

Abstract
Cancer is a leading cause of death worldwide. Cytochrome P450s (P450s) play an important role in the metabolism of endogenous as well as exogenous substances, especially drugs. Moreover, many P450s can serve as targets for disease therapy. Increasing reports of epidemiological, diagnostic, and clinical research indicate that P450s are enzymes that play a major part in the formation of cancer, prevention, and metastasis. The purposes of this review are to shed light on the current state of knowledge about the cancer molecular mechanism involving P450s and to summarize the link between the cancer effects and the participation of P450s.
1. Introduction
Functional genomics, transcriptomics, and proteomics have improved the speed of discovery in scientific research by adding a genome-wide perspective. In the majority of these studies, analyses of individual gene groupings and entire genomes have demonstrated that the extension of multigene families through gene duplication is a critical and repeating phenomenon in the advancement of new gene capacity and increasingly complex living things. As early as the 1930s, researchers hypothesized that repetitive duplicates of existing genes would be under decreased specific pressure and could change and ultimately develop new functions [1, 2]. Later, researchers would contend that gene and genome duplications were an essential, although not the only, mechanism by which new gene capacity could emerge. Because of repeated duplications throughout history, multigene super families, such as the serine proteases and the protein kinases, have come to represent an important portion of protein coding groupings in the genomes of complex life forms [3, 4]. The cytochrome P450s (P450s) comprise various hemoproteins and are one of the largest and most functionally versatile superfamilies. The functional range of P450 activity is remarkable from microscopic organisms to humans [5].
The history of “P450 investigation” began in the early 1950s, and originally, it was thought that “P450” was a solitary cytochrome that was present only in the liver and that its role was to process drugs and other synthetic exogenous substances. These protein interactions have been studied, and it was suggested that they were of clinical importance in medicine and treatment [6, 7]. With the explosion of molecular science in the 1980s, P450 genes were revealed to exist in practically all animals, from prokaryotes to rodents and humans, and the amino acid sequences prompted the main proposal of a transformative disparity-dependent gene classification system. This hypothesis proposed that all P450 genes arose today from a lonely precursor, most likely more than three billion years ago. Six vertebrates, namely, rodents, mice, humans, rabbits, dairy cattle and chickens, and yeast and Pseudomonas putida acquired the 30 genes originally announced in 1987. A quarter of a century later, the group had expanded to an Internet nomenclature that aggregates 18,687 protein-coding genes called P450s with putative tasks beginning in 2012 [5, 8]. P450s are helpfully organized into families and subfamilies in view of the percent amino acid similarity. Proteins sharing more than or approximately 40% identity are relegated to a specific family assigned by an Arabic numeral, while those sharing more than or approximately 55% identity make up a specific subfamily assigned by a letter. For instance, both sterol 27-hydroxylase and 25-hydroxy-D 1α-hydroxylase are assigned to the CYP27 family since they share more than 40% sequence identity [8, 9]. The human genome thus contains 18 P450 families, divided into 41 protein-coding subfamilies encoding 57 genes [10].
A growing amount of studies on P450s showed throughout the 1970s and 1980s that they were present in many species with numerous apparently unrelated life processes, including in crops; therefore, P450s were suggested to be significant upstream molecules in the synthesis and degradation of nearly all nonprotein ligands binding to receptors or activating second-messenger pathways governing growth, homeostasis, apoptosis, and neuroendocrine and differentiation functions [11]. As a rule, therapeutically relevant P450s affect compounds that are similar to essential endogenous substrates and can affect their functions. Numerous human P450 drugs are used to treat human maladies. Others are important for amalgamation of endogenous substances fundamental for human physiology. In a few occurrences, changes in explicit P450s can influence various processes and lead to serious human diseases [12]. Cancer is a complex disease mediated by many molecular and cellular processes. Cancer constitutes one of the world's major causes of death [13]. A global metabolic network expressed in many species, including phase I xenobiotic enzymes, CYPs, and phase II xenobiotic-conjugating enzymes, also uncovered a metabolic pathway that was discovered to bioactivate chemicals and cause cancer. Many researchers have found that P450s (1A1, 1A2, 1B1, 2A6, 2A13, 2E1, and 3A4) play a role in the activation of different carcinogenic compounds in the environment, including tobacco-related nitrosamines and polycyclic aromatic hydrocarbons (PAHs) [14–16]. Electrophilic intermediates from various tobacco-related nitrosamines and PAHs can form covalent bonds on DNA nucleotides, particularly protooncogenes or tumor suppressors, with nucleophilic areas. With the lack of precise DNA repair of adducts, these chemical changes can lead to changes in the encoded genes, which in turn can start the shift from an ordinary cell to a cancer cell [17]. P450s are the most important enzymes that catalyze reactions related to antineoplastic agents. The use of cytotoxic antineoplastic agents in chemotherapy remains a key part of the management of malignant tumors. The CYP2A, CYP2B, CYP2C, CYP2D, and CYP3A subfamilies are mainly responsible for the metabolism of anticancer drugs. One of the most important problems in the treatment of intracranial tumors is chemoresistance [18, 19]. Increased concentrations of P450s may result in intracellular drug inactivation. It has also been suggested that local expression of P450s in tumors is essential for cancer management because P450s expressed in tumors may be involved in chemotherapeutic drug activation and/or inactivation [20]. P450s may be present in tumor cells as part of a pleiotropic reaction to tumor growth. Moreover, they could partially weaken the production and migration of cells. For instance, CYP2W1, CYP1B1, CYP2C9, CYP2C8, CYP2J2, and CYP4A have been linked to tumor-specific expression. These findings have led to the identification of the part played by P450s in cancer growth and tumor formation. For that reason, P450s have a crucial role in cancer formation, chemoprevention, metastasis, and chemotherapy (Figure 1). The aim of this review was to summarize the detailed impact and involvement of P450s in human cancer.
Figure 1.
The potential role of P450 family proteins in cancer.
2. Role of P450s in Cancer Formation
As people reach their 6th decade, they face an exponentially expanded risk of developing cancer. Approximately 5% of human malignant growths are caused by infections, 5% by radiation, and 90% by chemicals. Of these, an expected 30% are caused by the utilization of tobacco and the rest by synthetic substances related to diet, lifestyle, and the environment [21]. The significance of these substances in the etiology of malignant growth is reflected by the finding that up to 8% of cases of every single human disease are related to synthetic factors [22]. Synthetic cancer-causing agents, or their metabolites, are exceptionally receptive electrophiles, which have electron-insufficient molecules that can respond to nucleophilic, electron-rich regions in the cell [23]. Deoxyribonucleic acid (DNA), specifically, comprises a variety of nucleophilic foci at which these DNA-damaging agents can form DNA adducts through at least one covalent bond [24, 25]. CYP-mediated reactions can be used to examine the potential of chemicals to be activated to form reactive species, leading to the formation of covalent DNA adducts [26–28]. In the large group of enzymes involved in the metabolism of carcinogens, P450s are the most important enzymes in the metabolism of PAHs [29–33].
2.1. Xenobiotic Metabolism
Procarcinogens are extremely hydrophobic compounds found in “xenobiotics.” Therefore, these procarcinogens are substrates for some P450 family 1 isoforms (CYP1A1, CYP1A2, and CYP1B1) [16, 34, 35]. The substrate-inducible CYP1A1 and CYP1A2 are responsible for xenobiotic metabolism, such as polycyclic aromatic hydrocarbon metabolism [36–40]. In this group, charred food and cigarette smoke catalyze N-oxidation of carcinogenic aromatic and heterocyclic amines [41–43]. The enzymes are selective for planar molecules, which are carcinogens, as substrates. The substrates are then oxygenated in conformationally delayed positions that result in the formation of highly reactive epoxides. These peptides that are not easily detoxified by glutathione transferase, epoxide hydrolase, and other detoxification enzymes are cytosolic (steroid-like) receptors. The AhR receptor [44] regulates peptides that bond with carcinogens and other planar molecules, resulting in the increased production of P450s, AhR protein, and other enzymes through genomic depression. This change also activates the protein kinase C cascade [45]. While carcinogenic chemicals are activated metabolically by P450 family 1 members, some small molecules, such as nitrosamines, are activated by CYP2E1. The majority of xenobiotic procarcinogens are characterized as highly hydrophobic. In this respect, these procarcinogens act as substrates for the enzymes CYP1A1, CYP1A2, and CYP1B1 [46]. In addition, the CYP1A1 and CYP1B1 enzymes metabolize PAHs that are exceptionally hydrophobic as well as polyhalogenated aromatic hydrocarbons (PHAHs) [47] (Table 1). For effective metabolism, at least two nearby unreplaced positions in the aromatic ring of PHAHs are essential. Those PHAHs that do not have this feature are metabolized very slowly, resulting in a chemical half-life varying from a few weeks to months and even years at times. The coplanar PHAHs and PAHs that degrade slowly are effective inducers of CYP1 member enzymes due to their strong attraction to the AhR [48]. AhR regulates the three CYP1 member genes, which in turn are activated due to the binding of coplanar PHAHs and PAHs. AhR can react with estrogen receptor-α [49], nuclear factor κB (NF-κB) [50], and retinoblastoma protein 1 (RB1) [51]. This in turn leads to gene transcription that participates in growth, apoptosis, and the cell cycle. AhR can therefore be considered a xenobiotic-metabolizing enzyme (XME) receptor that promotes CYP1 member enzymes that metabolize procarcinogens. Moreover, AhR may also promote environmental carcinogenesis by changing cell-cycle functions, including apoptosis, without relying on CYP-mediated ROMs [34]. The CYP1 member enzymes metabolize an endogenous ligand for the identified AhR [52].
Table 1.
P450s involved in the bioactivation of chemical carcinogens.
| Types | Carcinogens | Compound | Ref |
|---|---|---|---|
| CYP1A1 CYP1A2 CYP1B1 |
PAH, arylamine, heterocyclic amine, nitroarene, and estrogen | Benzo[a]pyrene (B[a]P); 7,12-dimethylbenz[a]anthracene (7,12-DMBA); benz[a]anthracene (B[a]A); benzo[c]phenanthrene; 5-methylchrysene; dibenzo[a,l]pyrene (DB[a,l]P); 3-methylcholanthrene (3-MC); fluoranthene; 2-Acetylaminofluorene; 2-amino-6-methyldipyrido[1,2-a: 3′,2′-d]imidazole (Glu-P-1); 3-amino-1,4-dimethyl-5H-pyrido[4,3-b]indole (Trp-P-1); 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP); 17β-estradiol; estrone; 4-hydroxyestradiol; 1-Nitropyrene; 2-nitropyrene; 6-nitrochrysene; 2-aminofluorene; 2-aminoanthracene; 6-aminochrysene; 2-Amino-3-methylimidazo[4,5-f]quinoline (IQ); 2-amino-3,5-dimethylimidazo[4,5-f]quinoline (MeIQ) | [131–136] |
| CYP2A6 | Mycotoxin, alkenylbenzene: occurs in a variety of foods including essential oils of tarragon, sweet basil, sweet fennel, tobacco-specific nitrosamine, and nitrosamine | 1, 2-Dibromoethane (ethylene dibromide); 1,3-butadiene, 2,6-dichlorobenzonitrile (dichlobenil); 3-(N-nitrosomethylamino) propiona aldehyde; benzhydrol; butadiene monoxide (1,2-epoxy-3-butene, methoxsalen); (8-methoxypsoralen, xanthotoxin), N-nitrosomethylbutylamine; N-nitrosopiperidine; N-nitrosopyrrolidine; p-benzoylphenol (4-hydroxybenzophenone) | [134, 137–139] |
| CYP2A13 | Nitrosamine | 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK); N-nitrosonornicotine (NNN); 3-(N-nitrosomethylamino) propionaldehyde; 3-methylindole (skatole); 3-N-nitrosoguvacoline; 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK); bergapten, 5 methoxypsoralen; N-nitrosopyrrolidine | [58, 140–142] |
| CYP3A4 | Difuranocoumarin, mycotoxin | Aflatoxin B1; aflatoxinG1; sterigmatocystin; dihydrodiol derivatives of PAHs | [143–146] |
| CYP2B6 | Haloalkane, azoaromatic amine, tobacco-specific nitrosamine, herbicide, chloroacetamide, PAH, hydrocarbon, and alkyl benzene | 1, 2-Dibromoethane (ethylene dibromide); 2, 2-dichloro-1,1,1-trifluoroethane (HCFC-123); 3-methoxy-4-aminoazobenzene; 4-vinyl-1-cyclohexene; (S)- and (R)-; N, N′, N″-triethylene thiophosphoramide (thioTEPA) | [29, 147–150] |
| CYP2C8 | Oxazaphosporine: anticancer, nitrogen mustard alkylating, tobacco-specific nitrosamine, nitrosamine, aromatic hydrocarbons, alkyl benzene | Ifosfamide; 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK); chloroform (trichloromethane); N-nitrosomethylpropylamine; styrene | [151–153] |
| CYP2C9 | Aliphatic epoxide, metabolite, oxazaphosphorine: anticancer, nitrogen mustard, alkylating, phenylpropene, from Rhizoma acorigramine, and nitrosamine | Butadiene monoxide (1, 2-epoxy-3-butene); cyclophosphamide; ifosfamide; methyleugenol; N-nitrosopyrrolidine | [134, 154–156] |
| CYP2D6 | Nitrosamine, tobacco-specific nitrosamine, difuranocoumarin; mycotoxin, produced by Aspergillus species on food products, pyrido-carbazole; antineoplastic, alkaloid, Apocynaceae plant compound, topoisomerase II inhibitor and DNA binding | 3-(N-nitrosomethylamino) propionaldehyde; NNK; NNAL; aflatoxin B1 (AFB1); ellipticine | [157–159] |
| CYP2E1 | Haloalkane, diene halobenzene, nitrile, herbicide, arylamine, and furanoterpene produced in sweet potatoes infected with Fusarium solani; pulmonary toxin, alkylating, organic solvents, alkylformamide, nitrosamine, o-methoxyaniline, cyclohexane derivative | 1, 2-Dichloroethane (ethylene dichloride); 1,3-butadiene; 1,4 and 2,3-dichlorobutane; 2,6-dichlorobenzonitrile (dichlobenil); 2-aminofluorene (2-AF); 4-ipomeanol; N, N-dimethylformamide (DMF), N-nitrosoethylbutylamine; o-anisidine 2-methoxyaniline; vinylcyclohexane | [160–164] |
| CYP2F1 | Indole, alkylating, pulmonary toxin; present in higher concentrations in mammalian digestive tract and coal tar, furanoterpene produced in sweet potatoes infected with Fusarium solani; pulmonary toxin, alkylating, aromatic hydrocarbon, alkyl benzene | 3-methylindole, skatole; 4-ipomeanol; styrene (vinyl benzene) | [165–167] |
| CYP2W1 | PAH, metabolite | Chrysene-1, 2-diol, dibenzo[a,l]pyrene-11,12-diol, sterigmatocystin | [168] |
| CYP3A4 | Nitroarene, triazole, heterocyclic amine, azoaromatic amine, N-heterocyclic aromatic hydrocarbon, dibenzocarbazole, estradiol derivative; estrogen, contraceptive, nitrosamine, triphenylethyleneamine; antiestrogen, estrogen receptor modulator | 1-Aminobenzotriazole (1-ABT); 1-aminopyrene; 1-nitropyrene; 2-aminofluorene; 3,6-dinitrobenzo[e]pyrene; 3-amino-1,4-dimethyl-5H-pyrido[4,3-b]indole (Trp-P-1); 3-methoxy-4-aminoazobenzene; 7H-dibenzo[c,g]carbazole; 17α-ethynylestradiol (ethi-nylestradiol 17α-); N-nitrosodibutylamine (N,N-dibutylnitrosamine); tamoxifen | [134, 149, 169] |
| CYP3A5 | Antimitotic, epipodophyllotoxin, topoisomerase II inhibitor, oxazaphosphorine; nitrogen mustard alkylating | Etoposide (VP-16); ifosfamide; tobacco-specific nitrosamine | [134, 149] |
2.1.1. P450s and 7,12-dimethylbenz[a]anthracene (DMBA)
The chemical 7,12-dimethylbenz[a]anthracene (DMBA), which is commonly used as a model chemical carcinogen, is a PAH. The compound has been applied in a rat mammary tumor model [30] and is the most studied PAH apart from benzo[a]pyrene (B(a)P). In vivo, CYP1B1, and not CYP1A1, was shown to be the main enzyme responsible for the metabolic activation of DMBA when exposed to carcinogenic metabolites [53]. While 3,4 diol-DMBA, 7 1,2-epoxide-3,4-diol-DMBA, and -OHM-12DMBA have all been proven to show genotoxic effects, the latter was found to be the ultimate carcinogen [54]. Substantial amounts of deoxyadenosine and deoxyguanosine adducts have been shown to be induced by the benzylic carbon of 1,2-epoxide-3,4-diol-DMBA [54]. In some cases, researchers have found minor adducts with the amino group of the 7-position of deoxyguanosine and deoxycytidine [55]. Some researchers believe that DMBA is a more effective carcinogen than B(a)P. The latter was found to commonly bind to guanine residues on DNA because it is less effective in tumor initiation than hydrocarbon-deoxyguanosine adducts [55]. Metabolites from DMBA under the activation of CYP1B1 have been shown to play a vital role in different tumors, where there are elevated levels of CYP1B1 compared to levels of normal nearby tissues, which include the breast, brain, ovary, colon, and lung [55] (Figure 2). In addition to its activity toward exogenous compounds, CYP1B1 is also responsible for the metabolism of endogenous substrates such as estrogen into reactive metabolites, e.g., 4-hydroxyestradiol (Table 1). Estrogen affects the carcinogenesis of endometrial and breast tissues by acting both as an initiator and a proliferator.
Figure 2.
P450s in cancer formation.
2.1.2. P450s and nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)
Nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is bioactivated in human lung cancer by CYP2A13. In this respect, the enzyme is more effective than CYP2A6. The Vmax/Km is 0.008 for NNK methylene hydroxylation, which is thought to be a significant step in ultimate carcinogen formation by CYP2A6. By comparison, that for CYP2A13 is 0.36 [56]. This elevated level of CYP2A13 bioactivation and the expression of CYP2A13 in the human liver indicate that CYP2A13 could play a major part in NNK activation, which is more effective in CYP2A13 in vitro metabolism than CYP2A6 [57]. Many different CYP2A subfamily members can effectively activate several carcinogenic nitrosamines in vitro [58]. These P450s play a significant role in NNK metabolic activation in vivo in the human liver and mouse lung. Studies also suggest that some P450 enzymes belonging to subfamilies play an important role in the metabolism of another tobacco-specific nitrosamine, N′-nitrosonornicotine, in both humans and rats [59–62]. Approximately 1 to 10% of the total P450 content in the human liver is composed of CYP2A6, which is a particularly efficient coumarin 7-hydroxylase (19). CYP2A6 is the primary enzyme responsible for nicotine metabolism in smokers, and the inactive metabolite in vivo is cotinine [11, 63]. CYP2A13 has approximately 94% sequence similarity to CYP2A6 and differs by just 32 amino acids [64]. Originally, CYP2A13 was cloned from the nasal mucosa of humans. The functional enzyme participates in the metabolism of several CYP2A6 substrates, such as NNK, coumarin, and N,N-dimethylaniline. However, the efficiencies of the two enzymes in affecting metabolism are significantly different [65]. NNK is particularly considered important for the formation of human lung cancer and is metabolized more effectively to potentially carcinogenic intermediates by CYP2A13 instead of CYP2A6. Nicotine metabolism by the two enzymes differs with the metabolism of cotinine. Despite the similarity in product distribution, the Km of nicotine 5′-oxidation by CYP2A6 is six times higher than that for CYP2A13 [66]. The CYP2 family plays a pivotal role in the activation and inactivation of precarcinogens. Both genetic and environmental factors result in interindividual differences in the actions driven by P450s.
3. Role of P450s in Cancer Metastasis
Metastasis is responsible for most malignant growth-related fatalities, yet it remains the least understood aspect of cancer biology. As metastasis study continues to evolve at a rapid pace, the biological mechanisms underlying the spread and metastatic output of malignant growth cells are beginning to be elucidated [67]. The P450s show promising results in the treatment of cancer due to varying epoxygenases, their impact in cancer progression, and their various expression patterns. EET therapy significantly increased the profiles of migration, invasion, and prometastatic gene expression in a range of cancers. P450 epoxygenases result in the conversion of arachidonic acid into four regioisomeric epoxyeicosatrienoic acids (REA) [68]. These enzymes exert different biological responses in different systems. Several studies found overexpression of CYP2J2 epoxygenase in human cancer cell lines and tissues and increased tumor growth due to EETs, reduced apoptosis of cancer cells, and enhanced proliferation of carcinoma cells [69–71] (Figure 3). The overexpression of CYP2J2 reduces the endothelial cell adhesion molecule expression induced by cytokines and prevents the adhesion of leukocytes to vessel walls [70]. The effects include suppression of IκB and NF-κB kinase, suggesting anti-inflammatory effects of EETs that are independent of the hyperpolarization of membranes. According to Ma et al., EETs are involved in the upregulation of endothelial nitric oxide synthase and the stimulation of the proliferation and angiogenesis of endothelial cells through the activation of the phosphatidylinositol 3-kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK) signaling pathways [72]. Addition of synthetic CYP2J2 or EET overexpression protected endothelial cells against injury due to hypoxia-reoxygenation in vitro and showed a fibrinolytic influence by enhancing the expression and activity of the tissue plasminogen activator (t-PA). Moreover, this treatment also prevents dysfunction of postischemic myocardial activity by stimulating the MAPK pathway [70]. These findings show that EETs derived from CYP2J2 play an important role in cardiovascular protection. Still, one recent study found potentially harmful effects of CYP2J2 expression and biosynthesis of EET [70]. CYP2J2 protein and mRNA levels were found to be high in certain human cancer tissues and human-derived cancer cell lines. However, they were not present in adjacent normal tissues and noncancer cell lines [73]. Hence, a number of specific CYP2J2 inhibitors have been developed, and their efficacy in inhibiting tumor progression has been actively studied. CYP2J2 inhibitors such as C26 (1-[4-(vinyl)phenyl]-4-[4-(diphenyl-hydroxymethyl)-piperidinyl]-butanone hydrochloride) caused a marked reduction in tumor proliferation and migration as well as promoted apoptosis in cancer cells [74].
Figure 3.
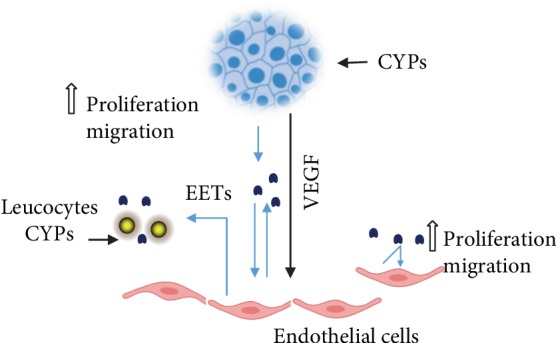
P450s in metastasis and the EET pathways. Blue depicts the crosstalk between the tumor epithelium, endothelial cells, and lymphocytes. This figure was adapted from Edson and Rettie [89].
The addition of recombinant adeno-associated viral vector- (rAAV-) mediated delivery of CYP2J2, exogenous EETs, or a selective 14,15-EET epoxygenase known as CYP102 F87V resulted in a high proliferation of cancer cells in vivo and in vitro [75]. The addition of EET or epoxygenase overexpression in neoplastic cell lines resulted in increased activation of the PI3K/Akt and MAPK pathways and increased phosphorylation of the epidermal growth factor receptor (EGFR). Carcinoma cell apoptosis was repressed by the upregulation of the antiapoptotic proteins Bcl-2 and Bcl-xL and downregulation of the proapoptotic protein Bax [76]. The results showed that the epoxygenase activity of P450s plays an important role in the neoplastic phenotype promotion as well as in the pathogenesis of different forms of human cancers.
CYP ω-hydroxylases, which mainly consist of CYP4F and CYP4A, promote the metabolism of arachidonic acid and the subsequent conversion to biologically active eicosanoids, such as 20-hydroxyeicosatetraenoic acid (20-HETE), which has different pathological and physiological functions [77, 78]. This molecule was found to serve as an additional messenger in the mitogenic-induced growth factor pathway and as an effective mediator in angiogenesis of vessel sprouting and vascular endothelial growth factor (VEGF). A selective inhibitor of 20-HETE synthesis, N-hydroxy-N-(4-butyl-2 methyl phenyl)-formamidine (HET0016), has been found to prevent the angiogenic responses to VEGF, FGF, EGF, and electrical stimulation in rats [79] [80] and inhibits angiogenesis in the cornea that is stimulated by the proliferation of human U251 glioblastoma cells in vitro [81]. The growth of renal adenocarcinoma in nude mice was inhibited by an antagonist of 20-HETE called WIT002 [82]. Moreover, the cell proliferation of U251 cells increased with the introduction of CYP4A1 via infection in vitro [83, 84]. In contrast, a stable 20-HETE agonist, WIT003, stimulated the cell proliferation of endothelial cells and VEGF expression in vitro [84]. CYP ω-hydroxylase affects tumor growth, metastasis, and angiogenesis [78].
Signaling pathways, including MAPK and PI3K/Akt, are considered important in invasion, proliferation, metastasis, and angiogenesis [85, 86]. Recent studies have suggested that CYP ω-hydroxylase-derived 20-HETE plays an important role in the activation of PI3K/Akt and ERK1/2 in endothelial cells by affecting cellular functions such as apoptosis [87] and proliferation [88]. Moreover, in 251 human gliomas, CYP4A1-20-HETE gene expression changed cell growth through a mechanism that involves ERK1/2 activation. According to Yu et al., CYP4A11 overexpression can result in an increase in phospho-Akt in A549 cells. Moreover, the overexpression resulted in WIT002 or HET0016 inhibition of endogenous 20-HETE stimulation of PI3K/Akt and ERK1/2. Furthermore, inhibitors of PI3K/Akt (wortmannin) and ERK1/2 (U0126) suppressed WIT003-induced MMP-9 and VEGF expression, which indicates that MMP-9 and VEGF induction by CYP ω-hydroxylase involves the ERK1/2 and PI3K signaling pathways [78]. Hydroxylation of AA by CYP4 enzymes such as CYP4F2, CYP4A11, and CYP4F3B at the omega position results in the formation of 20-HETE. This enzyme has been shown to have an effect on tumor angiogenesis and progression and inflammatory processes that are associated with metastasis and tumor growth [77, 89].
The role of CYP AA epoxygenase enzymes as either passengers or drivers of the carcinogenesis process is unknown due to a lack of genetic evidence implicating the enzymes in the natural history or the oncogenic transformation of specific tumor types. The evidence of CYP AA epoxygenase enzymes in cancer comes from research involving genetic analyses [90]. Primary tumor growth and metastasis are enhanced by the promotion of CYP AA epoxygenase enzymes as well as escape from dormancy [90]. Mammary tumor engraftment is inhibited through the knockdown of cancer cell intrinsic CYP3A4 [91]. The results derived through genetic approaches show the supporting role of CYP AA epoxygenase enzymes in the progression and metastasis of tumors, which can serve as an important strategy in cancer therapy. This view is supported by knockout and knockdown research on the tumor or the immediate microenvironment that shows the role of P450s in the spread of multiple types of tumors [74, 92]. Targeted therapies have shown restricted success in reducing the existing metastatic development and enhancing survival when used as monotherapies or combination therapies. There are, however, still minimal and contentious epidemiological data on this significant point, partially owing to the inherent problems in conducting epidemiological surveys of cancer metastasis CYPs. Nevertheless, future studies investigating the relationship between P450s and metastasis will shed more light on this very important aspect of cancer.
4. Role of P450s in Chemoprevention
Chemoprevention refers to the administration of a medication for the purpose of preventing disease. Cancer chemoprevention has long been acknowledged as a significant prophylactic approach for reducing the health care system's burden of cancer. Chemoprevention of cancer includes the use of one or more pharmacologically active agents to block, suppress, deter, or reverse invasive cancer growth. Nevertheless, comparatively little research has been performed to characterize the capacity of putative chemopreventive agents to modulate the expression of P450s or to comprehend the relationship between P450s and chemopreventive agents. P450s accelerate the process of activation of compounds to toxic products [44]. Moreover, they participate in many different functions, such as the oxidation of fatty acids and steroid hormones [44]. The modulation of enzyme expression can affect chemical toxicity, mutagenicity, and carcinogenicity. An interesting application of cancer chemoprevention is through the administration of a dietary nontoxic component that helps inhibit or prevent neoplastic disease. The chemopreventive agents are regulated by AhR and are known to be procarcinogenic PAHs. While the CYP1 family members are expressed in extrahepatic tissues, CYP1B1 is different due to it being overexpressed in different tumor types compared to expression in normal tissues [93]. This finding has attracted increased interest. Studies have found that this enzyme affects tumorigenesis due to its ability to activate different carcinogens in the PAH chemical classes, aromatic amines, heterocyclic amines, and nitropolycyclic hydrocarbons. In addition, recent studies have found a link between CYP1B1 polymorphisms and a reduced or enhanced risk of certain types of cancers [94, 95]. CYP1B1 and CYP1A1 may also affect the formation of advanced carcinoma and affect enzymes involved in the metabolism of chemotherapeutic agents that may help prevent tumor cytotoxicity [74]. The most important role played by CYP1B1 is in the metabolism of estradiol. The enzyme catalyzes the hydroxylation of estradiol mainly at the C-4 position. Moreover, C-2 hydroxylation can occur mainly through CYP3A4 and CYP1A2 [96, 97]. However, the preferred pathway outside the liver is C-4 hydroxylation, which may play a part in tumorigenesis induced by estrogen-related factors [98]. The reason for this is the strong ER agonist ability of 4-hydroxyestradiol and the binding affinity for the estrogen receptor that is approximately 1.5 times greater than that of estradiol [99]. Another reason is the subsequent conversion of 4-hydroxyestradiol to estradiol 3,4-quinone, which has been shown to bind with DNA and results in the formation of unstable adducts that cause gene mutations [100]. Researchers studying the effects of CYP1B1 knockout as well as CYP1A1 and CYP1A2 found that animals that lack these genes did not suffer from any deficiencies and displayed normal growth. Moreover, the CYP1B1 knockout mice displayed strong resistance to the formation of tumors induced by DMBA [101]. The studies show proof of the potential safety and efficacy of chemopreventive agents that inhibit the activity and expression of CYP1B1. The ability to induce CYP1 family expression by PAHs is illustrated by studies that show high levels of CYP1B1 and CYP1A1 expression in urothelial and lung tissue of smokers [79]. A practical chemopreventive strategy is treatment with AhR antagonists. The activation of different carcinogens that stimulate CYP1 expression through AhR is possible in the presence of CYP1B1 and CYP1A1 expressions.
4.1. Natural Compounds
Researchers have found many different natural products showing promising results in this respect. One recent study has shown that the flavonoid kaempferol prevents agonist binding to AhR. Moreover, researchers have found that the compound plays a role in the complex induction and formation of CYP1A1 expression [102, 103] (Figure 4). Another major discovery was that kaempferol prevented the growth of immortalized lung epithelial cells (BEAS-2B) caused by cigarette smoke condensate in a soft agar colony assay. This study found that the compound prevented AhR binding with an IC50 of 28 nM at a cellular concentration of 10 μM (~IC90) [102]. Other natural compounds that were found to prevent CYP1 family expression include the stilbene phytoestrogen resveratrol and the 5,7-dimethoxyflavone that prevents CYP1A1 expression and action. Many different compounds have been investigated to assess their ability to inhibit the enzymatic activity of CYP1B1 [35, 104]. Some of the molecules that were found to have potent inhibitory effects include coumarins, resveratrol, stilbenes, and flavonoids. Different compounds that inhibit the CYP1 family are provided in Figure 4. However, it remains to be seen whether CYP1 inhibition results in chemoprevention in vivo.
Figure 4.
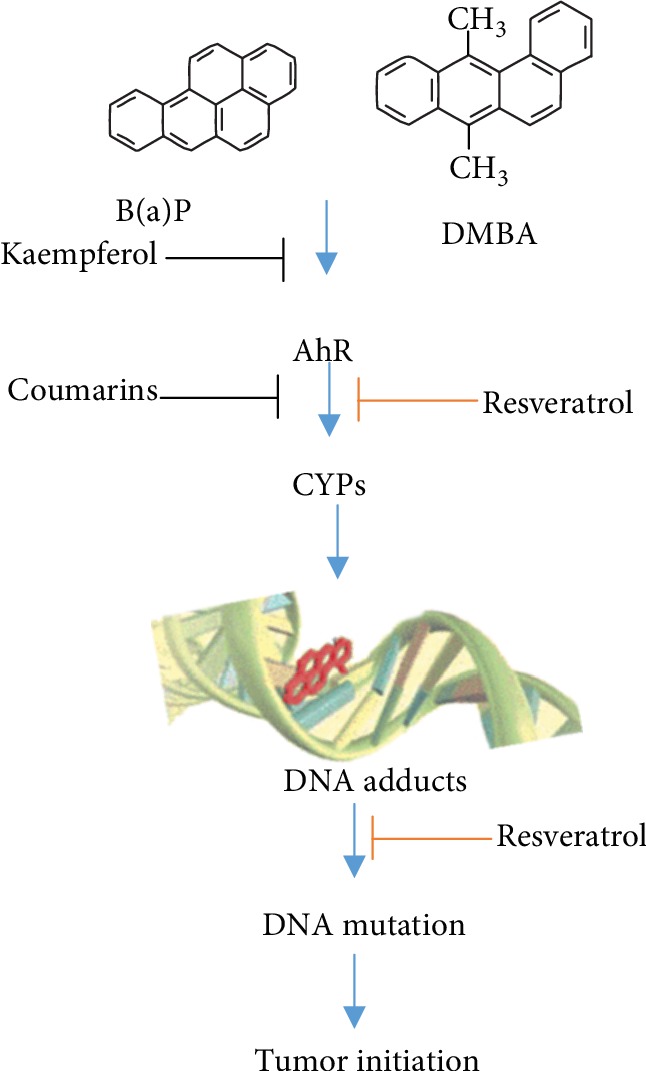
P450s in chemoprevention.
An alternative CYP1-based chemotherapy technique involves the introduction of an inactive prodrug to a cytotoxic compound. Studies have found that resveratrol can be metabolized to piceatannol through CYP1B1 within cancerous cells [105]. Different synthetic drugs that are specifically activated by CYP1 agents have been developed. One compound that shows promising results is phortress, which is a benzothiazole prodrug that has begun phase I clinical trials [106]. The hydrophilic lysine-amide compound does not undergo hydrolysis of the parent compound 5F203 except in the presence of cells [107]. This compound is then absorbed by sensitive cells and acts as a potent agonist of AhR, resulting in the induction of CYP1 family genes. Subsequently, CYP1A1 metabolizes 5F203, resulting in the production of reactive electrophilic species that cause cell death and DNA damage. The drug has shown strong benefits in preclinical trials both in vivo and in vitro against different types of tumors [108]. Since only AhR-expressing cancer cells are vulnerable to phortress, patients need to be properly screened to ensure that they will benefit from the drug. Recently, phase I clinical trials of aminoflavone (5-amino-2,3-fluorophenyl-6,8-difluoro-7-methyl-4H-1-benzopyran-4-1) have been completed, which is believed to function in a similar manner to 5F203. However, recent studies have shown that aminoflavone requires sulfotransferase A1 (SULTA1) expression for a cellular response [109].
This finding suggests that the activation of the aminoflavone may be complex, and the long-term effects of treating patients with aminoflavone or phortress are unclear. As mentioned earlier, induction of CYP1 family proteins and the activation of AhR will likely lead to the development of cancer and may do more harm in the long run. Nevertheless, the developers of phortress have highlighted the fact that the cell-specific activation of AhR by the drug is different than that of other carcinogens, such as PAHs. Preclinical studies have shown low toxicity levels, but further research is required to prove the clinical effectiveness of aminoflavone and phortress in cancer treatment [110]. An advantage of using a prodrug that is specifically designed to activate tumor-specific CYP1B1 is that it does not induce CYP1 enzymes. An example is DMU-135 (3,4-methylenedioxy-3′,4′,5′-trimethoxy chalcone) that is converted within tumors by CYP1B1 to form DMU-117, which is an effective nonselective tyrosine kinase inhibitor and a possible COX inhibitor [111]. Researchers have found that DMU-135 can act as a chemopreventive agent to prevent the formation of gastrointestinal tumors in mice without toxicity. It is the first drug that is specifically targeted for CYP1B1 activation [111]. Moreover, different CYP2 family members have been found in extrahepatic tissue, of which, the notable ones discovered by the Human Genome Project include CYP2R1, CYP2S1, and CYP2U1 [112]. These enzymes can produce important xenobiotic and endogenous metabolites. For instance, CYP2S1 and CYP2R1 can be used in the metabolism of ATRA and vitamin D hydroxylase, respectively [112]. However, the exact role of the enzymes in tumor progression is not clear. CYP2W1 has been identified in tumor-specific CYP24, particularly adrenal and gastric cancers [113]. Moreover, indole and arachidonic acid have recently been identified as potential substrates. Despite the lack of knowledge on the enzyme functions, the tumor-specific enzyme actions remain intriguing and require further research.
The roles of other P450 family members in vivo suggest their potential use in chemopreventive therapies specifically targeting P450 inhibition [114]. Selective P450 inhibitors, such as methoxylated or hydroxylated flavonols and flavones, methoxytrans-stilbenes, kaempferol, and berberine and rutaecarpine alkaloid derivatives, also serve as promising chemopreventive agents for both estrogen-related and environmental carcinogen-induced carcinogenesis to inhibit the formation of tumors in cancerous cells. In cancer prevention and treatment, targeting P450s with natural or synthetic small molecules provides potential benefits. Since the crystal structures are still to be determined for almost all CYPs, drug design strategies depend on the knowledge of the substrate structure and the mechanism of action of the enzyme. These enzyme-targeting techniques include the following: (i) designing enzyme-inhibiting molecules, (ii) the development of enzyme-activated prodrugs, (iii) immuno-based therapies targeting enzyme immune responses, and (iv) genetic therapy strategies to express different P450s in cancer cells.
5. Role of P450s in Chemotherapy
Chemotherapy with cytotoxic antineoplastics remains an important technique for the clinical treatment of patients with malignant tumors. The most important site for P450 metabolism is the liver, where the enzymes are omnipresent. Anticancer drugs are usually metabolized by a number of parallel and/or sequential reactions after absorption in the organism. Metabolism occurred in two distinct sequential phases called “Phase I” and “Phase II,” although this order is not exclusive (Phase I did not always accompany Phase II; Phase II was not always followed by Phase I) [115]. P450s are key players within the phase I-dependent metabolism and for the most part, catalyze the oxidations of drugs [116]. CYP1, CYP2, and CYP3 family members are involved in drug metabolism. P450s catalyze a number of different reactions, including hydroxylation, epoxidation, dealkylation, and deamination, of which hydroxylation is likely the most important. Phase II-dependent reactions make the products suitable for excretion through the kidneys as inactive polar products. With regard to anticancer agents, P450s are involved not only in the detoxification of cytotoxic products but also in the activation of drugs that make them therapeutically active. P450s metabolize many anticancer agents, shown in Table 2. Prodrugs are inactive agents that, upon exposure to tumor tissues, are converted to active cytotoxic drugs with high expression of activating enzymes. This targeting strategy minimizes toxicity to normal tissues while increasing the tumor tissue delivery of the active agent. Liver P450s metabolize cyclophosphamide, ifosfamide, dacarbazine, procarbazine, tegafur, and thiotepa [117]. Another example is 1,4-bis-([2-(dimethylamino-N-oxide)ethyl]amino)5,8-dihydroxy anthracene-9,10-dione (AQ4N), a bioreductive drug that requires CYP2S1 and CYP2W1 activation in tumor tissues to be transformed into an inhibitor of topoisomerase II [118].
Table 2.
P450s involved in cancer drug metabolism.
| Drugs | CYPs | Ref |
|---|---|---|
| Altretamine | 2B | [170] |
| Bexarotene | 2C9, 3A4 | [171] |
| Busulfan | 3A4 | [172] |
| Cisplatin | 2E1, 3A4 | [173] |
| Cyclophosphamide | 2B6, 2C9, 3A4 | [174] |
| Cytarabine | 3A4 | [175] |
| Dacarbazine | 1A1, 1A2, 2E1 | [176, 177] |
| Docetaxel | 1B1, 3A4, 3A5 | [176] |
| Doxorubicin | 2D6, 3A4 | [178] |
| Erlotinib | 1A1, 1A2, 3A4 | [179] |
| Etoposide | 1A2, 2E1, 3A4, 3A5 | [180] |
| Exemestane | 3A4 | [181] |
| Fulvestrant | 3A4 | [182] |
| Gefitinib | 3A4 | [183] |
| Idarubicin | 2D6, 2C9 | [184] |
| Ifosfamide | 2A6, 2B1, 2B6, 2C9, 2C18, 2C19, 3A4, 3A5 | [185] |
| Imatinib mesylate | 1A2, 2C9, 2C19, 2D6, 3A4 | [186] |
| Irinotecan | 3A4, 3A5 | [187] |
| Letrozole | 2A6, 3A4 | [188] |
| Paclitaxel | 2C8, 3A4, 3A5 | [189] |
| Tamoxifen | 1A1, 1A2, 1B1, 2B6, 2C9, 2C19, 2D6, 2E1, 3A4, 3A5 | [190] |
| Teniposide | 3A4, 3A5 | [180] |
| Thiotepa | 2B1, 2C11 | [191] |
| Topotecan | 3A4 | [192] |
| Toremifene | 1A2, 3A4 | [193] |
| Tretinoin | 2C8, 2C9, 2E, 3A4 | [194] |
| Vinblastine | 3A4 | [195] |
| Vincristine | 3A4 | [196] |
| Vinorelbine | 3A4 | [197] |
The use of immunohistochemistry, western blot analysis, and reverse transcriptase PCR has shown reliable expression of CYP3A types in kidney cancer cells. This study suggested that the expressed CYP3A may be involved in the development of renal cancer and that the multidrug resistance found in this cancer is caused by these types of CYP3A [119, 120]. CYP3A4 plays a major role in the metabolism of several anticancer agents (taxanes, vinca-alkaloids, and new drugs such as imatinib, gefitinib, and sorafenib). CYP3A4 metabolizes docetaxel into hydroxylated derivatives that are inactive. A high activity of CYP3A4 will result in the drug's poor therapeutic outcome. Thus, a 49% decrease in docetaxel clearance was observed in patients with cancer treated with docetaxel in conjunction with the active CYP3A4 inhibitor ketoconazole [121]. Low CYP3A4 expression in breast tumors improved the response to docetaxel. Likewise, hepatic CYP3A4 activity assessed by the erythromycin breath test and midazolam clearance predicted the clearance of docetaxel and showed lower toxicity in patients with the lowest CYP3A4 activity. Unlike docetaxel, irinotecan is inactivated by CYP3A4, and the induction of CYP3A4 results in a significant decrease in the development of the toxic metabolite of this drug in patients receiving irinotecan. In addition, the CYP3A4 phenotype is significantly associated with irinotecan pharmacokinetics as measured by midazolam clearance. A study recently indicated that the pregnane X-receptor (PXR) pathway also includes irinotecan resistance in the colon cancer cell line through the upregulation of drug-metabolizing genes such as CYP3A4 [122–124].
The main expression pattern of CYP4Z1 renders it a suitable candidate for cancer therapy. However, for prodrug therapy based on CYP4Z1 for the treatment of breast cancer, lung cancer, ovarian cancer, and prostate cancer, effective prodrugs must be identified. Previous studies have used CYP enzymes to activate different anticancer prodrugs [125]. Most researchers have investigated oxazaphosphorines, including ifosfamide and cyclophosphamide [126–128]. In clinical trials, CYP2B6-dependent activation of cyclophosphamide has shown positive results [129, 130]. Presently, researchers are working on new mutants of CYP2B6 to improve the activation of cyclophosphamide. For instance, Haque and Pattanayak found a superior ability of a triple mutant of CYP2B6 to fuse with NADPH cytochrome P450 reductase that can help in converting cyclophosphamide into its cytotoxic form [131].
Moreover, increasing the number of cancerous tissues results in low P450 activity compared to that of the nearby healthy tissues where the prodrugs are activated, resulting in cytotoxicity. An important cancer research objective is to develop therapeutic agents that specifically target tumor cells. Studies have found that P450s provide therapeutic options at higher levels in tumor cells compared to that in the surrounding tissues due to activation of the prodrugs in the cancer cells, which reduces side effects [20, 132]. As a result, there are opportunities for enhanced tumor-specific endogenous expression of P450s and CYP-mediated gene therapy. Most studies have extensively investigated CYP1B1 as opposed to other P450 family members. The enzymes have shown high expression, particularly in liver cells. CYP1B1 protein expression has been found in different tumors, and the protein was not detected in normal tissues [133]. Many different agents, such as phortress and resveratrol, are activated by CYP1B1 in preclinical studies. Moreover, a CYP1B1 vaccine known as Zyc300 is presently in phase I and II trials that aim to destroy cancer cells through T-cell response induction [117, 134]. These strategies can be initiated with other P450s that have been identified in tumor cells, such as CYP2J2, CYP2W1, and CYP4Z1. This requires the identification of an appropriate prodrug. However, additional research is required to ensure the success of CYP-based cancer therapy.
5.1. P450 Polymorphisms
Genetic polymorphisms of P450s can affect the catalytic activity of the enzyme and have been reported to be associated with different diseases and adverse drug reactions in different populations. In terms of drug metabolism, P450 polymorphism phenotypes vary from ultra-fast to weak metabolizers. Of the 50 known P450 isoenzymes catalyzing drug metabolism, more than 20 P450 genes are functionally polymorphic, such as CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP1B1, and CYP1A2. The polymorphic P450s catalyze about 40% of the drug metabolism [119, 135]. In addition, P450 polymorphisms were reported to confer susceptibility to disease and disease protection or reduced risk (Table 3). Inhibition of P450 enzymes by a new chemical entity (NCE) may decrease the metabolism of comedicated drugs. However, P450s were largely overlooked in the development of cancer drugs until recently, recognized only for their role in the chemotherapeutic metabolism of Phase I. The first successful strategy in cancer therapy to target P450s was the production of active CYP19 (aromatase) inhibitors for the treatment of breast cancer. Aromatase inhibitors have entered a new era in hormone ablation therapy for estrogen-dependent cancers, paving the way for similar strategies to combat androgen-dependent prostate cancer. Identification of the P450s involved in the inactivation of vitamin D3 and vitamin A anticancer metabolites has also triggered the development of agents targeting these enzymes. The discovery in cancer cells of the overexpression of exogenous metabolizing P450s, such as CYP1B1, has increased interest in the creation of chemoprevention inhibitors and prodrugs intended to be triggered by P450s in cancer cells only [74].
Table 3.
Diseases associated with P450 polymorphism.
| S. no. | CYPs | Diseases | Gene polymorphism | Country of population | Ref |
|---|---|---|---|---|---|
| 1 | CYP1A2 | Prostate cancer | T3801C at 3′UTR | Indian | [198] |
| 2 | CYP7A1 | Tuberculosis | rs3808607 | Moroccan | [199] |
| 3 | CYP1A2 | Cancers | rs762551 | Caucasians | [200] |
| 4 | CYP17A1 | Prostate cancer | Wild | General | [201] |
| 5 | CYP24A1∗ | Idiopathic infantile hypercalcemia | rs114368325 rs6068812 |
German, Russia, Turkey | [202] |
| 6 | CYP8A1 | A left main coronary artery disease | C1117 | Greece | [203] |
| 7 | CYP19A1 | Alzheimer disease | rs3751592 | Chinese | [204] |
| 8 | CYP1B1 | T2D | rs1056827 | Saudi Arabians | [205] |
| 9 | CYP4A11 | Hypertension | rs1126742 rs3890011 |
Chinese | [206] |
| 10 | CYP1B1 | Atherosclerosis | Wild | mice | [207] |
| 11 | CYP2C9 | Epistatic interactions to coronary heart disease susceptibility | rs9332242 and rs61886769 | Russian | [208] |
| 12 | CYP2J2 | Ischemic stroke | -50G/T | Chinese | [209] |
| 13 | CYP17 | Gallbladder cancer /breast cancer | rs743572 | Indian/Chinese | [127] |
Currently, decisions on which drugs to prescribe are made for many disorders on a trial-and-error basis. Genetically based screening methods would allow the tailoring of drug therapy, drug selection, and dosing according to the ability of an individual to metabolize a drug under the pharmacogenomic paradigm. The fact that two genomes are involved complicates cancer pharmacogenomics: the patient's germline genome and the tumor's somatic genome. Chemotherapeutic drugs are highly sensitive to genetic background, as they are generally unspecific drugs with narrow therapeutic indexes that often result in severe or fatal toxicity.
5.1.1. Cyclophosphamide
Cyclophosphamide (CPA), a prodrug used in cancer therapy, is activated by CYP2C19, CYP2C9, CYP3A4, and CYP2B6 to treat some autoimmune disorders. CYP2C19∗2 and CYP2B6∗5 carriers have been shown to have a significantly lower removal of CPA and worse therapeutic performance. Also in the liver, CYP2B6 enzyme metabolizes ifosfamide, tamoxifen, procarbazine, and thiotepa in the same way as it activates CPA [117].
5.1.2. Tamoxifen
Tamoxifen is a modulator of estrogen receptors used in hormone receptor-positive breast cancers. It has been suggested that CYP2D6, the active metabolizer of tamoxifen, is necessary for the formation of endoxifen. Several studies have shown that CYP2D6 PMs have decreased relapse-free time and disease-free survival rate, but they do not experience hot flashes of the same magnitude as patients with the wild-type allele. As a result of enzyme inhibition (serotonin reuptake inhibitors, antidepressants, and other inhibitors of CYP2D6), a similar loss of efficacy is observed [117].
5.1.3. Thalidomide
Thalidomide bioactivation depends on CYP2C19 (5-hydroxythalidomide) metabolism. Another pathway also exists that produces thalidomide arene oxide and is mediated by CYP1A1 and CYP2E1. The response to thalidomide and dexamethasone parallel treatment was reported to be higher in CYP2C19 EMs than in PMs in multiple myeloma. The lower response rate observed in PMs may be due to reduced angiogenesis inhibition activity. Notwithstanding this evidence, the CYP2C19 polymorphism does not have a major influence on the treatment outcome [136].
5.1.4. Tegafur
Tegafur is a drug that is converted by CYP2A6 to 5-fluorouracil. The drug is weakly metabolized in patients with CYP2A6∗4 or CYP2A6∗11. Since other P450s influence tegafur metabolism (CYP3A4, CYP3A5, glutathione S-transferases), it is difficult to calculate the effective dose [137].
5.1.5. Imatinib
Imatinib mesylate (IM), a specific inhibitor of the BCR-ABL tyrosine kinase, is a well-established first-line treatment for chronic myeloid leukemia (CML). IM is utilized for the most part by P450s in the liver, specifically the CYP3A4 and CYP3A5 catalysts. Polymorphisms in these genes can modify the protein action of IM and may influence its reaction. Yuan et al. reported the effect of two single-nucleotide polymorphisms (SNPs), namely, CYP3A5∗3 (6986A>G) and CYP3A4∗18 (878T>C), on IM treatment reaction in patients with CML (n = 270; 139 IM resistant and 131 IM responsive) [138].
6. Conclusion
P450s play a vital role in chemoprevention, carcinogenesis, cancer therapy, and metastasis through regulation. As a result, whether inhibition of P450s reduces the risk of cancer depends on the cancer type, its etiology, and the treatment. The literature review shows that much progress has been made in understanding the role of drug-metabolizing P450s in cancer treatment. The P450 family genes may play a role in the formation of different types of cancer, as demonstrated by their overexpression, which promotes carcinogenicity. Certain P450 family members are upregulated in cancer, making them potential targets for cancer treatment. Thus, by providing P450-mediated metabolism at the tumor site such as the site of anticancer drug action, individual P450s, which are overexpressed in tumor cells, may represent exciting and novel targets for cancer. In addition, patient-specific therapeutic regimens, including prodrugs, reversible inhibitors, and immunotherapy, can be customized to facilitate the management of a variety of human tumors by recognizing the complement of functionally active P450s within the tumor and nontumor tissues. However, whether the enzyme activities need to be inhibited or enhanced depends on different types of cancer and the important products that these P450s produce. Recent accomplishments in the use of polymorphic C as drug targets in cancer therapy are promising and could provide a new and effective alternative for future cancer therapy.
Acknowledgments
The authors are grateful to the Deanship of Scientific Research at King Faisal University, Saudi Arabia, for supporting this study under Grant No. 150164.
Abbreviations
- P450s:
Cytochrome P450s
- PAHs:
Polycylic aromatic hydrocarbon
- EETs:
Epoxyeicosatrienoic acids
- DHETs:
Dihydroxyeicosatrienoic acids
- GBMs:
Glioblastomas
- PHAH:
Polyhalogenated aromatic hydrocarbons
- AhR:
Aryl hydrocarbon receptor
- NF-κB:
Nuclear factor κB
- RB1:
Retinoblastoma protein 1
- DMBA:
7,12-Dimethylbenz[a]anthracene
- B(a)P:
Benzo[a]pyrene
- NNK:
Nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- MAPK:
Mitogen-activated protein kinase
- PI3K:
Phosphatidylinositol 3-kinase
- EGF:
Epidermal growth factor
- VEGF:
Vascular endothelial growth factor
- ARNT:
Aryl hydrocarbon nuclear translocator
- DMU-135:
3,4-Methylenedioxy-3′,4′,5′-trimethoxy chalcone
- ROM:
Reactive oxygenate intermediate
- NCE:
New chemical entity
- XME:
Xenobiotic-metabolizing enzyme
- REA:
Regioisomeric epoxyeicosatrienoic acids
- t-PA:
Tissue plasminogen activator
- rAAV:
Recombinant adeno-associated viral vector
- SULTA1:
Sulfotransferase A1
- PXR:
Pregnane X-receptor
- CML:
Chronic myeloid leukemia
- SNPs:
Single-nucleotide polymorphisms
- IM:
Imatinib mesylate.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1.Ohta T. Multigene families and the evolution of complexity. Journal of Molecular Evolution. 1991;33(1):34–41. doi: 10.1007/BF02100193. [DOI] [PubMed] [Google Scholar]
- 2.Muller H. J. The origination of chromatin deficiencies as minute deletions subject to insertion elsewhere. Genetica. 1935;17(3-4):237–252. doi: 10.1007/BF01985012. [DOI] [Google Scholar]
- 3.Hunter T. A thousand and one protein kinases. Cell. 1987;50(6):823–829. doi: 10.1016/0092-8674(87)90509-5. [DOI] [PubMed] [Google Scholar]
- 4.Kruse M., Muller I. M., Muller W. E. Early evolution of metazoan serine/threonine and tyrosine kinases: identification of selected kinases in marine sponges. Molecular biology and evolution. 1997;14(12):1326–1334. doi: 10.1093/oxfordjournals.molbev.a025742. [DOI] [PubMed] [Google Scholar]
- 5.Danielson B. S. P. P. B. The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Current drug metabolism. 2002;3(6):561–597. doi: 10.2174/1389200023337054. [DOI] [PubMed] [Google Scholar]
- 6.Estabrook R. W. A passion for P450s (remembrances of the early history of research on cytochrome P450) Drug metabolism and disposition. 2003;31(12):1461–1473. doi: 10.1124/dmd.31.12.1461. [DOI] [PubMed] [Google Scholar]
- 7.Nebert D. W., Wikvall K., Miller W. L. Human cytochromes P450 in health and disease. Philosophical Transactions of the Royal Society B: Biological Sciences. 2013;368(1612):p. 20120431. doi: 10.1098/rstb.2012.0431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson D. R., Goldstone J. V., Stegeman J. J. The cytochrome P450 genesis locus: the origin and evolution of animal cytochrome P450s. Philosophical Transactions of the Royal Society B: Biological Sciences. 2013;368(1612):p. 20120474. doi: 10.1098/rstb.2012.0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson D. R., Koymans L., Kamataki T., et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6(1):1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Fanni D., Ambu R., Gerosa C., et al. Cytochrome P450 genetic polymorphism in neonatal drug metabolism: role and practical consequences towards a new drug culture in neonatology. London, England: SAGE Publications Sage UK; 2014. [DOI] [PubMed] [Google Scholar]
- 11.Zanger U. M., Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & therapeutics. 2013;138(1):103–141. doi: 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- 12.Pikuleva I. A., Waterman M. R. Cytochromes p450: roles in diseases. Journal of Biological Chemistry. 2013;288(24):17091–17098. doi: 10.1074/jbc.R112.431916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Redig A. J., McAllister S. S. Breast cancer as a systemic disease: a view of metastasis. Journal of internal medicine. 2013;274(2):113–126. doi: 10.1111/joim.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Badawi A. F., Cavalieri E. L., Rogan E. G. Role of human cytochrome P450 1A1, 1A2, 1B1, and 3A4 in the 2-, 4-, and 16[alpha ]-hydroxylation of 17[beta ]-estradiol. Metabolism-Clinical and Experimental. 2001;50(9):1001–1003. doi: 10.1053/meta.2001.25592. [DOI] [PubMed] [Google Scholar]
- 15.Hecht S. S., Stepanov I., Carmella S. G. Exposure and metabolic activation biomarkers of carcinogenic tobacco-specific nitrosamines. Accounts of chemical research. 2016;49(1):106–114. doi: 10.1021/acs.accounts.5b00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hodges R. E., Minich D. M. Modulation of metabolic detoxification pathways using foods and food-derived components: a scientific review with clinical application. Journal of nutrition and metabolism. 2015;2015:23. doi: 10.1155/2015/760689.760689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue J., Yang S., Seng S. Mechanisms of cancer induction by tobacco-specific NNK and NNN. Cancers. 2014;6(2):1138–1156. doi: 10.3390/cancers6021138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujita K.-i. Cytochrome P450 and anticancer drugs. Current drug metabolism. 2006;7(1):23–37. doi: 10.2174/138920006774832587. [DOI] [PubMed] [Google Scholar]
- 19.Ando Y. Handbook of Anticancer Pharmacokinetics and Pharmacodynamics. Springer; 2004. Cytochrome P450 and anticancer drugs; pp. 215–229. [Google Scholar]
- 20.Molina-Ortiz D., Camacho-Carranza R., González-Zamora J. F., et al. Differential expression of cytochrome P450 enzymes in normal and tumor tissues from childhood rhabdomyosarcoma. PloS one. 2014;9(4):p. e93261. doi: 10.1371/journal.pone.0093261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parsa N. Environmental factors inducing human cancers. Iranian journal of public health. 2012;41(11):1–9. [PMC free article] [PubMed] [Google Scholar]
- 22.Malarkey D. E., Hoenerhoff M., Maronpot R. R. Haschek and Rousseaux's Handbook of Toxicologic Pathology. Third Edition. Elsevier; 2013. Carcinogenesis: mechanisms and manifestations; pp. 107–146. [Google Scholar]
- 23.Pitot H. C., Goldsworthy T., Moran S. The natural history of carcinogenesis: implications of experimental carcinogenesis in the genesis of human cancer. Journal of supramolecular structure and cellular biochemistry. 1981;17(2):133–146. doi: 10.1002/jsscb.380170204. [DOI] [PubMed] [Google Scholar]
- 24.Seo M. M. R. Y. R. An overview on DNA damage and DNA repair in cancer research. Cancer prevention research. 2008;13:237–246. [Google Scholar]
- 25.Monien B. H., Schumacher F., Herrmann K., Glatt H., Turesky R. J., Chesné C. Simultaneous detection of multiple DNA adducts in human lung samples by isotope-dilution UPLC-MS/MS. Analytical chemistry. 2014;87(1):641–648. doi: 10.1021/ac503803m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stiborová M., Černá V., Moserová M., Mrízová I., Arlt V., Frei E. The anticancer drug ellipticine activated with cytochrome P450 mediates DNA damage determining its pharmacological efficiencies: studies with rats, hepatic cytochrome P450 reductase null (HRN™) mice and pure enzymes. International journal of molecular sciences. 2015;16(1):284–306. doi: 10.3390/ijms16010284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hang B. Formation and repair of tobacco carcinogen-derived bulky DNA adducts. Journal of nucleic acids. 2010;2010:29. doi: 10.4061/2010/709521.709521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stiborová M., Moserová M., Černá V., et al. Cytochrome b5 and epoxide hydrolase contribute to benzo[a]pyrene-DNA adduct formation catalyzed by cytochrome P450 1A1 under low NADPH:P450 oxidoreductase conditions. Toxicology. 2014;318:1–12. doi: 10.1016/j.tox.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 29.White I. N. H., Razvi N., Gibbs A. H., et al. Neoantigen formation and clastogenic action of hydrochlorofluorocarbons-123 and perchloroethylene in human MCL-5 cells. Toxicology letters. 2001;124(1-3):129–138. doi: 10.1016/S0378-4274(00)00281-2. [DOI] [PubMed] [Google Scholar]
- 30.Rengarajan T., Rajendran P., Nandakumar N., Lokeshkumar B., Rajendran P., Nishigaki I. Exposure to polycyclic aromatic hydrocarbons with special focus on cancer. Asian Pacific Journal of Tropical Biomedicine. 2015;5(3):182–189. doi: 10.1016/S2221-1691(15)30003-4. [DOI] [Google Scholar]
- 31.Moorthy B., Chu C., Carlin D. J. Polycyclic aromatic hydrocarbons: from metabolism to lung cancer. Toxicological Sciences. 2015;145(1):5–15. doi: 10.1093/toxsci/kfv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao P. S. S., Kumar S. Polycyclic aromatic hydrocarbons and cytochrome P450 in HIV pathogenesis. Frontiers in microbiology. 2015;6:p. 550. doi: 10.3389/fmicb.2015.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agundez J. Cytochrome P450 gene polymorphism and cancer. Current drug metabolism. 2004;5(3):211–224. doi: 10.2174/1389200043335621. [DOI] [PubMed] [Google Scholar]
- 34.Nebert D. W., Dalton T. P. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nature Reviews Cancer. 2006;6(12):947–960. doi: 10.1038/nrc2015. [DOI] [PubMed] [Google Scholar]
- 35.Shiizaki K., Kawanishi M., Yagi T. Modulation of benzo [a] pyrene–DNA adduct formation by CYP1 inducer and inhibitor. Genes and Environment. 2017;39(1):p. 14. doi: 10.1186/s41021-017-0076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li D., Wang M., Cheng S., et al. CYP1A1 based on metabolism of xenobiotics by cytochrome P450 regulates chicken male germ cell differentiation. In Vitro Cellular & Developmental Biology-Animal. 2017;53(4):293–303. doi: 10.1007/s11626-016-0108-z. [DOI] [PubMed] [Google Scholar]
- 37.Mena-Ulecia K., MacLeod-Carey D. Interactions of 2-phenyl-benzotriazole xenobiotic compounds with human Cytochrome P450-CYP1A1 by means of docking, molecular dynamics simulations and MM-GBSA calculations. Computational biology and chemistry. 2018;74:253–262. doi: 10.1016/j.compbiolchem.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 38.Katoh M. Effects of xenobiotics on drug pharmacokinetics and safety. Yakugaku zasshi: Journal of the Pharmaceutical Society of Japan. 2015;135(10):1115–1122. doi: 10.1248/yakushi.15-00187. [DOI] [PubMed] [Google Scholar]
- 39.Altay A., Bozoğlu F. Salvia fruticosa modulates mRNA expressions and activity levels of xenobiotic metabolizing CYP1A2, CYP2E1, NQO1, GPx, and GST enzymes in human colorectal adenocarcinoma HT-29 cells. Nutrition and cancer. 2017;69(6):892–903. doi: 10.1080/01635581.2017.1339817. [DOI] [PubMed] [Google Scholar]
- 40.He X., Feng S. Role of metabolic enzymes P450 (CYP) on activating procarcinogen and their polymorphisms on the risk of cancers. Current drug metabolism. 2015;16(10):850–863. doi: 10.2174/138920021610151210164501. [DOI] [PubMed] [Google Scholar]
- 41.Kim D., Guengerich F. P. Cytochrome P450 activation of arylamines and heterocyclic amines. Annual Review of Pharmacology and Toxicology. 2005;45(1):27–49. doi: 10.1146/annurev.pharmtox.45.120403.100010. [DOI] [PubMed] [Google Scholar]
- 42.Turesky R. J., Le Marchand L. Metabolism and biomarkers of heterocyclic aromatic amines in molecular epidemiology studies: lessons learned from aromatic amines. Chemical research in toxicology. 2011;24(8):1169–1214. doi: 10.1021/tx200135s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jorge-Nebert L. F., Zhang G., Wilson K. M., et al. Head-and-neck squamous cell carcinoma risk in smokers: no association detected between phenotype and AHR, CYP1A1, CYP1A2, or CYP1B1 genotype. Human genomics. 2016;10(1):p. 39. doi: 10.1186/s40246-016-0094-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Henderson C., Sahraouei A., Wolf C. Cytochrome P450s and chemoprevention. Portland Press Limited; 2000. [DOI] [PubMed] [Google Scholar]
- 45.Ahmed S., Valen E., Sandelin A., Matthews J. Dioxin increases the interaction between aryl hydrocarbon receptor and estrogen receptor alpha at human promoters. Toxicological Sciences. 2009;111(2):254–266. doi: 10.1093/toxsci/kfp144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miron A., Aprotosoaie A. C., Trifan A., Xiao J. Flavonoids as modulators of metabolic enzymes and drug transporters. Annals of the New York Academy of Sciences. 2017;1398(1):152–167. doi: 10.1111/nyas.13384. [DOI] [PubMed] [Google Scholar]
- 47.Dutkiewicz Z., Mikstacka R. Structure-based drug design for cytochrome P450 family 1 inhibitors. Bioinorganic chemistry and applications. 2018;2018:21. doi: 10.1155/2018/3924608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nebert D. W. Aryl hydrocarbon receptor (AHR): "pioneer member" of the basic- helix/loop/helix per-Arnt-sim (bHLH/PAS) family of "sensors" of foreign and endogenous signals. Progress in lipid research. 2017;67:38–57. doi: 10.1016/j.plipres.2017.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Göttel M., le Corre L., Dumont C., Schrenk D., Chagnon M. C. Estrogen receptor α and aryl hydrocarbon receptor cross-talk in a transfected hepatoma cell line (HepG2) exposed to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicology reports. 2014;1:1029–1036. doi: 10.1016/j.toxrep.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu J., Nogueira S. V., Drongelen V. ., et al. Shared epitope–aryl hydrocarbon receptor crosstalk underlies the mechanism of gene–environment interaction in autoimmune arthritis. Proceedings of the National Academy of Sciences. 2018;115(18):4755–4760. doi: 10.1073/pnas.1722124115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tappenden D. M., Hwang H. J., Yang L., Thomas R. S., LaPres J. J. The aryl-hydrocarbon receptor protein interaction network (AHR-PIN) as Identified by tandem affinity purification (TAP) and mass spectrometry. Journal of toxicology. 2013;2013:12. doi: 10.1155/2013/279829.279829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hankinson O. The role of AHR-inducible cytochrome P450s in metabolism of polyunsaturated fatty acids. Drug metabolism reviews. 2016;48(3):342–350. doi: 10.1080/03602532.2016.1197240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Androutsopoulos V. P., Tsatsakis A. M., Spandidos D. A. Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC cancer. 2009;9(1) doi: 10.1186/1471-2407-9-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Badal S., Delgoda R. CYP1B1: friend or foe? A critical review. OA Biochemistry. 2013;1(1) doi: 10.13172/2052-9651-1-1-601. [DOI] [Google Scholar]
- 55.Chang H.-F., Huffer D. M., Chiarelli M. P., Cho B. P. Characterization of DNA adducts and tetraols derived from anti-benzo [ghi] fluoranthane-3, 4-dihydrodiol-5, 5a-epoxide. Chemical research in toxicology. 2002;15(2):187–197. doi: 10.1021/tx010133d. [DOI] [PubMed] [Google Scholar]
- 56.von Weymarn L. B., Chun J. A., Hollenberg P. F. Effects of benzyl and phenethyl isothiocyanate on P450s 2A6 and 2A13: potential for chemoprevention in smokers. Carcinogenesis. 2005;27(4):782–790. doi: 10.1093/carcin/bgi301. [DOI] [PubMed] [Google Scholar]
- 57.Murphy S. E., Raulinaitis V., Brown K. M. Nicotine 5′-oxidation and methyl oxidation by P450 2A enzymes. Drug Metabolism and Disposition. 2005;33(8):1166–1173. doi: 10.1124/dmd.105.004549. [DOI] [PubMed] [Google Scholar]
- 58.Liu X., Zhang J., Zhang C., Yang B., Wang L., Zhou J. The inhibition of cytochrome P450 2A13-catalyzed NNK metabolism by NAT, NAB and nicotine. Toxicology Research. 2016;5(4):1115–1121. doi: 10.1039/c6tx00016a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hollander M. C., Zhou X., Maier C. R., Patterson A. D., Ding X., Dennis P. A. A Cyp2a polymorphism predicts susceptibility to NNK-induced lung tumorigenesis in mice. Carcinogenesis. 2011;32(8):1279–1284. doi: 10.1093/carcin/bgr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shimada T. Inhibition of carcinogen-activating cytochrome P450 enzymes by xenobiotic chemicals in relation to antimutagenicity and anticarcinogenicity. Toxicological research. 2017;33(2):79–96. doi: 10.5487/TR.2017.33.2.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anttila S., Raunio H., Hakkola J. Cytochrome P450–mediated pulmonary metabolism of Carcinogens. American journal of respiratory cell and molecular biology. 2011;44(5):583–590. doi: 10.1165/rcmb.2010-0189RT. [DOI] [PubMed] [Google Scholar]
- 62.Tanner J.-A., Tyndale R. Variation in CYP2A6 activity and personalized medicine. Journal of personalized medicine. 2017;7(4):p. 18. doi: 10.3390/jpm7040018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patramurti C., Sugiyanto, Nurrochmad A., Martono S. Polymorphism of cytochrome P450 2A6 (CYP2A6∗1 AND CYP2A6∗4) among Javanese Indonesian smoker and non smoker. Indonesian Journal of Pharmacy. 2015;26(1):p. 11. doi: 10.14499/indonesianjpharm26iss1pp11. [DOI] [Google Scholar]
- 64.Patramurti C., Martono S., Sugiyanto S., Nurrochmad A. Inter-individual variability of cytochrome P450 2A6 activity in Javanese smokers’ urine. Media Penelitian dan Pengembangan Kesehatan. 2017;27(3):133–140. doi: 10.22435/mpk.v27i3.4777.133-140. [DOI] [Google Scholar]
- 65.Brown K. M., von Weymarn L. B., Murphy S. E. Identification ofN-(Hydroxymethyl) norcotinine as a major product of cytochrome P450 2A6, but not cytochrome P450 2A13-catalyzed cotinine metabolism. Chemical research in toxicology. 2005;18(12):1792–1798. doi: 10.1021/tx0501381. [DOI] [PubMed] [Google Scholar]
- 66.von Weymarn L. B., Chun J. A., Knudsen G. A., Hollenberg P. F. Effects of eleven isothiocyanates on P450 2A6-and 2A13-catalyzed coumarin 7-hydroxylation. Chemical research in toxicology. 2007;20(9):1252–1259. doi: 10.1021/tx700078v. [DOI] [PubMed] [Google Scholar]
- 67.Lambert A. W., Pattabiraman D. R., Weinberg R. A. Emerging biological principles of metastasis. Cell. 2017;168(4):670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharma M., McCarthy E. T., Reddy D. S., et al. 8,9-Epoxyeicosatrienoic acid protects the glomerular filtration barrier. Prostaglandins & other lipid mediators. 2009;89(1-2):43–51. doi: 10.1016/j.prostaglandins.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Node K., Huo Y., Ruan X., et al. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science. 1999;285(5431):1276–1279. doi: 10.1126/science.285.5431.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jiang J.-G., Ning Y. G., Chen C., et al. Cytochrome p450 epoxygenase promotes human cancer metastasis. Cancer research. 2007;67(14):6665–6674. doi: 10.1158/0008-5472.CAN-06-3643. [DOI] [PubMed] [Google Scholar]
- 71.Chen C., Wang D. W. CYP epoxygenase derived EETs: from cardiovascular protection to human cancer therapy. Current topics in medicinal chemistry. 2013;13(12):1454–1469. doi: 10.2174/1568026611313120007. [DOI] [PubMed] [Google Scholar]
- 72.Ma J., Zhang L., Han W., et al. Activation of JNK/c-Jun is required for the proliferation, survival, and angiogenesis induced by EET in pulmonary artery endothelial cells. Journal of lipid research. 2012;53(6):1093–1105. doi: 10.1194/jlr.M024398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei X., Zhang D., Dou X., et al. Elevated 14,15- epoxyeicosatrienoic acid by increasing of cytochrome P450 2C8, 2C9 and 2J2 and decreasing of soluble epoxide hydrolase associated with aggressiveness of human breast cancer. BMC cancer. 2014;14(1) doi: 10.1186/1471-2407-14-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bruno R. D., Njar V. C. Targeting cytochrome P450 enzymes: a new approach in anti-cancer drug development. Bioorganic & Medicinal Chemistry. 2007;15(15):5047–5060. doi: 10.1016/j.bmc.2007.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang S., Lin L., Chen J.-X., et al. CytochromeP-450 epoxygenases protect endothelial cells from apoptosis induced by tumor necrosis factor-α via MAPK and PI3K/Akt signaling pathways. American Journal of Physiology-Heart and Circulatory Physiology. 2007;293(1):H142–H151. doi: 10.1152/ajpheart.00783.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang J.-G., Chen C. L., Card J. W., et al. Cytochrome P450 2J2 promotes the neoplastic phenotype of carcinoma cells and is up-regulated in human tumors. Cancer research. 2005;65(11):4707–4715. doi: 10.1158/0008-5472.CAN-04-4173. [DOI] [PubMed] [Google Scholar]
- 77.Johnson A. L., Edson K. Z., Totah R. A., Rettie A. E. Advances in Pharmacology. Elsevier; 2015. Cytochrome P450 ω-hydroxylases in inflammation and cancer; pp. 223–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu W., Chen L., Yang Y. Q., et al. Cytochrome P450 ω-hydroxylase promotes angiogenesis and metastasis by upregulation of VEGF and MMP-9 in non-small cell lung cancer. Cancer chemotherapy and pharmacology. 2011;68(3):619–629. doi: 10.1007/s00280-010-1521-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Seki T., Wang M. H., Miyata N., Laniado-Schwartzman M. Cytochrome P450 4A isoform inhibitory profile of N-hydroxy-N′-(4-butyl-2-methylphenyl)-formamidine (HET0016), a selective inhibitor of 20-HETE synthesis. Biological and Pharmaceutical Bulletin. 2005;28(9):1651–1654. doi: 10.1248/bpb.28.1651. [DOI] [PubMed] [Google Scholar]
- 80.Guo M., Roman R. J., Fenstermacher J. D., et al. 9L gliosarcoma cell proliferation and tumor growth in rats are suppressed byN-Hydroxy-N′-(4-butyl-2-methylphenol) formamidine (HET0016), a selective inhibitor of CYP4A. Journal of Pharmacology and Experimental Therapeutics. 2006;317(1):97–108. doi: 10.1124/jpet.105.097782. [DOI] [PubMed] [Google Scholar]
- 81.Borin T. F., Zuccari D. A. P. C., Jardim-Perassi B. V., et al. HET0016, a selective inhibitor of 20-HETE synthesis, decreases pro-angiogenic factors and inhibits growth of triple negative breast cancer in mice. PLoS One. 2014;9(12) doi: 10.1371/journal.pone.0116247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alexanian A., Rufanova V. A., Miller B., Flasch A., Roman R. J., Sorokin A. Down-regulation of 20-HETE synthesis and signaling inhibits renal adenocarcinoma cell proliferation and tumor growth. Anticancer research. 2009;29(10):3819–3824. [PMC free article] [PubMed] [Google Scholar]
- 83.Guo A. M., Sheng J., Scicli G. M., et al. Expression of CYP4A1 in U251 human glioma cell induces hyperproliferative phenotype in vitro and rapidly growing tumors in vivo. Journal of Pharmacology and Experimental Therapeutics. 2008;327(1):10–19. doi: 10.1124/jpet.108.140889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo M., Roman R. J., Falck J. R., Edwards P. A., Scicli A. G. Human U251 glioma cell proliferation is suppressed by HET0016 [N-hydroxy-N′-(4-butyl-2-methylphenyl) formamidine], a selective inhibitor of CYP4A. Journal of Pharmacology and Experimental Therapeutics. 2005;315(2):526–533. doi: 10.1124/jpet.105.088567. [DOI] [PubMed] [Google Scholar]
- 85.Jiao D., Wang J., Lu W., et al. Curcumin inhibited HGF-induced EMT and angiogenesis through regulating c-Met dependent PI3K/Akt/mTOR signaling pathways in lung cancer. Molecular Therapy-Oncolytics. 2016;3(3) doi: 10.1038/mto.2016.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.He Q., Ren X., Chen J., et al. miR-16 targets fibroblast growth factor 2 to inhibit NPC cell proliferation and invasion via PI3K/AKT and MAPK signaling pathways. Oncotarget. 2016;7(3):3047–3058. doi: 10.18632/oncotarget.6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rocic P., Schwartzman M. L. 20-HETE in the regulation of vascular and cardiac function. Pharmacology & therapeutics. 2018;192:74–87. doi: 10.1016/j.pharmthera.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Borin T., Angara K., Rashid M., Achyut B., Arbab A. Arachidonic acid metabolite as a novel therapeutic target in breast cancer metastasis. International journal of molecular sciences. 2017;18(12):p. 2661. doi: 10.3390/ijms18122661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Edson K. Z., Rettie A. E. CYP4 enzymes as potential drug targets: focus on enzyme multiplicity, inducers and inhibitors, and therapeutic modulation of 20-hydroxyeicosatetraenoic acid (20-HETE) synthase and fatty acid ω-hydroxylase activities. Current topics in medicinal chemistry. 2013;13(12):1429–1440. doi: 10.2174/15680266113139990110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guo Z., Johnson V., Barrera J., et al. Targeting cytochrome P450-dependent cancer cell mitochondria: cancer associated CYPs and where to find them. Cancer and Metastasis Reviews. 2018;37(2-3):409–423. doi: 10.1007/s10555-018-9749-6. [DOI] [PubMed] [Google Scholar]
- 91.Guo Z., Norris B., Henriksen J., et al. CYP3A4 promotes mammary carcinoma angiogenesis in a cell iIntrinsic fashion. AACR; 2013. [Google Scholar]
- 92.Mitra R., Guo Z., Milani M., et al. CYP3A4 mediates growth of estrogen receptor-positive breast cancer cells in part by inducing nuclear translocation of phospho-Stat3 through biosynthesis of (±)-14, 15-epoxyeicosatrienoic acid (EET) Journal of Biological Chemistry. 2011;286(20):17543–17559. doi: 10.1074/jbc.M110.198515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murray G. I., Taylor M. C., McFadyen M., et al. Tumor-specific expression of cytochrome P450 CYP1B1. Cancer research. 1997;57(14):3026–3031. [PubMed] [Google Scholar]
- 94.He X.-F., Wei J., Liu Z. Z., et al. Association between CYP1A2 and CYP1B1 polymorphisms and colorectal cancer risk: a meta-analysis. PloS one. 2014;9(8):p. e100487. doi: 10.1371/journal.pone.0100487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Thyagarajan B., Brott M., Mink P., et al. CYP1B1 and CYP19 gene polymorphisms and breast cancer incidence: no association in the ARIC study. Cancer letters. 2004;207(2):183–189. doi: 10.1016/j.canlet.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 96.Yu A.-M., Fukamachi K., Krausz K. W., Cheung C., Gonzalez F. J. Potential role for human cytochrome P450 3A4 in estradiol homeostasis. Endocrinology. 2005;146(7):2911–2919. doi: 10.1210/en.2004-1248. [DOI] [PubMed] [Google Scholar]
- 97.Nishida C. R., Everett S., Ortiz de Montellano P. R. Specificity determinants of CYP1B1 estradiol hydroxylation. Molecular pharmacology. 2013;84(3):451–458. doi: 10.1124/mol.113.087700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kamel M., Shouman S., el-Merzebany M., et al. Effect of tumour necrosis factor-alpha on estrogen metabolic pathways in breast cancer cells. Journal of Cancer. 2012;3:310–321. doi: 10.7150/jca.4584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.van Aswegen C. H., Purdy R. H., Wittliff J. L. Binding of 2-hydroxyestradiol and 4-hydroxyestradiol to estrogen receptors from human breast cancers. Journal of steroid biochemistry. 1989;32(4):485–492. doi: 10.1016/0022-4731(89)90380-4. [DOI] [PubMed] [Google Scholar]
- 100.Cavalieri E. L. Estrogen-induced depurination of DNA: a novel target for breast cancer prevention. NEBRASKA UNIV AT OMAHA; 2007. [Google Scholar]
- 101.Zhu B. T. On the general mechanism of selective induction of cytochrome P450 enzymes by chemicals: some theoretical considerations. Expert opinion on drug metabolism & toxicology. 2010;6(4):483–494. doi: 10.1517/17425250903578642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Puppala D., Gairola C. G., Swanson H. I. Identification of kaempferol as an inhibitor of cigarette smoke-induced activation of the aryl hydrocarbon receptor and cell transformation. Carcinogenesis. 2007;28(3):639–647. doi: 10.1093/carcin/bgl169. [DOI] [PubMed] [Google Scholar]
- 103.Zhang S., Qin C., Safe S. H. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: effects of structure and cell context. Environmental health perspectives. 2003;111(16):1877–1882. doi: 10.1289/ehp.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Busbee P. B., Rouse M., Nagarkatti M., Nagarkatti P. S. Use of natural AhR ligands as potential therapeutic modalities against inflammatory disorders. Nutrition reviews. 2013;71(6):353–369. doi: 10.1111/nure.12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.D'Uva G., Baci D., Albini A., Noonan D. M. Cancer chemoprevention revisited: cytochrome P450 family 1B1 as a target in the tumor and the microenvironment. Cancer treatment reviews. 2018;63:1–18. doi: 10.1016/j.ctrv.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 106.Bradshaw T. Phortress: the smart antitumour agent which induces its own metabolism. Pharmaceutical Journal. 2010;284(7584):p. 23. [Google Scholar]
- 107.Perez A. L., Bradshaw T. Ovarian Cancer. IntechOpen; 2018. Exploring new molecular targets in advanced ovarian cancer: the arylhydrocarbon receptor (AhR) and antitumor benzothiazole ligands as potential therapeutic candidates. [Google Scholar]
- 108.McLean L. S., Watkins C. N., Campbell P., et al. Aryl hydrocarbon receptor ligand 5F 203 induces oxidative stress that triggers DNA damage in human breast cancer cells. Chemical research in toxicology. 2015;28(5):855–871. doi: 10.1021/tx500485v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Huang X., Cao M., Wang L., et al. Expression of sulfotransferase SULT1A1 in cancer cells predicts susceptibility to the novel anticancer agent NSC-743380. Oncotarget. 2015;6(1):345–354. doi: 10.18632/oncotarget.2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Luzzani G. A., Callero M. A., Kuruppu A. I., et al. In vitro antitumor effects of AHR ligands aminoflavone (AFP 464) and benzothiazole (5F 203) in human renal carcinoma cells. Journal of cellular biochemistry. 2017;118(12):4526–4535. doi: 10.1002/jcb.26114. [DOI] [PubMed] [Google Scholar]
- 111.Ruparelia K. C., Zeka K., Ijaz T., et al. The synthesis of chalcones as anticancer prodrugs and their bioactivation in CYP1 expressing breast cancer cells. Medicinal Chemistry. 2018;14(4):322–332. doi: 10.2174/1573406414666180112120134. [DOI] [PubMed] [Google Scholar]
- 112.Karlgren M., Miura S., Ingelmansundberg M. Novel extrahepatic cytochrome P450s. Toxicology and applied pharmacology. 2005;207(2):57–61. doi: 10.1016/j.taap.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 113.Hrycay E. G., Bandiera S. M. Advances in Pharmacology. Elsevier; 2015. Involvement of cytochrome P450 in reactive oxygen species formation and cancer; pp. 35–84. [DOI] [PubMed] [Google Scholar]
- 114.Rajendran P., Ekambaram G., Sakthisekaran D. Protective role of mangiferin against benzo (a) pyrene induced lung carcinogenesis in experimental animals. Biological and Pharmaceutical Bulletin. 2008;31(6):1053–1058. doi: 10.1248/bpb.31.1053. [DOI] [PubMed] [Google Scholar]
- 115.Akhdar H., Legendre C., Aninat C., More F. Topics on Drug Metabolism. IntechOpen; 2012. Anticancer drug metabolism: chemotherapy resistance and new therapeutic approaches. [Google Scholar]
- 116.Sim S. C., Ingelman-Sundberg M. The Human Cytochrome P450 (CYP) Allele Nomenclature website: a peer-reviewed database of CYP variants and their associated effects. Human genomics. 2010;4(4):278–281. doi: 10.1186/1479-7364-4-4-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rodriguez-Antona C., Ingelman-Sundberg M. Cytochrome P 450 pharmacogenetics and cancer. Oncogene. 2006;25(11):1679–1691. doi: 10.1038/sj.onc.1209377. [DOI] [PubMed] [Google Scholar]
- 118.Nishida C. R., Lee M., de Montellano P. R. O. Efficient hypoxic activation of the anticancer agent AQ4N by CYP2S1 and CYP2W1. Molecular pharmacology. 2010;78(3):497–502. doi: 10.1124/mol.110.065045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Engels F. K., Sparreboom A., Mathot R. A. A., Verweij J. Potential for improvement of docetaxel-based chemotherapy: a pharmacological review. British journal of cancer. 2005;93(2):173–177. doi: 10.1038/sj.bjc.6602698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mathijssen R. H. J., de Jong F. A., van Schaik R. H. N., et al. Prediction of irinotecan pharmacokinetics by use of cytochrome P450 3A4 phenotyping probes. Journal of the National Cancer Institute. 2004;96(21):1585–1592. doi: 10.1093/jnci/djh298. [DOI] [PubMed] [Google Scholar]
- 121.Basseville A., Preisser L., de Carné Trécesson S., et al. Irinotecan induces steroid and xenobiotic receptor (SXR) signaling to detoxification pathway in colon cancer cells. Molecular cancer. 2011;10(1):p. 80. doi: 10.1186/1476-4598-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Goh B.-C., Lee S.-C., Wang L.-Z., et al. Explaining interindividual variability of docetaxel pharmacokinetics and pharmacodynamics in Asians through phenotyping and genotyping strategies. Journal of clinical oncology. 2002;20(17):3683–3690. doi: 10.1200/JCO.2002.01.025. [DOI] [PubMed] [Google Scholar]
- 123.Zhang X., Li X., You Q., Zhang X. Prodrug strategy for cancer cell-specific targeting: a recent overview. European journal of medicinal chemistry. 2017;139:542–563. doi: 10.1016/j.ejmech.2017.08.010. [DOI] [PubMed] [Google Scholar]
- 124.Chun Y. J., Kim S. Discovery of cytochrome P450 1B1 inhibitors as new promising anti-cancer agents. Medicinal research reviews. 2003;23(6):657–668. doi: 10.1002/med.10050. [DOI] [PubMed] [Google Scholar]
- 125.Stavrinou P., Mavrogiorgou M.-C., Polyzoidis K., et al. Expression profile of genes related to drug metabolism in human brain tumors. PloS one. 2015;10(11, article e0143285) doi: 10.1371/journal.pone.0143285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Elfaki I., Mir R., Almutairi F. M., Duhier F. M. A. Cytochrome P450: polymorphisms and roles in cancer, diabetes and atherosclerosis. Asian Pacific journal of cancer prevention: APJCP. 2018;19(8):2057–2070. doi: 10.22034/APJCP.2018.19.8.2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sun J., Zhang H., Gao M., et al. Association between CYP17 T-34C rs743572 and breast cancer risk. Oncotarget. 2018;9(3):4200–4213. doi: 10.18632/oncotarget.23688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vangsted A. J., Søeby K., Klausen T. W., et al. No influence of the polymorphisms CYP2C19 and CYP2D6 on the efficacy of cyclophosphamide, thalidomide, and bortezomib in patients with multiple myeloma. BMC cancer. 2010;10(1):p. 404. doi: 10.1186/1471-2407-10-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Daigo S., Takahashi Y., Fujieda M., et al. A novel mutant allele of the CYP2A6 gene (CYP2A6∗11) found in a cancer patient who showed poor metabolic phenotype towards tegafur. Pharmacogenetics and Genomics. 2002;12(4):299–306. doi: 10.1097/00008571-200206000-00005. [DOI] [PubMed] [Google Scholar]
- 130.Maddin N., Husin A., Gan S. H., Aziz B. A., Ankathil R. Impact of CYP3A4∗18 and CYP3A5∗3 polymorphisms on imatinib mesylate response among chronic myeloid leukemia patients in Malaysia. Oncology and Therapy. 2016;4(2):303–314. doi: 10.1007/s40487-016-0035-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Haque M. W., Pattanayak S. P. Taxifolin inhibits 7, 12-dimethylbenz (a) anthracene-induced breast carcinogenesis by regulating AhR/CYP1A1 signaling pathway. Pharmacognosy magazine. 2018;13:p. S749. doi: 10.4103/pm.pm_315_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Phalen L. J., Köllner B., Hogan N. S., van den Heuvel M. R. Transcriptional response in rainbow trout (Oncorhynchus mykiss) B cells and thrombocytes following in vivo exposure to benzo[a]pyrene. Environmental toxicology and pharmacology. 2017;53:212–218. doi: 10.1016/j.etap.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 133.Jiang W., Wang L., Kondraganti S. R., et al. Disruption of the gene for CYP1A2, which is expressed primarily in liver, leads to differential regulation of hepatic and pulmonary mouse CYP1A1 expression and augmented human CYP1A1 transcriptional activation in response to 3-methylcholanthrene in vivo. Journal of Pharmacology and Experimental Therapeutics. 2010;335(2):369–379. doi: 10.1124/jpet.110.171173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rendic S., Guengerich F. P. Contributions of human enzymes in carcinogen metabolism. Chemical research in toxicology. 2012;25(7):1316–1383. doi: 10.1021/tx300132k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Koda M., Iwasaki M., Yamano Y., Lu X., Katoh T. Association between NAT2, CYP1A1, and CYP1A2 genotypes, heterocyclic aromatic amines, and prostate cancer risk: a case control study in Japan. Environmental health and preventive medicine. 2017;22(1):p. 72. doi: 10.1186/s12199-017-0681-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Iyer S., Wang Y., Xiong W., et al. Significant interactions between maternal PAH exposure and single nucleotide polymorphisms in candidate genes on B [a] P–DNA adducts in a cohort of non-smoking Polish mothers and newborns. Carcinogenesis. 2016;37(11):1110–1115. doi: 10.1093/carcin/bgw090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zhang N.-Y., Qi M., Zhao L., et al. Curcumin prevents aflatoxin B1 hepatoxicity by inhibition of cytochrome P450 isozymes in chick liver. Toxins. 2016;8(11):p. 327. doi: 10.3390/toxins8110327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yuan J.-M., Nelson H. H., Carmella S. G., et al. CYP2A6 genetic polymorphisms and biomarkers of tobacco smoke constituents in relation to risk of lung cancer in the Singapore Chinese Health Study. Carcinogenesis. 2017;38(4):411–418. doi: 10.1093/carcin/bgx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zarth A. T., Upadhyaya P., Yang J., Hecht S. S. DNA adduct formation from metabolic 5′-hydroxylation of the tobacco-specific CarcinogenN′-Nitrosonornicotine in human enzyme systems and in rats. Chemical research in toxicology. 2016;29(3):380–389. doi: 10.1021/acs.chemrestox.5b00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chiang H.-c., Wang C.-Y., Lee H.-L., Tsou T.-C. Metabolic effects of CYP2A6 and CYP2A13 on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK)-induced gene mutation--A mammalian cell-based mutagenesis approach. Toxicology and applied pharmacology. 2011;253(2):145–152. doi: 10.1016/j.taap.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 141.Weems J. M., Lamb J. G., D’Agostino J., Ding X., Yost G. S. Potent mutagenicity of 3-methylindole requires pulmonary cytochrome P450-mediated bioactivation: a comparison to the prototype cigarette smoke mutagens B (a) P and NNK. Chemical research in toxicology. 2010;23(11):1682–1690. doi: 10.1021/tx100147z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ji M., Zhang Y., Li N., et al. Nicotine component of cigarette smoke extract (cse) decreases the cytotoxicity of CSE in BEAS-2B cells stably expressing human cytochrome P450 2A13. International journal of environmental research and public health. 2017;14(10):p. 1221. doi: 10.3390/ijerph14101221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jiang H., Wu J., Zhang F., Wen J., Jiang J., Deng Y. The critical role of porcine cytochrome P450 3A46 in the bioactivation of aflatoxin B1. Biochemical pharmacology. 2018;156:177–185. doi: 10.1016/j.bcp.2018.08.030. [DOI] [PubMed] [Google Scholar]
- 144.Cho T. M., Rose R. L., Hodgson E. The effect of chlorpyrifos-oxon and other xenobiotics on the human cytochrome P450-dependent metabolism of naphthalene and deet. Drug metabolism and drug interactions. 2007;22(4):235–262. doi: 10.1515/dmdi.2007.22.4.235. [DOI] [PubMed] [Google Scholar]
- 145.Fasullo M., Freedland J., St John N., et al. An in vitro system for measuring genotoxicity mediated by human CYP3A4 in Saccharomyces cerevisiae. Environmental and molecular mutagenesis. 2017;58(4):217–227. doi: 10.1002/em.22093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bren U., Fuchs J. E., Oostenbrink C. Cooperative binding of aflatoxin B1 by cytochrome P450 3A4: a computational study. Chemical research in toxicology. 2014;27(12):2136–2147. doi: 10.1021/tx5004062. [DOI] [PubMed] [Google Scholar]
- 147.Wormhoudt L. W., Ploemen J. H. T. M., de Waziers I., et al. Inter-individual variability in the oxidation of 1,2-dibromoethane: use of heterologously expressed human cytochrome P450 and human liver microsomes. Chemico-biological interactions. 1996;101(3):175–192. doi: 10.1016/0009-2797(96)03723-4. [DOI] [PubMed] [Google Scholar]
- 148.Coleman S., Linderman R., Hodgson E., Rose R. L. Comparative metabolism of chloroacetamide herbicides and selected metabolites in human and rat liver microsomes. Environmental health perspectives. 2000;108(12):1151–1157. doi: 10.1289/ehp.001081151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nock N. L., Tang D., Rundle A., et al. Associations between smoking, polymorphisms in polycyclic aromatic hydrocarbon (PAH) metabolism and conjugation genes and PAH-DNA adducts in prostate tumors differ by race. Cancer Epidemiology Biomarkers & Prevention. 2007;16(6):1236–1245. doi: 10.1158/1055-9965.EPI-06-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Piipari R., Savela K., Nurminen T., et al. Expression of CYP1A1, CYP1B1 and CYP3A, and polycyclic aromatic hydrocarbon-DNA adduct formation in bronchoalveolar macrophages of smokers and non-smokers. International journal of cancer. 2000;86(5):610–616. doi: 10.1002/(SICI)1097-0215(20000601)86:5<610::AID-IJC2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 151.Chang T. K. H., Yu L., Maurel P., Waxman D. J. Enhanced cyclophosphamide and ifosfamide activation in primary human hepatocyte cultures: response to cytochrome P-450 inducers and autoinduction by oxazaphosphorines. Cancer research. 1997;57(10):1946–1954. [PubMed] [Google Scholar]
- 152.Fujita K.-i., Kamataki T. Predicting the mutagenicity of tobacco‐related N‐nitrosamines in humans using 11 strains of Salmonella typhimurium YG7108, each coexpressing a form of human cytochrome P450 along with NADPH–cytochrome P450 reductase. Environmental and molecular mutagenesis. 2001;38(4):339–346. doi: 10.1002/em.10036. [DOI] [PubMed] [Google Scholar]
- 153.Ruiz-Pinto S., Pita G., Patiño-García A., et al. Identification of genetic variants in pharmacokinetic genes associated with Ewing Sarcoma treatment outcome. Annals of Oncology. 2016;27(9):1788–1793. doi: 10.1093/annonc/mdw234. [DOI] [PubMed] [Google Scholar]
- 154.Kamataki T., Fujita K.-i., Nakayama K., Yamazaki Y., Miyamoto M., Ariyoshi N. Role of human cytochrome P450 (CYP) in the metabolic activation of nitrosamine derivatives: application of genetically engineered Salmonella expressing human CYP. Drug metabolism reviews. 2002;34(3):667–676. doi: 10.1081/DMR-120005668. [DOI] [PubMed] [Google Scholar]
- 155.Calinski D. M., Zhang H., Ludeman S., Dolan M. E., Hollenberg P. F. Hydroxylation and N-dechloroethylation of Ifosfamide and deuterated ifosfamide by the human cytochrome p450s and their commonly occurring polymorphisms. Drug Metabolism and Disposition. 2015;43(7):1084–1090. doi: 10.1124/dmd.115.063628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Schirmer J. H., Bremer J. P., Moosig F., et al. Cyclophosphamide treatment-induced leukopenia rates in ANCA-associated vasculitis are influenced by variant CYP450 2C9 genotypes. Pharmacogenomics. 2016;17(4):367–374. doi: 10.2217/pgs.15.176. [DOI] [PubMed] [Google Scholar]
- 157.Miyazaki M., Sugawara E., Yoshimura T., Yamazaki H., Kamataki T. Mutagenic activation of betel quid-specific N-nitrosamines catalyzed by human cytochrome P450 coexpressed with NADPH-cytochrome P450 reductase in Salmonella typhimurium YG7108. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 2005;581(1-2):165–171. doi: 10.1016/j.mrgentox.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 158.Zhu A. Z. X., Binnington M. J., Renner C. C., et al. Alaska Native smokers and smokeless tobacco users with slower CYP2A6 activity have lower tobacco consumption, lower tobacco-specific nitrosamine exposure and lower tobacco-specific nitrosamine bioactivation. Carcinogenesis. 2013;34(1):93–101. doi: 10.1093/carcin/bgs306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Satoh D., Iwado S., Abe S., et al. Establishment of a novel hepatocyte model that expresses four cytochrome P450 genes stably via mammalian-derived artificial chromosome for pharmacokinetics and toxicity studies. PloS one. 2017;12(10, article e0187072) doi: 10.1371/journal.pone.0187072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sun Q., Wang G., Gao L., et al. Roles of CYP2e1 in 1, 2-dichloroethane-induced liver damage in mice. Environmental toxicology. 2016;31(11):1430–1438. doi: 10.1002/tox.22148. [DOI] [PubMed] [Google Scholar]
- 161.Hartman J. H., Miller G. P., Caro A. A., et al. 1,3-Butadiene-induced mitochondrial dysfunction is correlated with mitochondrial CYP2E1 activity in Collaborative Cross mice. Toxicology. 2017;378:114–124. doi: 10.1016/j.tox.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kim A., Parry E. M., Parry J. M. Effects of chlorinated aliphatic hydrocarbons on the fidelity of cell division in human CYP2E1 expressing cells. Experimental & molecular medicine. 2002;34(1):83–89. doi: 10.1038/emm.2002.12. [DOI] [PubMed] [Google Scholar]
- 163.Huffman M. P., Høie A. H., Svendsen C., et al. An in vitro study on the genotoxic effect of substituted furans in cells transfected with human metabolizing enzymes: 2, 5-dimethylfuran and furfuryl alcohol. Mutagenesis. 2016;31(5):597–602. doi: 10.1093/mutage/gew025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Doerr-Stevens J. K., Liu J., Stevens G. J., et al. Induction of cytochrome P-450 enzymes after repeated exposure to 4-vinylcyclohexene in B6C3F1 mice. Drug metabolism and disposition. 1999;27(2):281–287. [PubMed] [Google Scholar]
- 165.Weems J. M., Yost G. S. 3-Methylindole metabolites induce lung CYP1A1 and CYP2F1 enzymes by AhR and non-AhR mechanisms, respectively. Chemical research in toxicology. 2010;23(3):696–704. doi: 10.1021/tx9004506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lanza D. L., Code E., Crespi C. L., Gonzalez F. J., Yost G. S. Specific dehydrogenation of 3-methylindole and epoxidation of naphthalene by recombinant human CYP2F1 expressed in lymphoblastoid cells. Drug Metabolism and Disposition. 1999;27(7):798–803. [PubMed] [Google Scholar]
- 167.Cruzan G., Bus J., Hotchkiss J., et al. Studies of styrene, styrene oxide and 4-hydroxystyrene toxicity in CYP2F2 knockout and CYP2F1 humanized mice support lack of human relevance for mouse lung tumors. Regulatory Toxicology and Pharmacology. 2013;66(1):24–29. doi: 10.1016/j.yrtph.2013.02.008. [DOI] [PubMed] [Google Scholar]
- 168.Karlgren M., Ingelman-Sundberg M. Tumour-specific expression of CYP2W1: its potential as a drug target in cancer therapy. Expert opinion on therapeutic targets. 2006;11(1):61–67. doi: 10.1517/14728222.11.1.61. [DOI] [PubMed] [Google Scholar]
- 169.Luckert C., Ehlers A., Buhrke T., Seidel A., Lampen A., Hessel S. Polycyclic aromatic hydrocarbons stimulate human CYP3A4 promoter activity via PXR. Toxicology letters. 2013;222(2):180–188. doi: 10.1016/j.toxlet.2013.06.243. [DOI] [PubMed] [Google Scholar]
- 170.Yanev S., Stoyanova T. Manipulating cytochrome P450 enzymes: new perspectives for cancer treatment. Biomedical Reviews. 2018;28(0):120–124. doi: 10.14748/bmr.v28.4458. [DOI] [Google Scholar]
- 171.Kuan H., Loewen G., Geiser R. Lack of effect of ketoconazole on the pharmacokinetics of oral bexarotene in healthy subjects. Clinical Pharmacology & Therapeutics. 2005;77(2):p. P74. doi: 10.1016/j.clpt.2004.12.174. [DOI] [Google Scholar]
- 172.Gulbis A. M., Culotta K. S., Jones R. B., Andersson B. S. Busulfan and metronidazole: an often forgotten but significant drug interaction. Annals of Pharmacotherapy. 2011;45(7-8, article e39):p. 1024. doi: 10.1345/aph.1Q087. [DOI] [PubMed] [Google Scholar]
- 173.Quintanilha J. C. F., de Sousa V. M., Visacri M. B., et al. Involvement of cytochrome P450 in cisplatin treatment: implications for toxicity. Cancer chemotherapy and pharmacology. 2017;80(2):223–233. doi: 10.1007/s00280-017-3358-x. [DOI] [PubMed] [Google Scholar]
- 174.Pinto N., Ludeman S. M., Dolan M. E. Drug focus: pharmacogenetic studies related to cyclophosphamide-based therapy. Pharmacogenomics. 2009;10(12):1897–1903. doi: 10.2217/pgs.09.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Panneerselvam S., Yesudhas D., Durai P., Anwar M., Gosu V., Choi S. A combined molecular docking/dynamics approach to probe the binding mode of cancer drugs with cytochrome P450 3A4. Molecules. 2015;20(8):14915–14935. doi: 10.3390/molecules200814915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Preissner S., Simmaco M., Gentile G., Preissner R. Advances in Pharmacology. Elsevier; 2015. Personalized Cancer Therapy Considering Cytochrome P450 Variability; pp. 113–130. [DOI] [PubMed] [Google Scholar]
- 177.May J. E., Xu J., Morse H. R., Avent N. D., Donaldson C. Toxicity testing: the search for anin vitroalternative to animal testing. British journal of biomedical science. 2009;66(3):160–165. doi: 10.1080/09674845.2009.11730265. [DOI] [PubMed] [Google Scholar]
- 178.Mach C. M., Fugii H., Wakame K., Smith J. Evaluation of active hexose correlated compound hepatic metabolism and potential for drug interactions with chemotherapy agents. Journal of the Society for Integrative Oncology. 2008;6(3):105–109. [PubMed] [Google Scholar]
- 179.Zhou S.-F., Yang L.-P., Zhou Z.-W., Liu Y.-H., Chan E. Insights into the substrate specificity, inhibitors, regulation, and polymorphisms and the clinical impact of human cytochrome P450 1A2. The AAPS journal. 2009;11(3):481–494. doi: 10.1208/s12248-009-9127-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Ruggiero A., Cefalo M. G., Coccia P., Mastrangelo S., Maurizi P., Riccardi R. The role of diet on the clinical pharmacology of oral antineoplastic agents. European journal of clinical pharmacology. 2012;68(2):115–122. doi: 10.1007/s00228-011-1102-8. [DOI] [PubMed] [Google Scholar]
- 181.Brueggemeier R. W. Overview of the pharmacology of the aromatase inactivator exemestane. Breast cancer research and treatment. 2002;74(2):177–185. doi: 10.1023/A:1016121822916. [DOI] [PubMed] [Google Scholar]
- 182.Robertson J. F. R., Harrison M. Fulvestrant: pharmacokinetics and pharmacology. British journal of cancer. 2004;90(S1):S7–S10. doi: 10.1038/sj.bjc.6601630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Norden A. D., Raizer J. J., Abrey L. E., et al. Phase II trials of erlotinib or gefitinib in patients with recurrent meningioma. Journal of neuro-oncology. 2010;96(2):211–217. doi: 10.1007/s11060-009-9948-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Colburn D. E., Giles F. J., Oladovich D., Smith J. A. In VitroEvaluation of cytochrome P450-mediated drug interactions between cytarabine, idarubicin, itraconazole and caspofungin. Hematology. 2004;9(3):217–221. doi: 10.1080/10245330410001701585. [DOI] [PubMed] [Google Scholar]
- 185.Ortiz de Montellano P. R. Cytochrome P450-activated prodrugs. Future medicinal chemistry. 2013;5(2):213–228. doi: 10.4155/fmc.12.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee V. W. Y., Hu M., Tomlinson B. Integrating pharmacogenetics for assessment of herb-drug interactions: has the time come? Current Pharmacogenomics and Personalized Medicine. 2012;10(3):247–253. doi: 10.2174/187569212802509988. [DOI] [Google Scholar]
- 187.Santos A., Zanetta S., Cresteil T., et al. Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans. Clinical Cancer Research. 2000;6(5):2012–2020. [PubMed] [Google Scholar]
- 188.Murai K., Yamazaki H., Nakagawa K., Kawai R., Kamataki T. Deactivation of anti-cancer drug letrozole to a carbinol metabolite by polymorphic cytochrome P450 2A6 in human liver microsomes. Xenobiotica. 2009;39(11):795–802. doi: 10.3109/00498250903171395. [DOI] [PubMed] [Google Scholar]
- 189.Rahman A., Korzekwa K. R., Grogan J., Gonzalez F. J., Harris J. W. Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2C8. Cancer research. 1994;54(21):5543–5546. [PubMed] [Google Scholar]
- 190.Notley L. M., de Wolf C. J. F., Wunsch R. M., Lancaster R. G., Gillam E. M. J. Bioactivation of tamoxifen by recombinant human cytochrome P450 enzymes. Chemical research in toxicology. 2002;15(5):614–622. doi: 10.1021/tx0100439. [DOI] [PubMed] [Google Scholar]
- 191.Jacobson P., Green K., Birnbaum A., Remmel R. Cytochrome P450 isozymes 3A4 and 2B6 are involved in the in vitro human metabolism of thiotepa to TEPA. Cancer chemotherapy and pharmacology. 2002;49(6):461–467. doi: 10.1007/s00280-002-0453-3. [DOI] [PubMed] [Google Scholar]
- 192.Mounier N., Katlama C., Costagliola D., Chichmanian R.-M., Spano J.-P. Drug interactions between antineoplastic and antiretroviral therapies: Implications and management for clinical practice. Critical reviews in oncology/hematology. 2009;72(1):10–20. doi: 10.1016/j.critrevonc.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 193.Styles J. A., Davies A., Lim C. K., et al. Genotoxicity of tamoxifen epoxide and toremifene in human lymphoblastoid cells containing human cytochrome P450s. Carcinogenesis. 1994;15(1):5–9. doi: 10.1093/carcin/15.1.5. [DOI] [PubMed] [Google Scholar]
- 194.Javed F., Al-Hezaimi K., Al-Rasheed A., Almas K., Romanos G. E. Implant survival rate after oral cancer therapy: a review. Oral oncology. 2010;46(12):854–859. doi: 10.1016/j.oraloncology.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 195.Ohnishi A., Matsuo H., Yamada S., et al. Effect of furanocoumarin derivatives in grapefruit juice on the uptake of vinblastine by Caco-2 cells and on the activity of cytochrome P450 3A4. British journal of pharmacology. 2000;130(6):1369–1377. doi: 10.1038/sj.bjp.0703433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Groninger E., Meeuwsen-de Boer T., Koopmans P., et al. Pharmacokinetics of vincristine monotherapy in childhood acute lymphoblastic leukemia. Pediatric Research. 2002;52(1):113–118. doi: 10.1203/00006450-200207000-00021. [DOI] [PubMed] [Google Scholar]
- 197.Gebbia V., Puozzo C. Oral versus intravenous vinorelbine: clinical safety profile. Expert opinion on drug safety. 2005;4(5):915–928. doi: 10.1517/14740338.4.5.915. [DOI] [PubMed] [Google Scholar]
- 198.Vijayalakshmi K., Vettriselvi V., Krishnan M., Shroff S., Jayanth V. R., Paul S. F. D. Cytochrome p4501A1 gene variants as susceptibility marker for prostate cancer. Cancer Biomarkers. 2005;1(4-5):251–258. doi: 10.3233/CBM-2005-14-508. [DOI] [PubMed] [Google Scholar]
- 199.Qrafli M., Amar Y., Bourkadi J., et al. The CYP7A1 gene rs3808607 variant is associated with susceptibility of tuberculosis in Moroccan population. Pan African Medical Journal. 2014;18 doi: 10.11604/pamj.2014.18.1.3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Wang H., Zhang Z., Han S., Lu Y., Feng F., Yuan J. CYP1A2 rs762551 polymorphism contributes to cancer susceptibility: a meta-analysis from 19 case-control studies. BMC cancer. 2012;12(1):p. 528. doi: 10.1186/1471-2407-12-528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Gomez L., Kovac J. R., Lamb D. J. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids. 2015;95:80–87. doi: 10.1016/j.steroids.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Schlingmann K. P., Kaufmann M., Weber S., et al. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. New England Journal of Medicine. 2011;365(5):410–421. doi: 10.1056/NEJMoa1103864. [DOI] [PubMed] [Google Scholar]
- 203.Bousoula E., Kolovou V., Vasiliadis I., et al. CYP8A1 gene polymorphisms and left main coronary artery disease. Angiology. 2011;63(6):461–465. doi: 10.1177/0003319711425230. [DOI] [PubMed] [Google Scholar]
- 204.Zheng J., Yan H., Shi L., et al. The CYP19A1 rs3751592 variant confers susceptibility to Alzheimer disease in the Chinese Han population. Medicine. 2016;95(35):p. e4742. doi: 10.1097/md.0000000000004742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Elfaki I., Almutairi F. M., Mir R., Khan R., Abu-Duhier F. Cytochrome P450 CYP1B1∗2 gene and its association with T2D in Tabuk population, northwestern region of Saudi Arabia. Asian Journal of Pharmaceutical and Clinical Research. 2018;11(1):55–59. doi: 10.22159/ajpcr.2018.v11i1.21657. [DOI] [Google Scholar]
- 206.Zhang H., Jin L., Mu T., et al. Associations of CYP4A11 gene–gene and gene–smoking interactions with essential hypertension in the male eastern Chinese Han population. Clinical and Experimental Hypertension. 2017;39(5):448–453. doi: 10.1080/10641963.2016.1267201. [DOI] [PubMed] [Google Scholar]
- 207.Song C. Y., Ghafoor K., Ghafoor H. U., et al. Cytochrome P450 1B1 contributes to the development of atherosclerosis and hypertension in apolipoprotein E–deficient mice. Hypertension. 2016;67(1):206–213. doi: 10.1161/HYPERTENSIONAHA.115.06427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Polonikov A., Kharchenko A., Bykanova M., et al. Polymorphisms of CYP2C8, CYP2C9 and CYP2C19 and risk of coronary heart disease in Russian population. Gene. 2017;627:451–459. doi: 10.1016/j.gene.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 209.Wang S.-Y., Xing P.-F., Zhang C.-Y., Deng B.-Q. Association of CYP2J2 gene polymorphisms with ischemic stroke and stroke subtypes in Chinese population. Medicine. 2017;96(10):p. e6266. doi: 10.1097/md.0000000000006266. [DOI] [PMC free article] [PubMed] [Google Scholar]