta potential were observed among bare NSs (−35.1 ± 0.7) to NS–PEG (−31.8 ± 0.7) and NS–PS (−29.3 ± 0.2) (Figure 2b). Scanning electron microscopy (SEM) images indicated that the NS–PS had a highly monodisperse size distribution (Figure 2c), as did bare NSs and NS–PEG (Figure S6). The photosensitizer loading on NSs was quantified using an established standard curve (Figure S5) based on the absorbance of the photosensitizer at 488 nm. The loading was determined to be 39 600 ± 2260 per NS (Figure 2d). Notably, spectrophotometry analysis of the NS–PS conjugates demonstrated that they were extremely stable when stored in water. The extinction profile of the conjugates was maintained up to 15 months (Figure S7), and no precipitation was visible to the eye.
Figure 2.

Characterization of NS–PS conjugates. (a) Spectrophotometry showing the optical characteristics of bare NSs and NS–PS conjugates in Millipore water. (b) Hydrodynamic diameter and zeta potential measurements of bare NSs, NS–PEG, and NS–PS. (c) Scanning electron micrographs of NS–PS. Scale bar = 500 nm. (d) Quantification of PS loading on NSs. N = 3.
NS–PS Generate 1O2 and Heat upon Excitation with Pulsed NIR Light
We examined the ability of NS–PS to generate 1O2 in water using a SOSG assay (Figures 3, S8). In this assay, reaction of SOSG with 1O2 produces a more emissive endoperoxide,51 enabling a qualitative comparison of the relative amounts of 1O2 produced by different photoactive agents. The changes in integrated emission intensity of SOSG over time as NS–PEG, Pd[DMBil1]–PEG5000–SH, the NS–PS conjugates, or a mixture of NS–PEG and Pd[DMBil1]–PEG750 in ultrapure water were irradiated are shown in Figure S8. Irradiation with 550 nm continuous wave (CW) light via a LED lightbox overlaid with a yellow filter (Figures S8a and S9), which directly activates the biladiene PS, drove the largest increase in SOSG emission intensity in the presence of Pd[DMBil1]–PEG5000–SH or the mixture of NS–PEG and Pd[DMBil1]–PEG750. The change in SOSG emission intensity of these two solutions was not significantly different, and this result is in agreement with our previous observations that mixing NSs with photosensitizers without physically connecting them does not compromise the PS′ ability to generate 1O2 upon irradiation with visible light.29 The solution of NS–PEG alone and the ultrapure water control did not show any increase in SOSG emission upon 550 nm CW irradiation, and therefore did not produce 1O2. Conversely, the SOSG emission intensity from the solution of NS–PS conjugates did show a significant increase compared to the control and NS–PEG solutions, but the enhancement was much less pronounced than that observed for Pd[DMBil1]–PEG5000–SH alone or the mixture of NS–PEG and Pd[DMBil1]–PEG750. This result suggests that conjugating the biladiene PS to the NS surface may slightly attenuate the 1O2 quantum yield, but the NS–PS conjugates can still function as PDT agents using visible light as 1O2 production was not eliminated completely.
Figure 3.

Emission profile of SOSG in an aqueous solution (OD1) of (a) NS–PS conjugates and (b) NS–PEG measured after 0, 5, 10, 15, and 20 min of irradiation with an 800 nm pulsed laser. Emission spectra were smoothed with Savitzky–Golay functions (polynomial order = 3, 20 points per window). (c) Increase in integrated emission intensity of SOSG with NS–PEG vs NS–PS under 800 nm pulsed laser irradiation as a function of time. *p < 0.05, **p < 0.01 by a Student’s t-test. (d) Heating profile of NS–PS at different concentrations upon irradiation with an 800 nm pulsed laser (1 W/cm2) after 24 h of incubation in complete cell culture media.
When pulsed 800 nm laser light was used to activate the NSs and induce visible photoluminescence as shown in Scheme 1, the NS–PS conjugates produced the greatest enhancement in SOSG emission intensity (Figures 3a and S8b), consistent with 1O2 photosensitization using NIR light. Solutions of Pd[DMBil1]–PEG5000–SH do not absorb 800 nm light and, unsurprisingly, did not induce an increase in SOSG emission intensity upon direct NIR excitation. Based on these results, it is clear that the 1O2 produced by the NS–PS conjugates cannot be the result of direct excitation of the biladiene PS. We note that gold nanoparticles have been reported to produce 1O2,55 which may partially explain the increase in SOSG emission intensity observed upon irradiation of the NS–PEG with 800 nm pulsed laser light (Figures 3b and S8b). Additionally, SOSG and the endoperoxide it forms following reaction with 1O2 can also photosensitize 1O2.51,56 Although SOSG (and the formed endoperoxide) do not absorb 800 nm light, the absorption spectra of these fluorescein derivatives overlap with the wavelengths of upconverted emission produced by the NSs, which may contribute to the SOSG emission enhancement observed in the presence of the NS–PEG.
Although there is some evidence of 1O2 production upon 800 nm irradiation of NS–PEG, the observed increase in SOSG emission intensity produced by the NS–PS conjugates was significantly higher (Figure 3c) at all sampling times, indicating that the 1O2 formed by the conjugates cannot simply be attributed to the NSs or SOSG. Furthermore, the increase in SOSG emission intensity observed for solutions of NS–PEG + Pd[DMBil1]–PEG750 (where the NSs and PSs are together in solution, but not physically tethered) is essentially identical to that detected for NS–PEG alone (Figure S8b). This result implies that negligible amounts of PS are activated by photoluminescence emitted from the NSs when they are not physically conjugated to one another. This finding is not surprising given that LRET is extremely sensitive to donor–acceptor distance. Proximity is required for efficient energy transfer. Overall, these observations support the conclusion that the 1O2 generated by the NS–PS conjugates under 800 nm pulsed laser light is the result, at least in part, of activation of the biladiene PS by the photoluminescence emitted from the NSs upon NIR exposure. Based on these results, the NS–PS conjugates have the capability of serving as PDT agents using deeply penetrating NIR light.
In addition to producing upconverted photoluminescence, NSs can also generate heat upon NIR laser irradiation because of their surface plasmon resonance and high photothermal conversion efficiency.57−59 To confirm that NS–PS retain the photothermal properties of the underlying NSs, we measured the temperature of NS–PEG and NS–PS suspended in water at concentrations of OD1, OD2, or OD3 while exposed to 800 nm pulsed laser light (1 W/cm2). Thermal images were taken with an FLIR forward-looking infrared E6 camera once per minute for the first 5 min of irradiation and then once every 5 min for up to 20 min. We found that the temperature of all NS–PS and NS–PEG solutions increased rapidly during the first 5 min, then reached a plateau (Figure S10a). In contrast, no obvious temperature increase was observed for water irradiated with the laser that did not contain NSs (Figure S10a, black line). We also evaluated the photothermal profile of NS–PS after they were incubated in complete cell culture media for 24 h. Similar to the studies performed in water, the temperature increased rapidly during the first 5 min, then climbed slowly for the next 5 min until reaching a plateau (Figure 3d). The overall temperature increase for NS–PS in media was less than that for NS–PS in water, which is expected due to the interaction of serum proteins and nanoparticles. Finally, the photothermal stability of NS–PS was also studied by irradiating NS–PS in a cyclic fashion, which revealed that the photothermal conversion abilities of the NS–PS conjugates are not compromised within four irradiation cycles (Figure S10b). Together, these results demonstrate that NS–PS are capable of generating heat sufficiently, in addition to generating 1O2 upon NIR irradiation, making it an excellent candidate for dual PDT/PTT.
MDA-MB-231 TNBC Cells Internalize NS–PS Conjugates
Darkfield microscopy was used to examine the interaction between NS–PS and MDA-MB-231 TNBC cells. In darkfield microscopy images, NS–PS appear as reddish spots owing to the NS′ strong light-scattering properties. The results reveal that NS–PS (Figure 4, right, pink-red spots) are bound to or internalized by MDA-MB-231 cells following a 24 h incubation period. In contrast, cells that are not treated with NS–PS appear blue, as a result of the intrinsic scattering from intracellular structures (Figure 4, left). The ability of MDA-MB-231 cells to engage NS–PS is important as the distance between photosensitizers and genomic DNA is critical to the efficacy of PDT treatment. As the half-life of 1O2 is short, the effects of photosensitizers are felt only within an approximately 100 nm radius of the source.1 The NS–PS conjugates appear to be well dispersed within the cells. This is an important observation, as the aggregation of gold NSs can reduce their upconversion efficiency and impair 1O2 sensitization and/or photothermal conversion processes critical to PDT/PTT efficacy. Based on these data, we further evaluated NS–PS as agents for combined PDT/PTT of TNBC (vide infra).
Figure 4.

Darkfield images show NS–PS interacting with MDA-MB-231 cells following a 24 h incubation period. Cells exposed to media only (no treatment) exhibit an intrinsic blue scattering signal. NS–PS (red) are visible against the blue cell background in samples treated with NS–PS owing to their enhanced light-scattering properties.
NS–PS Produce ROS and Induce DNA Damage upon NIR Activation
To assess in vitro generation of ROS by NS–PS under NIR irradiation, 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) was used as a fluorogenic probe. As shown in Figures 5a and S11, MDA-MB-231 cells incubated with NS–PS or NS–PEG and carboxy-H2DCFDA but not exposed to light did not exhibit green fluorescence indicative of ROS formation. Similarly, cells that were irradiated with 800 nm pulsed laser light, in the absence or presence of NS–PEG, did not show the green fluorescence signal that is the hallmark of ROS production. By contrast, cells incubated with NS–PS and then treated with λex > 500 nm light irradiation using a lightbox with a longpass filter (Figure S9) to activate PDT, but not PTT, showed a slight increase in green fluorescence, signaling modest production of ROS (Figure 5a). This weak PDT effect is likely due to the relatively low power density of the LED lightbox and short irradiation time, which was set based on the viability effect of PDT/PTT. Importantly, cells incubated with NS–PS and then exposed to 800 nm pulsed light to induce both PDT and PTT emitted brighter green fluorescence than all other control groups, indicating efficient ROS generation under these conditions (Figure 5a).
Figure 5.

(a) Image-iT live green analysis of ROS production in MDA-MB-231 cells that were untreated, or subjected to laser only, NS–PS only, NS–PS + λ > 500 nm lightbox (PDT only, direct activation of PS), or NS–PS + 800 nm laser (PDT/PTT, indirect activation of PS via upconverted photoluminescence). Samples exposed to the lightbox or laser were irradiated for 5 min each. Blue indicates nuclei and green indicates ROS. Scale bar = 50 μm. (b) Western blot showing γ-H2AX expression in MDA-MB-231 cells that were untreated or subjected to laser only, NS–PS only, NS–PS + lightbox, or NS–PS + laser with 5 min lightbox/laser exposure.
ROS are known to cause DNA damage.60,61 To investigate the impact of PDT/PTT mediated by NS–PS on the extent of MDA-MB-231 cellular DNA damage, we probed the expression level of γ-H2AX, which is a marker for DNA damage and genomic instability,62 by western blot analysis. As shown in Figure 5b, MDA-MB-231 cells treated with NS–PS in the dark showed negligible γ-H2AX expression, indicating that NS–PS alone do not induce DNA damage. Cells treated with 800 nm pulsed light, but not incubated with NS–PS, exhibited a slight increase in γ-H2AX expression, which is in agreement with previous studies63,64 describing DNA damage effects of different laser types, as a function of wavelengths and power. In comparison, cells treated with NS–PS and then exposed to λ > 500 nm light to induce PDT exhibited an increased level of γ-H2AX, indicating DNA damage because of successful generation of 1O2. Importantly, the highest expression of γ-H2AX was observed in cells treated with NS–PS and pulsed 800 nm irradiation to induce dual PDT/PTT, which is consistent with the ROS levels determined by the SOSG assay (Figure 3a,c) and carboxy-H2DCFDA imaging (Figure 5a) experiments.
PDT/PTT Mediated by NS–PS Reduces TNBC Cell Metabolic Activity and Induces Apoptosis
To benchmark the efficacy of NS–PS as photochemotherapeutic agents of cancer via tandem PDT/PTT, we examined the impact of phototreatment on cellular metabolic activity. MDA-MB-231 TNBC cells were treated with 8 × 109 NS–PS/mL for 24 h, then irradiated with either the 800 nm laser at 1 W/cm2 for 5 min/well to activate PDT and PTT or with λ > 500 nm light using the LED lightbox for 5 or 10 min/well to activate PDT only. Control groups were included that were not incubated with NS–PS but were exposed to the NIR or visible light treatments. After the light treatments, the TNBC cells were incubated for 24 h and cellular metabolic activity was assessed using an Alamar Blue assay. Cells treated with laser irradiation only (Figure S12) or with NS–PS without laser irradiation showed minimal losses in metabolic activity, demonstrating that neither the pulsed laser conditions nor the concentration of NS–PS utilized are inherently harmful to TNBC cells (Figure 6). Cells incubated with NS–PS and irradiated using a lightbox (λ > 500 nm) to induce PDT via direct activation of the biladiene PS on conjugates’ surface experienced ∼5% loss in metabolic activity following 5 min irradiation and ∼10% loss in metabolic activity following 10 min irradiation (Figures 6 and S13). By comparison, cells incubated with NS–PS and irradiated with the 800 nm laser for 5 min to induce dual PDT/PTT experienced a nearly 80% reduction in metabolic activity, which is significantly lower than that of the cells that received PDT alone. These results demonstrate that dual PDT/PTT can be achieved with NS–PS following excitation with a single wavelength NIR laser, and that the therapeutic effect is improved versus PDT alone.
Figure 6.

Viability of MDA-MB-231 cells that were untreated or treated with only the 800 nm pulsed laser, only NS–PS, NS–PS + lightbox (PDT only), or NS–PS + 800 nm pulsed laser (PDT/PTT). The duration of laser or lightbox exposure was 5 min for all samples. *p < 0.001 compared to cells treated with laser only, NS–PS only, NS–PS + lightbox, or no treatment by one-way ANOVA with post hoc Tukey.
After showing that PDT/PTT mediated by NS–PS can potently reduce cellular metabolic activity, we next evaluated the mechanism by which cell death proceeds. Understanding the mechanism of cell death is important as apoptosis versus necrosis can produce very different responses in the tumor microenvironment, which ultimately dictate whether phototreatment is successful.65 When necrosis occurs, cells lose their plasma membrane integrity, which causes intracellular contents including damage-associated molecular patterns (DAMPs) to be released into the extracellular milieu and trigger local inflammatory and immunogenic responses. These responses can be detrimental to treatment success unless the primary therapy is combined with a form of immunotherapy that can take advantage of the ability of DAMPs and released tumor-associated antigens to stimulate an immune response. Conversely, during apoptosis, cell membrane integrity is maintained resulting in an anti-inflammatory mechanism of cell death that may be more appealing when PDT/PTT is applied as a standalone treatment.29,65
To clarify the mechanism of cell death induced by NS–PS under the dosage and laser irradiation conditions employed in this work, we performed AnnexinV-FITC (AnnV-FITC) and PI staining followed by flow cytometry after TNBC cells were treated as described in the Experimental Methods. AnnV binds phosphatidylserine on apoptotic cells, whereas PI is excluded from cells with an intact membrane. Accordingly, cells that die by different pathways appear in distinct regions of flow cytometry density quadrant plots. Representative data are shown in Figure 7a, where live cells are AnnV–/PI– (bottom left quadrant), early apoptotic cells are AnnV+/PI– (bottom right quadrant), late apoptotic cells are AnnV+/PI+ (top right quadrant), and necrotic cells are AnnV–/PI+ (top left quadrant). Analysis of these data across experiments reveals that TNBC cells incubated with NS–PS but not exposed to light are equally viable as compared to cells that received no treatment (Figure 7a,b), supporting the NS–PS biocompatibility studies described above (Figure 6). Cells that underwent laser irradiation alone were ∼15% apoptotic, compared to ∼5% in control cells (Figure 7a,b). This slight increase may be due to the minor DNA damage that was observed following NIR laser irradiation (Figure 5b). Interestingly, this slight apoptotic profile change did not influence the cells’ metabolic activity (Figure 6). By comparison, cells incubated with NS–PS that were subjected to lightbox irradiation (i.e., cells that received PDT) demonstrated ∼15% apoptosis (Figure 7b) and also experienced a modest decrease in metabolic activity (Figure 6). This disparity illustrates the complexity of cell homeostasis under external insults. Further studies will be needed to determine the specific repair/survival mechanisms governing the stress response and the relationship between apoptosis and metabolic activity under different treatment conditions.
Figure 7.

Flow cytometry analysis of MDA-MB-231 cells treated with laser only, NS–PS only, NS–PS + lightbox (PDT), or NS–PS + laser (PDT/PTT) following 5 min of lightbox/laser exposure. Untreated cells were also examined as a control group. (a) Representative flow cytometry density plot of cells following treatment. Bottom left quadrants = live cells, bottom right quadrants = early apoptotic cells, top right quadrants = late apoptotic cells, top left quadrant = necrotic cells. (b) Percentage of apoptotic cells and (c) percentage of necrotic cells following each treatment. ***p < 0.001 by one-way ANOVA with post hoc Tukey.
Notably, cells treated with NS–PS in combination with NIR light (i.e., tandem PDT/PTT) demonstrate a nearly 40% apoptotic population, which is significantly higher than observed in all other treatment groups (Figure 7b). The pronounced increase in apoptotic cell population observed upon NIR activation of NS–PS is another indication that the nanoshell–biladiene conjugates can mediate dual PDT/PTT. Importantly, no increase of necrotic cell population was observed across all treatment groups (Figure 7c). These results are encouraging given that apoptosis is the preferred mechanism of cell death for photoresponsive therapies. Taken together, the results of the metabolic activity and AnnV/PI assays indicate that NS–PS can feasibly mediate dual PDT/PTT upon excitation with NIR light to trigger proapoptotic cell death. Further, these assays indicate that NIR activation of NS–PS is more effective than use of visible light under the conditions evaluated here.
Conclusions and Future Directions
PDT makes use of light, a photoresponsive agent, and molecular oxygen to kill cells in a minimally invasive, spatially controlled manner. Although this type of treatment offers advantages over traditional treatment strategies, the challenge of developing a photosensitizer with optimal photophysical and pharmacological properties has been an obstacle to widespread clinical adoption of PDT. Whereas 650–850 nm light is ideal for photoactivated therapies because it can more deeply penetrate biological tissues than other wavelengths of light, many photosensitizers absorb at shorter visible wavelengths, thwarting their effective use as phototherapeutic agents. Pd[DMBil1]–PEG750 is an example of a photosensitizer that exhibits many favorable attributes such as straightforward synthesis and purification, excellent water solubility and biocompatibility, high 1O2 quantum yield, very low dark toxicity to cells, and an impressively high phototoxicity index. Nonetheless, this biladiene scaffold is impractical as a mediator of PDT in solid tumors because it does not absorb within the therapeutic NIR window (λ = 650–850 nm). In this study, we sought to address this shortcoming by conjugating Pd[DMBil1]-based PSs to photoluminescent NSs that efficiently absorb 800 nm light and emit shorter wavelengths of light that can activate the attached biladienes.
To enable the conjugation, a new biladiene derivative (Pd[DMBil1]–PEG5000–SH) was prepared via installation of a longer thiol-terminated PEG-chain in place of the shorter methoxy-terminated water-solubilizing group employed in Pd[DMBil1]–PEG750. This strategy takes advantage of the modular nature of biladiene syntheses, and ensured that the desirable photophysical and triplet photochemical properties of the Pd[DMBil1] PS were unperturbed (Figure 1, Table 1). Preparation of the NS–PS conjugates was accomplished by simple incubation of NSs with the Pd[DMBil1]–PEG5000–SH derivative.
The NS–PS conjugates can sensitize production of 1O2 upon irradiation with 550 nm light via direct excitation of the biladiene PS, or under 800 nm pulsed laser light exposure where the NSs are activated and emit upconverted photoluminescence that is reabsorbed by the attached PS (Figure 3). The NS–PS conjugates also retain the ability to generate heat (Figure 3), which is characteristic of the gold NSs and endows the conjugates with auxiliary PTT properties. The NS–PS were found to be essentially nontoxic to MDA-MB-231 TNBC cells in the dark at the optical densities tested (Figures 5–7), but caused ROS production, DNA damage, loss of metabolic activity, and, ultimately, apoptotic death in TNBC cells subjected to NIR laser treatment (Figures 5–7). In total, this work demonstrates that conjugating visible light-absorbing Pd[DMBil1] photosensitizers to upconverting nanoparticles is an effective way to use this compound for NIR-activated PDT without making major synthetic modifications to its structure. Additionally, the use of upconverting materials, such as NSs, that also produce heat upon NIR irradiation yields constructs capable of mediating both PDT and PTT in response to excitation with a single wavelength of light. The potential benefits of multimodal therapy, including the possibility of achieving improved therapeutic outcomes with lower treatment doses, may warrant continued investigations into hybrid photosensitizer/nanoshell constructs or alternative materials with analogous properties.
Although this study demonstrates that the NS–PS conjugates may potentially serve as powerful agents for cancer treatment, they will also require further optimization. For example, future studies could vary the length of the thiol-terminated PEG functionality to examine the relationship between the distance of the PS from the NS surface and the efficiency of LRET-based PS activation. Additionally, the NS surfaces could be passivated with methoxy–PEG–thiols that have larger average molecular weights than the PEG substituent linking the Pd[DMBil1] PS to the NSs to help shield the hydrophobic tetrapyrrole chromophores from the surrounding aqueous environment and reduce NS–PS aggregation that may diminish the stability and 1O2 production of the conjugates. Exploring various ratios of methoxy–PEG–thiol to PS–PEG–thiol may also permit studies of the relationship between PS loading and 1O2 production. Ongoing research in our groups is focused on developing new NS–PS conjugates for improved NIR-activated dual PDT and PTT. As this research progresses, we will perform additional studies to understand the intracellular trafficking of NS–PS and to evaluate their performance in the in vivo setting.
In conclusion, we have developed NS/Pd[DMBil1]–PEG5000 conjugates as new tools to enable NIR-activated tandem PDT and PTT. These materials are a substantial addition to the growing portfolio of nanotechnologies developed for photoactivated cancer therapy. One key advantage of this system over prior technologies created for PDT (or tandem PDT/PTT) is that it incorporates designer biladiene-based PSs that are much more efficient and potent than commercially available PSs that exhibit modest 1O2 quantum yields. This allows the NS–PS conjugates to elicit successful PDT/PTT with much lower nanoparticle/photosensitizer doses than prior systems, with less intense irradiation conditions and with reduced irradiation times.66−68 A second distinguishing feature of our system is that it exploits two-photon-induced photoluminescence emitted from the NSs to excite the PS, a mechanism that is distinct from that of traditional upconverting nanoparticles that have been tethered to PSs. Finally, in this study, we provide an in-depth examination of the mechanism of cell death induced by tandem PDT/PTT with NS–PS conjugates. We show that NS–PS conjugates produce ROS in cells upon NIR irradiation to induce DNA damage and trigger apoptosis. This is a desirable mechanism of cell death, as apoptosis can promote a beneficial immune response that may enhance tumor response to therapy. Thus, NS–PS conjugates are not only potent mediators of NIR-activated PDT/PTT, but they also initiate cell death through a preferred biological pathway. Given the many advantages of this system, with further development, it will offer a robust platform for NIR-activated tandem PDT/PTT of cancer.
Experimental Methods
General Materials and Methods
Air-sensitive reactions were carried out on a Schlenk line using flasks fitted with Suba-Seal rubber septa and solvents that were dried via passage through activated alumina.69 Solvents used were of reagent grade or better. Chemicals were purchased from Strem, VWR, Sigma-Aldrich, Fisher, Matrix Scientific, Acros, Decon Laboratories, Inc., Cambridge Isotopes Laboratories, or Alfa Aesar. Column chromatography was carried out with 40–63 μm silica gel purchased from SiliCycle and converted to C2–silica following a published procedure.22,70 Thin-layer chromatography was carried out with precoated glass plates from SiliCycle and visualized with UV light. Pd[DMBil1]–SCH2CO2H was synthesized following published procedures.20−22,45,71 Ultrapure water (MQ water) was obtained from a Milli-Q Century Academic water purification system from Millipore.
Synthesis of Pd[DMBil1]–PEG5000–SH
Pd[DMBil1]–PEG5000–SH (palladium 10,10-dimethyl-5-[para(N-(mercaptoethylpolyethyleneglycol)mercaptoacetamide)tetrafluorophenyl]-15-(pentafluorophenyl)biladiene) was synthesized using the carbo-diimide coupling strategy we have previously used to make other PEGylated derivatives of this photosensitizer.22 A Schlenk flask was charged with Pd[DMBil1]–SCH2CO2H (38 mg, 45 μmol), NHS (11 mg, 96 μmol), and EDC (18 mg, 94 μmol) and placed under vacuum for 1 h. Dry CH2Cl2 (4 mL) was added and the reaction was stirred for 20 h at room temperature under N2. The crude reaction mixture was diluted with additional CH2Cl2, washed four times with deionized water and once with brine, and dried over Na2SO4. The solvent was removed under reduced pressure and thiol–PEG5000–amine (203 mg, 40 μmol) was added to the flask containing the biladiene–NHS ester. The flask was placed under vacuum for 45 min, and then dry CH2Cl2 (5 mL) and triethylamine (52 μL, 373 μmol) were added. The reaction was stirred for 15 h at room temperature under N2, following which, the solvent was removed by rotary evaporation. The resulting crude product was redissolved in CH2Cl2, washed four times with deionized water and once with brine, and then dried over Na2SO4. The product was further purified by column chromatography on C2–silica by eluting in sequence with 1:1 hexane/CH2Cl2, 1:2 hexane/CH2Cl2, pure CH2Cl2 and 3% methanol in CH2Cl2 to remove several impurities. The product was recovered from the C2–silica column using 5–10% methanol in CH2Cl2 as the mobile phase and was obtained as a red, waxy solid (167 mg, 63% yield). 1H NMR (600 MHz, CDCl3, 25 °C) δ/ppm: 7.34 (d, J = 6.6 Hz, 2H), 7.27 (s, 1H), 6.62 (dd, J = 23.8, 4.4 Hz, 2H), 6.58–6.46 (m, 4H), 6.45–6.39 (m, 2H), 3.84 (s, 1H), 3.58 (s, 487H), 3.09 (d, J = 6.3 Hz, 1H), 2.82 (t, J = 6.7 Hz, 1H), 1.74 (s, 6H). 13C NMR (151 MHz, CDCl3, 25 °C) δ/ppm: 193.49, 167.14, 166.73, 166.53, 152.12, 151.98, 147.44, 145.69, 145.26, 144.03, 143.62, 134.85, 134.55, 134.23, 133.96, 131.14, 130.89, 128.99, 128.94, 128.71, 128.17, 118.23, 118.18, 117.63, 116.53, 114.16, 112.20, 76.44, 73.49, 70.53, 70.24, 69.59, 69.51, 69.33, 49.83, 43.91, 43.14, 41.60, 39.74, 39.20, 38.41, 37.50, 30.93, 29.48. 19F NMR (565 MHz, CDCl3, 25 °C) δ/ppm: −132.30 (dd, J = 24.5, 11.7 Hz, 1F), −132.66 (dd, J = 24.3, 11.2 Hz, 2F), −137.86 to −138.35 (m, 5F), −151.89 to −152.43 (m, 1F), −160.69 (dd, J = 29.0, 14.2 Hz, 2F).
Characterization of Pd[DMBil1]–PEG5000–SH
1H NMR, 13C NMR, and 19F NMR spectra (Figures S1–S3) were measured at 25 °C on a Bruker 600 MHz spectrometer with a 5 mm Bruker SMART probe. Proton spectra are referenced to the residual proton resonance of the deuterated solvent (CDCl3 = δ 7.26) and carbon spectra are referenced to the carbon resonances of the deuterated solvent (CDCl3 = δ 77.16).72 Fluorine spectra are referenced to an external trifluoroacetic acid standard (TFA = δ −76.55 in CD3CN).73 Chemical shifts are reported in parts-per-million using standard δ notation.
Analysis of UV–Visible Absorption of Pd[DMBil1]–PEG5000–SH
UV–visible absorbance spectra of Pd[DMBil1]–PEG5000–SH were measured with a StellarNet Silver-NoVA CCD array UV–vis spectrometer. Samples were prepared in quartz cuvettes (6I) of 1.0 cm pathlength manufactured by Starna Cells, Inc. Room-temperature absorption spectra were collected from 8, 12, 16, 20, 24, and 40 μM solutions of Pd[DMBil1]–PEG5000–SH in methanol or ultrapure water.
Measurement of Pd[DMBil1]–PEG5000–PEG5000–SH Emission Spectra
Pd[DMBil1]–PEG5000–SH emission spectra were collected on an automated Photon Technology International (PTI) QuantaMaster 40 fluorometer equipped with an LPS-220B lamp power supply, a 75 W Xenon arc lamp, and a Hamamatsu R2658 photomultiplier tube. A 25 μM solution of Pd[DMBil1]–PEG5000–SH in methanol was prepared under a nitrogen atmosphere in a 1 cm pathlength quartz cuvette with a screw cap closure manufactured by Firefly Scientific. An air-saturated solution of [Ru(bpy)3](PF6)2 in acetonitrile was prepared such that its absorptivity at λ = 460 nm closely matched that of the 25 μM solution of Pd[DMBil1]–PEG5000–SH at 460 nm, and was used as an emission standard (Φref = 0.018 under air).74 The sample and standard were excited at λex = 460 nm, and emission was monitored from 480–900 nm using a step size of 1 nm and an integration time of 0.25 s. The emission spectrum of Pd[DMBil1]–PEG5000–SH was re-measured after exposure to air. All reported spectra are the average of five individual acquisitions. Emission quantum yields were calculated using the following expression
where Φs and Φref are the emission quantum yields of the sample and reference, respectively, Is and Iref are the integrated emission intensities of the sample and reference, respectively, As and Aref are the absorbances of the sample and reference at λex = 460 nm, and ηs and ηref are the refractive indices of the solvents in which the sample and reference were dissolved (methanol and acetonitrile, respectively).
Examination of Singlet Oxygen Sensitization by Pd[DMBil1]–PEG5000–SH and NS–PS Conjugates
The following procedures were used to determine 1O2 production capabilities of Pd[DMBil1]–PEG5000–SH. Data were collected with an automated PTI QuantaMaster 40 fluorometer equipped with an LPS-220 lamp power supply, a 150 W xenon arc lamp, and a PTI 810 Photomultiplier Detection System with an R1527 photomultiplier tube. All 1O2 experiments were conducted in 1.0 cm pathlength quartz cuvettes (6I) manufactured by Starna Cells, Inc. 1O2 quantum yields were calculated using the following equation
where Φs and Φref are the 1O2 quantum yields of the sample and a reference photosensitizer, respectively, ms and mref represent the rates of consumption of a 1O2 trapping probe in the presence of the sample or reference photosensitizer, respectively, and εs and εref are the extinction coefficients of the sample and reference photosensitizer at the wavelength of irradiation. All reported 1O2 quantum yields are averages obtained from a minimum of three trials.
For singlet oxygen studies in methanol, cuvettes were prepared to contain 2 mL of 10.0 μM H2N–PEG5000–SH in methanol, 10.0 μM Pd[DMBil1]–PEG5000–SH in methanol, or 10.0 μM H2N–PEG5000–SH + 10.0 μM [Ru(bpy)3](PF6)2 (used as a reference photosensitizer with Φref = 0.81),51 in methanol. Addition of H2N–PEG5000–SH to the methanol control and [Ru(bpy)3](PF6)2 standard solutions was intended to correct for any environmental effects of the long PEG chain attached to the Pd[DMBil1]–PEG5000–SH photosensitizer. DPBF in methanol (20 μL of a 100 μM solution) was added to each cuvette. 1O2 generation was measured by tracking the attenuation of the DPBF fluorescence signal50 following 0, 5, 10, 15, and 20 s of irradiation with light from a 150 W halogen lamp (Nikon, Inc., model MKII) set at half power and fitted with a 10 nm full width at half-maximum (fwhm) bandpass filter centered at 500 nm (Thor Labs, FB500-10). To obtain emission spectra of DPBF, samples were excited at λex = 405 nm and scanned from λem = 400–600 nm using a step size of 1 nm and an integration time of 0.25 s.
To correct for absorption of the DPBF emission by the photosensitizers, calibration curves plotting the integrated emission intensity as a function of the concentration of DPBF in the presence of Pd[DMBil1]–PEG5000–SH or [Ru(bpy)3](PF6)2 were generated, as has been reported previously.21 Emission spectra used in the calibration curves were collected from 10.0 μM solutions of Pd[DMBil1]–PEG5000–SH or the reference standard, which contained 0, 0.25, 0.50, 0.75, 1.00, 1.25, or 1.50 μM concentrations of DPBF and had not endured exposure to light. Linear regression analyses of the data from each solution enabled conversion of the integrated emission intensity values obtained during the 1O2 sensitization experiments into the corresponding concentrations of unreacted DPBF. Linear regression analyses of plots of the concentration of unreacted DPBF versus irradiation time for Pd[DMBil1]–PEG5000–SH and [Ru(bpy)3](PF6)2 enabled ms and mref, respectively, to be obtained as the slopes of the lines.
Similar methods were employed for singlet oxygen sensitization studies of Pd[DMBil1]–PEG5000–SH in MQ water. Cuvettes were prepared to contain 2 mL of 10.0 μM H2N–PEG5000–SH, 10.0 μM Pd[DMBil1]–PEG5000–SH, or 10.0 μM H2N–PEG5000–SH + 10.0 μM methylene blue (used as a reference photosensitizer with Φref = 0.52).53,54 As indicated above, addition of H2N–PEG5000–SH to the solvent control and methylene blue standard solutions was intended to correct for any environmental effects of the PEG chain attached to the Pd[DMBil1]–PEG5000–SH photosensitizer. A 20 μL aliquot of 250.0 μM SOSG in 5% methanol in MQ water was then added to each cuvette in accordance with instructions from the manufacturer. 1O2 production was monitored by observing the enhancement in SOSG emission intensity52,56 following 0, 30, 60, 90, and 120 s of irradiation with the 150 W halogen lamp (Nikon, Inc., model MKII) used at full power and fitted with a 10 nm fwhm bandpass filter centered at 550 nm (Thor Labs, FB550-10). To obtain emission spectra of SOSG, samples were excited at λex = 480 nm and scanned from λem = 500–650 nm using a step size of 1 nm and an integration time of 0.25 s. Linear regression analyses of plots of the integrated emission intensity of SOSG versus irradiation time for Pd[DMBil1]–PEG5000–SH and methylene blue enabled ms and mref, respectively, to be obtained as the slopes of the lines.
For singlet oxygen studies of the NS–PS conjugates in MQ water, cuvettes were prepared to contain 1.5 mL of MQ water, a solution of NS–PEG with an optical density (OD) of 1, or an OD 1 solution of NS–PS conjugates (procedures for synthesis of NS–PEG and NS–PS are detailed in the following section; OD 1 corresponds to ∼2.7 × 109 NS/mL). A 15 μL aliquot of 250.0 μM SOSG in 5% methanol in MQ water was then added to each cuvette. 1O2 production was monitored by observing the enhancement in SOSG emission intensity, as mentioned above, after 0, 5, 10, 15, and 20 min of irradiation with either the 150 W halogen lamp (Nikon, Inc., model MKII) used at full power and fitted with a 10 nm fwhm bandpass filter centered at 550 nm (Thor Labs, FB550-10) or a laser (Coherent Legend-Elite regenerative amplifier seeded by a Coherent Mantis Ti:sapphire oscillator), which produced a train of 35 fs pulses centered at 800 nm with a repetition rate of 10 kHz and 100 μJ of energy. Emission spectra of SOSG were obtained by exciting the samples at λex = 480 nm and scanning from λem = 500–650 nm using a step size of 2 nm and an integration time of 3 s. Plots of the integrated emission intensity of SOSG as a function of irradiation time for the various samples were compared qualitatively.
NS Synthesis and Functionalization
NSs were synthesized according to published methods.30 Briefly, 3–4 nm diameter colloidal gold NPs formed by the Duff et al. method75 were combined with 120 nm diameter aminated silica NPs (Nanocomposix, San Diego, CA, USA) and reacted for several days to form “seed.” The seed solution was centrifuged (2000g, 20 min, twice) and the supernatant containing unreacted gold colloid was removed. The purified seed, which was diluted in water, was reacted with potassium carbonate containing HAuCl4 (Sigma-Aldrich, St. Louis, MO, USA) and formaldehyde to create NSs, which were then purified by centrifugation (1500g, 5 min, twice). Next, NSs at OD 1.5 (corresponding to 4 × 109 NSs/mL) were combined at an initial ratio of 7 × 105 ligands/NS with 5 μM Pd[DMBil1]–PEG5000–SH to form NS–PS or with 5 μM mPEG5000–SH (Laysan Bio, Arab, AL, USA) to form NS–PEG, which serves as an experimental control. After reacting at room temperature overnight, NS–PS or NS–PEG were purified by centrifugation (1500g, 7 min, thrice) to remove unbound PS or PEG, then stored in ultrapure water at 4 °C before use.
Characterization of NS Conjugates’ Optical and Structural Properties
The extinction of bare NSs or functionalized NSs suspended in water was determined by UV–visible spectrophotometry (Cary60 spectrophotometer, Agilent, Santa Clara, CA, USA). Bare NSs, NS–PEG, or NS–PS (suspended in water to OD 1, corresponding to 2.7 × 109 NSs/mL) were characterized by DLS and zeta potential (Litesizer500, Anton Paar, Graz, Austria), and the reported hydrodynamic diameter (using z-average mean) and zeta potential are the mean from three sample measurements. Bare NSs, NS–PEG, or NS–PS samples for SEM were diluted to 2.7 × 109 s/mL (OD 1) in 200 proof ethanol and dried directly onto a polished carbon stub prior to imaging (S4700, Hitachi, Chiyoda, Tokyo, Japan).
Quantification of Pd[DMBil1]–PEG5000–SH Loading on NS–PS
To quantify the number of Pd[DMBil1]–PEG5000–SH loaded per NS, we first prepared NS–PS by incubating bare NSs (OD 5) with 10 μM Pd[DMBil1]–PEG5000–SH overnight at room temperature. A separate 10 μM Pd[DMBil1]–PEG5000–SH was prepared as a total sample and kept the same way as the NS–PS sample. After incubation, the NS–PS were centrifuged at 15 000g for 10 min to form a pellet and the unbound Pd[DMBil1]–PEG5000–SH was removed with the supernatant. The absorbances of the PS-containing supernatant and the total PS sample were recorded by UV–visible spectrophotometry (Cary60). To calculate the concentration of Pd[DMBil1]–PEG5000–SH in each sample, a standard curve was established based on the OD at 490 nm (OD490) from a serial dilution of Pd[DMBil1]–PEG5000–SH solutions in water up to 50 μM. The concentration of NS-bound Pd[DMBil1]–PEG5000–SH was calculated by subtraction of the concentration of free Pd[DMBil1]–PEG5000–SH in the supernatant from the concentration in the total sample. The concentration of NS particles was calculated based on Beer’s law with OD recorded from UV–visible spectrophotometry. Final loading data were averaged based on data collected from three individual samples.
Characterization of NS Conjugates’ Photothermal Properties
To analyze the ability of NS–PS to produce heat upon NIR irradiation under different conditions, samples of NS–PS or NS–PEG at different concentrations (OD 1, OD 2, or OD 3) were dispersed in (i) water at room temperature or (ii) complete cell culture media, with samples in cell culture media incubated in a 96-well plate in a 37 °C humidified incubator for 24 h prior to testing. After incubation, each sample was irradiated using an 800 nm femtosecond (fs) pulsed laser at 1 W/cm2 and the temperature was recorded using an FLIR E6 thermal camera (Wilsonville, OR, USA). A photothermal cycling study of NS–PS was carried out by exposing NS–PS (at a concentration of OD 3) loaded in a 1.5 mL Eppendorf tube to laser irradiation at 1 W/cm2 for an on (5 min) and off (15 min) cycle four times. The laser (coherent) utilized in the entire study was equipped with a mode-locked Ti/Sapphire oscillator (Coherent Mantis) and a regenerative amplifier (Coherent Legend Elite). The output of the amplifier is a train of 35 fs pulses centered at 800 nm with a repetition rate of 10 kHz.
Cell Culture
MDA-MB-231 TNBC cells were purchased from American Type Culture Collection (ATCC) and cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin (i.e., complete medium). Cells were cultured in T75 cell culture flasks and incubated at 37 °C in a 5% CO2 humidified environment. Cells were passed between flasks or into sample plates by detaching the cells from the flasks with TrypLE Express Enzyme, diluting the cells with complete medium, and counting the cells with a hemocytometer before transferring to a new flask or well plate.
Imaging the Cellular Interactions of NS–PS
To analyze the interactions between MDA-MB-231 TNBC cells and NS–PS by darkfield microscopy, cells were plated at 25 000 cells/well in glass bottom eight-well plates with removable well chambers, and incubated overnight. Then, 8 × 109 NS–PS/mL diluted in complete cell culture medium was added to the cells for 24 h protected from light. The cells were then fixed with 4% formaldehyde for 15 min and rinsed three times with 1× PBS. Well chambers were removed and cells were imaged with a Zeiss AxioObserver Z1 Inverted Fluorescent Microscope (Oberkochen, Germany) using a darkfield condenser (to visualize scattering from NS–PS). Cells that did not receive NS–PS were used as a control for background scattering signal.
Detection of ROS In Vitro
To confirm that NS–PS can elicit PDT in vitro, an Image-iT LIVE Green ROSs Detection Kit (Invitrogen) was used to detect the intracellular generation of ROSs by NS–PS or control treatments following irradiation with light or without exposure to light. MDA-MB-231 cells were seeded in a Lab-Tek 8-well chambered coverglass at a density of 50 000 cells/well and incubated overnight. Then, the cell culture medium was replaced with fresh complete medium containing either 0 or 8 × 109 NS–PEG or NS–PS/mL, and the samples incubated for 24 h protected from light. The samples were then irradiated at 1 W/cm2 with the 800 nm fs-pulsed laser for 5 min. Control samples received either no irradiation or irradiation with λ > 500 nm CW light (which directly activates Pd[DMBil1]–PEG5000–SH on the NSs) via a LED lightbox overlaid with a yellow filter (Figure S9). After 24 h of incubation following each treatment, the cells were stained with fresh medium containing 25 μM carboxy-H2DCFDA (5-(and-6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate), which indicates the presence of ROS, for 30 min at 37 °C, protected from light. During the last 10 min of carboxy-H2DCFDA incubation, 1 μM Hoechst 33342 was added to stain cell nuclei. The cells were then washed with 1× PBS and imaged with a Zeiss AxioObserver Z1 Inverted Fluorescent Microscope equipped with an apotome using the EGFP (ROS) (excitation, 495 nm; emission, 525/50 nm) and DAPI (nuclei) (excitation, 395 nm; emission, 445/50 nm) fluorescence channels.
Western Blot Analysis for H2AX Expression
As PDT and PTT can induce DNA damage, we performed western blotting to assess the expression of H2AX, a DNA damage marker, in MDA-MB-231 cells exposed to no treatment, NS–PS only, 800 nm light only, NS–PS + 800 nm fs pulsed light (which triggers dual PDT and PTT), or NS–PS + λ > 500 nm CW light (which triggers only PDT via direct activation of Pd[DMBil1]–PEG5000–SH on the NSs). After the treatments were performed as described above, the MDA-MB-231 cells were washed 1× with PBS and lysed in RIPA buffer (Amresco) supplemented with Halt Protease Inhibitor Cocktail (Life Technologies) on ice. Protein concentration was determined using a DC Protein Assay (Bio-Rad, Hercules, CA) relative to a BSA standard. A 30 μg aliquot of protein was loaded into each well of 4–12% Bis–Tris gels (Thermo Fisher Scientific, Waltham, MA) and separated at 135 V for 60 min. The protein was then transferred to a 0.45 μm nitrocellulose membrane for 10 min using the Pierce Power System (Thermo Scientific). Membranes were blocked for 60 min in 5% nonfat milk in tris-buffered saline containing 0.1% Tween-20 (TBST). Membranes were probed with rabbit antihuman phospho-Histone H2AX (1:1000) or mouse antihuman β-actin (1:2000) primary antibodies (Cell Signaling Technology, Danvers, MA) at 4 °C overnight, followed by incubation with goat antirabbit (1:12 500) or antimouse (1:25 000) horseradish peroxidase-conjugated secondary antibodies (Kirkegaard & Perry Laboratories, Inc., Gaithersburg, MD) for 1 h at room temperature. Protein bands were detected by chemiluminescence using VisiGlo Select Chemiluminescent Substrate (Amresco, Solon, OH) and imaged on a ChemiDoc-It2 Imager (UVP, Upland, CA). The blot shown represents the average band density across experiments.
Assessment of Cellular Metabolic Activity
MDA-MB-231 cells were seeded into 96-well plates at 50 000 cells/well, treated with media containing 0 or 8 × 109 NS–PS/mL for 24 h, and then subjected to no irradiation, or irradiation for 5 min/well using the 800 nm pulsed laser with a beam diameter of approximately 1 cm (to cover the entire well) and a power density of 1 W/cm2 or using the LED lightbox with a 500 nm longpass filter. The plate was prewarmed and kept at 37 °C in a digital 2 block heater (VWR) until the end of irradiation. After irradiation, the cells were incubated for 24 h, then the media was removed and an Alamar blue viability reagent (Thermo Fisher, Waltham, MA, USA; diluted 1:10 in complete cell culture media) was added per manufacturer recommendations. Sample fluorescence was measured on a Synergy H1M plate reader with excitation and emission wavelengths of 560 and 590 nm, respectively. To analyze the data, background (Alamar blue reagent without cells) was subtracted from the fluorescence reading in each well.
Analysis of the Mechanism of Cell Death Induced by PDT/PTT with NS–PS
MDA-MB-231 cells seeded in 96-well plates were treated with media containing 0 or 8 × 109 NS–PS/mL for 24 h and then subjected to no irradiation, or irradiation for 5 min/well using the 800 nm pulsed laser with a beam diameter approximately 1 cm and a power density of 1 W/cm2 or using the LED lightbox with a 500 nm longpass filter. After treatment, samples were incubated for 24 h, and then an AnnexinV–propidium iodide (PI) stain (Cayman Chemicals, Ann Arbor, MI, USA) was conducted via manufacturer instructions to analyze cellular apoptosis versus necrosis. Briefly, cells were lifted with trypsin, washed once with 1× binding buffer (300g, 5 min), and resuspended in 50 μL of binding buffer containing 1:500 AnnexinV-FITC and 1:2000 PI stains for 10 min protected from light. The samples were then diluted with 150 μL 1× binding buffer and analyzed using an ACEA NovoCyte 2060 flow cytometer with the FITC (excitation, 488 nm; emission, 530/30 nm) and PerCP (excitation, 488 nm; emission, 675/30 nm) channels. Data analysis was performed in NovoExpress software (ACEA Biosciences, San Diego, CA, USA), and positive stained gates were based off of unstained cells. Single stained controls were used for compensation. Data shown are averaged amongst three independent experiments.
Statistical Analysis
All experiments were performed in triplicate, and data represent means ± standard deviations from three independent replicates unless otherwise indicated. Groups with significant differences were identified using one-way ANOVA with a post hoc Tukey test (or Student’s t-test when only two groups were compared), and differences were considered significant at p < 0.05. Statistical tests were performed in Minitab software (Minitab, State College, PA), and flow cytometry data were analyzed using NovoExpress software (ACEA Biosciences, San Diego, CA).
Acknowledgments
The authors thank Prof. Lars Gundlach, Baxter Abraham, Joseph Avenoso, and Han Yan for providing access to and assistance with the near-infrared laser used in this study.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b04150.
1H NMR of Pd[DMBil1] and Pd[DMBil1]–PEG5000–SH; 13C NMR of Pd[DMBil1] and Pd[DMBil1]–PEG5000–SH; 19F NMR of Pd[DMBil1] and Pd[DMBil1]–PEG5000–SH; aqueous absorption and emission of Pd[DMBil1]–PEG5000–SH; Beer’s Law plot of Pd[DMBil1]–PEG5000–SH; SEM images of bare NSs and NS–PEG; storage stability of NS–PS in water; change in SOSG emission intensity over time for MQ water, NS–PEG, Pd[DMBil1]–PEG5000–SH, NS–PS conjugates or NS–PEG + Pd[DMBil1]–PEG750 with 550 or 800 nm light; absorption spectrum of LED lightbox with yellow filter; photothermal properties of NS–PS and NS–PEG in water; image-iT live green analysis of ROS production in MDA-MB-231 cells that were untreated, treated with NS–PEG only, or treated with NS–PEG + 800 nm light (PTT); viability of MDA-MB-231 cells following laser-only treatment; and viability of MDA-MB-231 cells following NS–PS + light treatment (PDF)
Author Contributions
J.W. and A.M.P. contributed equally. The paper was written through contributions of all the authors. All the authors have given approval to the final version of the paper.
A Strategic Initiatives Grant from the Univ. of Delaware (UD) Research Foundation supported portions of this work. Microscopy access was supported by grants from the NIH-NIGMS (P20-GM103446), the NSF (IIA-1301765), and the State of Delaware. SEM was acquired with the Delaware INBRE Grant P20GM103446. Work in JR’s lab was supported through NSF CAREER award CHE1352120 and NIH P20GM104316. NMR and other data were acquired at UD using instrumentation obtained with assistance from the NSF and NIH (NSF-MIR 0421224, NSF-MIR 1048367, NSF-CRIF MU CHE-0840401 and CHE-0541775, NIH P20RR017716, and P20GM10431). Work in ED’s lab was supported by a grant from the W.M. Keck Foundation.
The authors declare no competing financial interest.
Supplementary Material
References
- Hatz S.; Poulsen L.; Ogilby P. R. Time-resolved singlet oxygen phosphorescence measurements from photosensitized experiments in single cells: Effects of oxygen diffusion and oxygen concentration. Photochem. Photobiol. 2008, 84, 1284–1290. 10.1111/j.1751-1097.2008.00359.x. [DOI] [PubMed] [Google Scholar]
- Henderson B. W.; Dougherty T. J. How does photodynamic therapy work?. Photochem. Photobiol. 1992, 55, 145–157. 10.1111/j.1751-1097.1992.tb04222.x. [DOI] [PubMed] [Google Scholar]
- Dougherty T. J.; Gomer C. J.; Henderson B. W.; Jori G.; Kessel D.; Korbelik M.; et al. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. 10.1093/jnci/90.12.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli J. P.; Spring B. Q.; Rizvi I.; Evans C. L.; Samkoe K. S.; Verma S.; et al. Imaging and photodynamic therapy: Mechanisms, monitoring, and optimization. Chem. Rev. 2010, 110, 2795–2838. 10.1021/cr900300p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin M. R.; Newman E. L. On the mechanism of the tumour-localising effect in photodynamic therapy. J. Photochem. Photobiol. B Biol. 1994, 23, 3–8. 10.1016/S1011-1344(94)80018-9. [DOI] [PubMed] [Google Scholar]
- Plaetzer K.; Krammer B.; Berlanda J.; Berr F.; Kiesslich T. Photophysics and photochemistry of photodynamic therapy: Fundamental aspects. Lasers Med. Sci. 2009, 24, 259–268. 10.1007/s10103-008-0539-1. [DOI] [PubMed] [Google Scholar]
- Vandongen G.; Visser G.; Vrouenraets M. Photosensitizer-antibody conjugates for detection and therapy of cancer. Adv. Drug Delivery Rev. 2004, 56, 31–52. 10.1016/j.addr.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Reddi E. Role of delivery vehicles for photosensitizers in the photodynamic therapy of tumours. J. Photochem. Photobiol. B Biol. 1997, 37, 189–195. 10.1016/s1011-1344(96)07404-0. [DOI] [PubMed] [Google Scholar]
- Wang H.; Xu Y.; Shi J.; Gao X.; Geng L. Photodynamic therapy in the treatment of basal cell carcinoma: A systematic review and meta-analysis. Photodermatol. Photoimmunol. Photomed. 2015, 31, 44–53. 10.1111/phpp.12148. [DOI] [PubMed] [Google Scholar]
- Triesscheijn M.; Baas P.; Schellens J. H. M.; Stewart F. A. Photodynamic therapy in oncology. The Oncologist 2006, 11, 1034–1044. 10.1634/theoncologist.11-9-1034. [DOI] [PubMed] [Google Scholar]
- Nowis D.; Stokłosa T.; Legat M.; Issat T.; Jakóbisiak M.; Gołąb J. The influence of photodynamic therapy on the immune response. Photodiagn. Photodyn. Ther. 2005, 2, 283–298. 10.1016/s1572-1000(05)00098-0. [DOI] [PubMed] [Google Scholar]
- Castano A. P.; Mroz P.; Hamblin M. R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison R. R.; Sibata C. H. Oncologic photodynamic therapy photosensitizers: A clinical review. Photodiagn. Photodyn. Ther. 2010, 7, 61–75. 10.1016/j.pdpdt.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Bonnett R. Photosensitizers of the porphyrin and phthalocyanine series for photodynamic therapy. Chem. Soc. Rev. 1995, 24, 19–33. 10.1039/cs9952400019. [DOI] [Google Scholar]
- Sternberg E. D.; Dolphin D.; Brückner C. Porphyrin-based photosensitizers for use in photodynamic therapy. Tetrahedron 1998, 54, 4151–4202. 10.1016/s0040-4020(98)00015-5. [DOI] [Google Scholar]
- Nyman E. S.; Hynninen P. H. Research advances in the use of tetrapyrrolic photosensitizers for photodynamic therapy. J. Photochem. Photobiol. B Biol. 2004, 73, 1–28. 10.1016/j.jphotobiol.2003.10.002. [DOI] [PubMed] [Google Scholar]
- Ormond A.; Freeman H. Dye sensitizers for photodynamic therapy. Materials 2013, 6, 817. 10.3390/ma6030817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamse H.; Hamblin M. R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. 10.1042/bj20150942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor A. E.; Gallagher W. M.; Byrne A. T. Porphyrin and nonporphyrin photosensitizers in oncology: Preclinical and clinical advances in photodynamic therapy. Photochem. Photobiol. 2009, 85, 1053–1074. 10.1111/j.1751-1097.2009.00585.x. [DOI] [PubMed] [Google Scholar]
- Pistner A. J.; Pupillo R. C.; Yap G. P. A.; Lutterman D. A.; Ma Y.-Z.; Rosenthal J. Electrochemical, spectroscopic, and 1o2 sensitization characteristics of 10,10-dimethylbiladiene complexes of zinc and copper. J. Phys. Chem. A 2014, 118, 10639–10648. 10.1021/jp506412r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocny A. M.; Pistner A. J.; Yap G. P. A.; Rosenthal J. Electrochemical, Spectroscopic, and 1O2 Sensitization Characteristics of Synthetically Accessible Linear Tetrapyrrole Complexes of Palladium and Platinum. Inorg. Chem. 2017, 56, 12703–12711. 10.1021/acs.inorgchem.7b00796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potocny A. M.; Riley R. S.; O’Sullivan R. K.; Day E. S.; Rosenthal J. Photochemotherapeutic properties of a linear tetrapyrrole palladium(ii) complex displaying an exceptionally high phototoxicity index. Inorg. Chem. 2018, 57, 10608–10615. 10.1021/acs.inorgchem.8b01225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Chung K.; Lee S.; Kim D. H.; Lee H. Near-infrared light-responsive nanomaterials for cancer theranostics. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2016, 8, 23–45. 10.1002/wnan.1347. [DOI] [PubMed] [Google Scholar]
- Qiu H.; Tan M.; Ohulchanskyy T.; Lovell J.; Chen G. Recent progress in upconversion photodynamic therapy. Nanomaterials 2018, 8, 344. 10.3390/nano8050344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y.; Shuhendler A. J.; Ye D.; Xu J.-J.; Chen H.-Y. Two-photon excitation nanoparticles for photodynamic therapy. Chem. Soc. Rev. 2016, 45, 6725–6741. 10.1039/c6cs00442c. [DOI] [PubMed] [Google Scholar]
- Hamblin M. R. Upconversion in photodynamic therapy: Plumbing the depths. Dalton Trans. 2018, 47, 8571–8580. 10.1039/c8dt00087e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Meng X.; Bu W. Upconversion-based photodynamic cancer therapy. Coord. Chem. Rev. 2019, 379, 82–98. 10.1016/j.ccr.2017.09.006. [DOI] [Google Scholar]
- Rastinehad A. R.; Anastos H.; Wajswol E.; Winoker J. S.; Sfakianos J. P.; Doppalapudi S. K.; et al. Gold nanoshell-localized photothermal ablation of prostate tumors in a clinical pilot device study. Proc. Natl. Acad. Sci. U.S.A. 2019, 116, 18590–18596. 10.1073/pnas.1906929116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley R.; O’Sullivan R.; Potocny A.; Rosenthal J.; Day E. Evaluating nanoshells and a potent biladiene photosensitizer for dual photothermal and photodynamic therapy of triple negative breast cancer cells. Nanomaterials 2018, 8, 658. 10.3390/nano8090658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldenburg S. J.; Averitt R. D.; Westcott S. L.; Halas N. J. Nanoengineering of optical resonances. Chem. Phys. Lett. 1998, 288, 243–247. 10.1016/s0009-2614(98)00277-2. [DOI] [Google Scholar]
- Bickford L.; Sun J.; Fu K.; Lewinski N.; Nammalvar V.; Chang J.; et al. Enhanced multi-spectral imaging of live breast cancer cells using immunotargeted gold nanoshells and two-photon excitation microscopy. Nanotechnology 2008, 19, 315102. 10.1088/0957-4484/19/31/315102. [DOI] [PubMed] [Google Scholar]
- Park J.; Estrada A.; Sharp K.; Sang K.; Schwartz J. A.; Smith D. K.; et al. Two-photon-induced photoluminescence imaging of tumors using near-infrared excited gold nanoshells. Opt. Express 2008, 16, 1590–1599. 10.1364/oe.16.001590. [DOI] [PubMed] [Google Scholar]
- Riley R. S.; Day E. S. Gold nanoparticle-mediated photothermal therapy: applications and opportunities for multimodal cancer treatment. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2017, 9, e1449 10.1002/wnan.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal S.; Clare S. E.; Halas N. J. Nanoshell-enabled photothermal cancer therapy: Impending clinical impact. Acc. Chem. Res. 2008, 41, 1842–1851. 10.1021/ar800150g. [DOI] [PubMed] [Google Scholar]
- Gobin A. M.; Lee M. H.; Halas N. J.; James W. D.; Drezek R. A.; West J. L. Near-infrared resonant nanoshells for combined optical imaging and photothermal cancer therapy. Nano Lett. 2007, 7, 1929–1934. 10.1021/nl070610y. [DOI] [PubMed] [Google Scholar]
- Abadeer N. S.; Murphy C. J. Recent progress in cancer thermal therapy using gold nanoparticles. J. Phys. Chem. C 2016, 120, 4691–4716. 10.1021/acs.jpcc.5b11232. [DOI] [Google Scholar]
- Wei A.; Thomas M.; Mehtala J.; Wang J.. Biomaterials for Cancer Therapeutics; Woodhead Publishing, 2013; pp 349–389e. [Google Scholar]
- Jin C. S.; Lovell J. F.; Chen J.; Zheng G. Ablation of hypoxic tumors with dose-equivalent photothermal, but not photodynamic, therapy using a nanostructured porphyrin assembly. ACS Nano 2013, 7, 2541–2550. 10.1021/nn3058642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q.; Zheng X.; Bu W.; Ge W.; Zhang S.; Chen F.; et al. A core/satellite multifunctional nanotheranostic for in vivo imaging and tumor eradication by radiation/photothermal synergistic therapy. J. Am. Chem. Soc. 2013, 135, 13041–13048. 10.1021/ja404985w. [DOI] [PubMed] [Google Scholar]
- Fan W.; Huang P.; Chen X. Overcoming the achilles’ heel of photodynamic therapy. Chem. Soc. Rev. 2016, 45, 6488–6519. 10.1039/c6cs00616g. [DOI] [PubMed] [Google Scholar]
- García Calavia P.; Bruce G.; Pérez-García L.; Russell D. A. Photosensitiser-gold nanoparticle conjugates for photodynamic therapy of cancer. Photochem. Photobiol. Sci. 2018, 17, 1534–1552. 10.1039/c8pp00271a. [DOI] [PubMed] [Google Scholar]
- Kim H.; Lee D. Near-infrared-responsive cancer photothermal and photodynamic therapy using gold nanoparticles. Polymers 2018, 10, 961. 10.3390/polym10090961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistner A. J.; Lutterman D. A.; Ghidiu M. J.; Walker E.; Yap G. P. A.; Rosenthal J. Factors controlling the spectroscopic properties and supramolecular chemistry of an electron deficient 5,5-dimethylphlorin architecture. J. Phys. Chem. C 2014, 118, 14124–14132. 10.1021/jp5016824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistner A. J.; Lutterman D. A.; Ghidiu M. J.; Ma Y.-Z.; Rosenthal J. Synthesis, electrochemistry, and photophysics of a family of phlorin macrocycles that display cooperative fluoride binding. J. Am. Chem. Soc. 2013, 135, 6601–6607. 10.1021/ja401391z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistner A. J.; Yap G. P. A.; Rosenthal J. A tetrapyrrole macrocycle displaying a multielectron redox chemistry and tunable absorbance profile. J. Phys. Chem. C 2012, 116, 16918–16924. 10.1021/jp3059382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal J.; Schuster D. I. The anomalous reactivity of fluorobenzene in electrophilic aromatic substitution and related phenomena. J. Chem. Educ. 2003, 80, 679. 10.1021/ed080p679. [DOI] [Google Scholar]
- Nguyen H. T.; Shen H. The effect of PEGylation on the stimulation of IL-1β by gold (Au) nanoshell/silica core nanoparticles. J. Mater. Chem. B 2016, 4, 1650–1659. 10.1039/c5tb01553g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q.; Ren Y.; Qi H.; Ruan L. Influence of peg coating on optical and thermal response of gold nanoshperes and nanorods. J. Quant. Spectrosc. Radiat. Transfer 2018, 212, 1–9. 10.1016/j.jqsrt.2018.03.012. [DOI] [Google Scholar]
- Kolate A.; Baradia D.; Patil S.; Vhora I.; Kore G.; Misra A. PEG—A versatile conjugating ligand for drugs and drug delivery systems. J. Controlled Release 2014, 192, 67–81. 10.1016/j.jconrel.2014.06.046. [DOI] [PubMed] [Google Scholar]
- Young R. H.; Wehrly K.; Martin R. L. Solvent effects in dye-sensitized photooxidation reactions. J. Am. Chem. Soc. 1971, 93, 5774–5779. 10.1021/ja00751a031. [DOI] [Google Scholar]
- Bhattacharyya K.; Das P. K. Quantitative aspects of all-trans-retinol singlet and triplet quenching by oxygen. Chem. Phys. Lett. 1985, 116, 326–332. 10.1016/0009-2614(85)80178-0. [DOI] [Google Scholar]
- Ragàs X.; Jiménez-Banzo A.; Sánchez-García D.; Batllori X.; Nonell S. Singlet oxygen photosensitisation by the fluorescent probe Singlet Oxygen Sensor Green. Chem. Commun. 2009, 2920–2922. 10.1039/b822776d. [DOI] [PubMed] [Google Scholar]
- Nemoto M.; Kokubun H.; Koizumi M. Determination of s*-t transition probabilities of some xanthene and thiazine dyes on the basis of t-energy transfer. Ii. Results in the aqueous solution. Bull. Chem. Soc. Jpn. 1969, 42, 2464–2470. 10.1246/bcsj.42.2464. [DOI] [Google Scholar]
- Usui Y.; Kamogawa K. A standard system to determine the quantum yield of singlet oxygen formation in aqueous solution. Photochem. Photobiol. 1974, 19, 245–247. 10.1111/j.1751-1097.1974.tb06506.x. [DOI] [Google Scholar]
- Vankayala R.; Sagadevan A.; Vijayaraghavan P.; Kuo C.-L.; Hwang K. C. Metal nanoparticles sensitize the formation of singlet oxygen. Angew. Chem., Int. Ed. 2011, 50, 10640–10644. 10.1002/anie.201105236. [DOI] [PubMed] [Google Scholar]
- Gollmer A.; Arnbjerg J.; Blaikie F. H.; Pedersen B. W.; Breitenbach T.; Daasbjerg K.; et al. Singlet Oxygen Sensor Green: Photochemical Behavior in Solution and in a Mammalian Cell. Photochem. Photobiol. 2011, 87, 671–679. 10.1111/j.1751-1097.2011.00900.x. [DOI] [PubMed] [Google Scholar]
- Day E. S.; Thompson P. A.; Zhang L.; Lewinski N. A.; Ahmed N.; Drezek R. A.; et al. Nanoshell-mediated photothermal therapy improves survival in a murine glioma model. J. Neuro Oncol. 2011, 104, 55–63. 10.1007/s11060-010-0470-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day E. S.; Zhang L.; Thompson P. A.; Zawaski J. A.; Kaffes C. C.; Gaber M. W.; et al. Vascular-targeted photothermal therapy of an orthotopic murine glioma model. Nanomedicine 2012, 7, 1133–1148. 10.2217/nnm.11.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day E.; Fay B.; Melamed J. Nanoshell-mediated photothermal therapy can enhance chemotherapy in inflammatory breast cancer cells. Int. J. Nanomed. 2015, 10, 6931–6941. 10.2147/ijn.s93031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J.; Wagner J. R. DNA base damage by reactive oxygen species, oxidizing agents, and uv radiation. Cold Spring Harbor Perspect. Biol. 2013, 5, a012559. 10.1101/cshperspect.a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke M. S.; Evans M. D.; Dizdaroglu M.; Lunec J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- Nagelkerke A.; Span P. N.. Advances in Experimental Medicine and Biology; Springer, 2016; pp 1–10. [DOI] [PubMed] [Google Scholar]
- Kong X.; Mohanty S. K.; Stephens J.; Heale J. T.; Gomez-Godinez V.; Shi L. Z.; et al. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res. 2009, 37, e68 10.1093/nar/gkp221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrando-May E.; Tomas M.; Blumhardt P.; Stöckl M.; Fuchs M.; Leitenstorfer A. Highlighting the DNA damage response with ultrashort laser pulses in the near infrared and kinetic modeling. Front. Genet. 2013, 4, 135. 10.3389/fgene.2013.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed J. R.; Edelstein R. S.; Day E. S. Elucidating the fundamental mechanisms of cell death triggered by photothermal therapy. ACS Nano 2015, 9, 6–11. 10.1021/acsnano.5b00021. [DOI] [PubMed] [Google Scholar]
- Seo S.-H.; Kim B.-M.; Joe A.; Han H.-W.; Chen X.; Cheng Z.; et al. Nir-light-induced surface-enhanced raman scattering for detection and photothermal/photodynamic therapy of cancer cells using methylene blue-embedded gold nanorod@sio2 nanocomposites. Biomaterials 2014, 35, 3309–3318. 10.1016/j.biomaterials.2013.12.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L.; Fei J.; Zhao J.; Li H.; Cui Y.; Li J. Hypocrellin-loaded gold nanocages with high two-photon efficiency for photothermal/photodynamic cancer therapy in vitro. ACS Nano 2012, 6, 8030–8040. 10.1021/nn302634m. [DOI] [PubMed] [Google Scholar]
- Wang S.; Huang P.; Nie L.; Xing R.; Liu D.; Wang Z.; et al. Single continuous wave laser induced photodynamic/plasmonic photothermal therapy using photosensitizer-functionalized gold nanostars. Adv. Mater. 2013, 25, 3055–3061. 10.1002/adma.201204623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangborn A. B.; Giardello M. A.; Grubbs R. H.; Rosen R. K.; Timmers F. J. Safe and convenient procedure for solvent purification. Organometallics 1996, 15, 1518–1520. 10.1021/om9503712. [DOI] [Google Scholar]
- Drolen C.; Conklin E.; Hetterich S. J.; Krishnamurthy A.; Andrade G. A.; Dimeglio J. L.; et al. Ph-driven mechanistic switching from electron transfer to energy transfer between [ru(bpy)3]2+ and ferrocene derivatives. J. Am. Chem. Soc. 2018, 140, 10169–10178. 10.1021/jacs.8b03933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien A. Y.; McGann J. P.; Geier G. R. Dipyrromethane + dipyrromethanedicarbinol routes to an electron deficient meso-substituted phlorin with enhanced stability. J. Org. Chem. 2007, 72, 4084–4092. 10.1021/jo070216k. [DOI] [PubMed] [Google Scholar]
- Gottlieb H. E.; Kotlyar V.; Nudelman A. Nmr chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- Filipovich G.; Tiers G. V. D. Fluorine N.s.r. Spectroscopy. I. Reliable Shielding Values, ø, by Use of CCl3F as Solvent and Internal Reference. J. Phys. Chem. 1959, 63, 761–763. 10.1021/j150575a040. [DOI] [Google Scholar]
- Brouwer A. M. Standards for photoluminescence quantum yield measurements in solution (iupac technical report). Pure Appl. Chem. 2011, 83, 2213. 10.1351/pac-rep-10-09-31. [DOI] [Google Scholar]
- Duff D. G.; Baiker A.; Edwards P. P. A new hydrosol of gold clusters. 1. Formation and particle size variation. Langmuir 1993, 9, 2301–2309. 10.1021/la00033a010. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.