

Abstract
Osteosarcoma is a malignant tumor in children and adolescents. Previous studies showed that ATG4A is an autophagy-related gene involved in cancers. In this study, we aimed to identify the biological role of ATG4A in osteosarcoma. The expression levels of ATG4A were analyzed in osteosarcoma tissues by using reverse transcription-quantitative polymerase chain reaction (qRT-PCR) and western blotting. ATG4A was knocked-down or overexpressed in SAOS2 and HOS cell lines by transfection. Cell counting kit-8 (CCK-8) and clone formation assay were used to assess the effects of ATG4A on cell proliferation. Wound healing and Transwell assays were performed to evaluate the effects of ATG4A on cell migration and invasion, respectively. Epithelial-mesenchymal transition (EMT) markers and Notch signaling pathway targeting molecules were examined by western blotting. The results indicated that ATG4A was up-regulated in osteosarcoma tissues. In SAOS2 cells, knockdown of ATG4A inhibited the proliferation, migration and invasion, up-regulated the expression of E-cadherin and down-regulated the expression of vimentin, Notch1 and Hes1. In HOS cells, overexpression of ATG4A promoted the proliferation, migration and invasion, up-regulated the expression of vimentin, Notch1 and Hes1 and down-regulated the expression of E-cadherin. In conclusion, these findings demonstrate that ATG4A is up-regulated in osteosarcoma tissues. In osteosarcoma cells, ATG4A promotes the EMT process partly by the Notch signaling pathway. These results suggest that ATG4A might represent a potential therapeutic target for patients with osteosarcoma.
Keywords: ATG4A, epithelial-mesenchymal transition, osteosarcoma
Introduction
As the most common malignant tumor of bone, osteosarcoma occurs predominantly in children and adolescents [1]. Osteosarcoma derives from mesenchymal stem cells and could directly develop into the immature bone or osteoid tissue [2,3]. It occurs mostly in the distal femur and proximal tibia region and metastasizes to the lung [3,4]. With the advancements in treatment therapies, the prognosis of patients with localized osteosarcoma was improved [5]. However, the therapeutic effects are insufficient in patients with metastasis or recurrence, leading to a poor 5-year survival rate [6]. The epithelial-mesenchymal transition (EMT) is a crucial process for the metastasis of cancer cells, thus investigation about the aberrant EMT process in osteosarcoma is helpful to develop effective therapeutic strategies.
ATG4A is a redox-regulated cysteine protease, which is well-known as an autophagy regulator. In the autophagic process, ATG4A removes the C-terminal arginine of ATG8 which allow the conjugation of ATG8 to phosphatidylethanolamine (PE), and then releases ATG8-PE to the autophagosomal membrane [7]. Besides the autophagy, several studies have reported the function of ATG4A in tumorigenesis, and the results indicated that ATG4A might contribute to the stem-like phenotype, drug resistance and tumor metastasis [8-10]. However, to our knowledge, no research has reported the role of ATG4A in osteosarcoma. In this study, we focused on determining the functional mechanism of ATG4A in osteosarcoma.
The present study detected the expression levels of ATG4A in 22 pairs of osteosarcoma and matched non-tumor tissue. Furthermore, the effects of ATG4A on cell proliferation, migration and invasion were assessed. In addition, the EMT markers and the Notch signal pathway targeting molecules were detected.
Materials and methods
Tissue samples
22 cases of osteosarcoma patient were recruited in this study from March 2012 to June 2016. The osteosarcoma patients were diagnosed by postoperative histopathological examination. This study was approved by the Ethical Committee of Nanjing Medical University Affiliated Wuxi Second Hospital and consent forms were obtained from all the patients. All the tissue samples were frozen in liquid nitrogen after resection.
Cell lines
The osteosarcoma cell lines were purchased from Cell Bank of Chinese Academy of Sciences (Shanghai, China). SAOS2 cells were cultured in McCoy’s 5A Media (Invitrogen Life Technologies, Carlsbad, CA, USA) supplemented with 15% FBS (Invitrogen), and HOS cells were cultured in EMEM medium supplemented with 10% FBS (Invitrogen). All mediums contain 100 U/ml penicillin and streptomycin (Invitrogen). All the cells were incubated at 37°C with 5% CO2.
qRT-PCR
Total RNA was isolated using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), and reversed-transcripted using a reverse-transcription kit SYBR Premix Ex Taq II (Takara Bio, Inc., Otsu, Japan) according to the manufacturer’s instructions. The PCR reaction was performed on an ABI 7500 system (Applied Biosystems; Thermo Fisher Scientific, Inc, Waltham, MA, USA). β-actin was used as an internal control. The primer sequences were: ATG4A sense, 5’-TTGGCCCAGGATGACAGCTG-3’, antisense, 5’-AGGGCCCGTTCCACCAATTG-3’; β-actin sense, 5’-TGGATGATGATGATATCGCC-3’, antisense, 5’-GTGATGACCTGGCCGTCAGG-3’. Amplification was performed as follows: initiation at 94°C for 30 s, amplification for 32 cycles with 95°C for 5 s, 62°C for 30 s, 72°C for 30 s.
Western blotting
Cells and tissues were lysed in radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China) for 5 min on ice. Supernatant containing proteins was isolated by centrifuging on 1, 2000 g for 5 min at 4°C. The protein quantification was determined using the BCA method (BCA Protein Assay Kit, Beyotime Institute of Biotechnology). Equal amount of proteins were separated on 10% SDS-PAGE and target proteins were transferred to PVDF membranes. After blocking in non-fat milk for 1 h, the membranes were incubated in the primary antibodies of β-actin (A5441, mouse monoclonal, 1:8,000, Sigma-Aldrich, St. Louis, MO, USA), ATG4A (ab108322, Rabbit monoclonal, 1:500, Abcam, Cambridge, UK), E-cadherin (#14472; mouse monoclonal, 1:500, Cell Signaling Technology, Inc., Beverly, MA, USA), vimentin (#3390, mouse monoclonal, 1:500, Cell Signaling Technology, Inc.), Notch1 (ABS90, rabbit polyclonal, 1:500, EMD Millipore, Billerica, MA, USA), Hes1 (#11988, rabbit monoclonal, Cell Signaling Technology, Inc.) overnight. After being washed in TBST, the membranes were incubated in secondary antibodies (#BA1054, goat anti-rabbit and #BA1051, goat anti-mouse 1:10,000; Wuhan Boster Biological Technology, Ltd.) for 2 h. After washing with TBST, protein bands were detected by using BeyoECL Plus kit (Beyotime Institute of Biotechnology).
Cell transfection
ATG4A small interfering (si)RNA/negative control (NC) and pcDNA3.1-ATG4A vector/empty vector were purchased from Genepharma (Shanghai, China). The ATG4A siRNA sequence was 5’-CCCGGAAAGAAATAGAACAAT-3’. The NC sequence was 5’-CCTCCAGGTCTATTCGACTTAAGCA-3’. Cells were transfected using Lipofectamine® 2000 (Invitrogen). The concentration of siRNA/NC was 50 nM, plasmid was 2 μg. The medium was replaced with fresh medium containing 10% FBS without antibiotics 6 h post-transfection. For the stable transfection of pcDNA3.1-ATG4A vector, cells were cultured in selected medium containing 400 μg/ml G418 (Invivogen) for 14 days.
Cell proliferation assay
Cell proliferation was measured by cell counting kit-8 (CCK-8, Dojindo Molecular Technologies, Kumamoto, Japan) after transfection. Cells were seeded in triplicate into the 96-well plate at a density of 3×103 cells per well. At different time-points (24, 48 and 72 h), the OD value was measured on a microplate reader (iMark; Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 450 nm after 2 h incubation with CCK-8.
Clone formation assay
500 cells were seeded into the 6-well plate after transfection, and the medium was replaced every 4 days. Cells were cultured for 12-14 days and stained with 0.3% crystal violet (Beyotime Institute of Biotechnology) for 30 min. Numbers of cell colony were counted and captured using a Nikon camera (Nikon, Tokyo, Japan).
Wound healing assay
Cells at 3×105 per well were planted into six-well plate (Corning). A scratch wound was made by a 20 μl pipette tip on the confluent cell monolayer after serum starvation for 24 h. The images were captured at 0 and 48 h under an inverted microscope (Nikon).
Transwell assay
Cells were starved for 8 h, trypsinized and suspended in serum-free medium. 100 μl cell suspension (3×105 cells) was added into the upper chamber which was pre-coated with matrigel (BD Biosciences, San Jose, CA, USA), and 600 μl RPMI-1640 medium containing 10% FBS was added into the lower well and. After incubation for 48 h, invaded cells to the lower surface were fixed with methanol and stained with 0.3% crystal violet (Beyotime Institute of Biotechnology) for 30 min. Cells were counted and captured under a microscope (Nikon, magnification 200×).
Statistical analysis
Statistical analysis was performed with SPSS Statistics 17 (SPSS Inc., Armonk, NY, USA). Numerical data are presented as the mean ± standard deviation (SD) from at least three independent experiments. Student’s t test was used to calculate the compare the results. P<0.05 was considered statistically significant.
Results
The expression level of ATG4A in osteosarcoma tissues
To elucidate the role of ATG4A in osteosarcoma, we analyzed the ATG4A expression levels in 22 pair of osteosarcoma tissues by qRT-PCR and western blotting. The results indicated that both the mRNA (Figure 1A) and protein (Figure 1B and 1C) levels of ATG4A in osteosarcoma tissues were much higher than that in matched non-tumor tissues, and the up-regulation of ATG4A was 68.18% (15/22). The results suggested that ATG4A might be an onco-protein in osteosarcoma.
Figure 1.
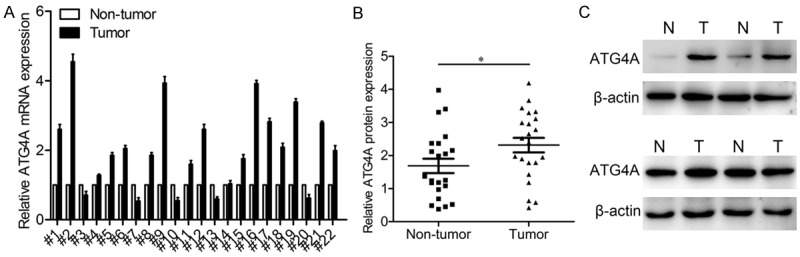
ATG4A is up-regulated in osteosarcoma tissues. A. The mRNA expression levels of ATG4A in 22 pairs of osteosarcoma tissues. B. Scatter plots of ATG4A protein expression in osteosarcoma tissues and matched non-tumor tissues. C. The protein expression levels of ATG4A in 4 pairs of representative osteosarcoma tissues. *P<0.05. T, Tumor tissues; N, adjacent non-tumor tissues.
Effects of ATG4A on proliferation of osteosarcoma cells
To determine the effects of ATG4A on osteosarcoma, ATG4A was knocked-down or over-expressed in osteosarcoma cells. SAOS2 cells transfected with ATG4A siRNA and negative control were termed SAOS-siATG4A and SAOS2-NC, respectively. Similarly, HOS cells transfected with ATG4A vector and negative control were termed HOS-ovATG4A and HOS-NC, respectively. The transfection efficacy was measured by qRT-PCR and western blotting analysis (Figure 2A). Then we investigated the role of ATG4A in cell proliferation. CCK-8 assay indicated that knockdown of ATG4A in SAOS2 cells inhibited the growth rates as compared with control cells, while overexpression of ATG4A in HOS cells promoted the growth rates as compared with control cells (Figure 2B and 2C). Additionally, to detect the long-term effects of ATG4A on cell proliferation, colony formation assay was further performed. The results showed that ATG4A down-regulation by its inhibitor reduced the number of formed colonies, adverse results were observed in ATG4A up-regulation cells (Figure 2D and 2E).
Figure 2.
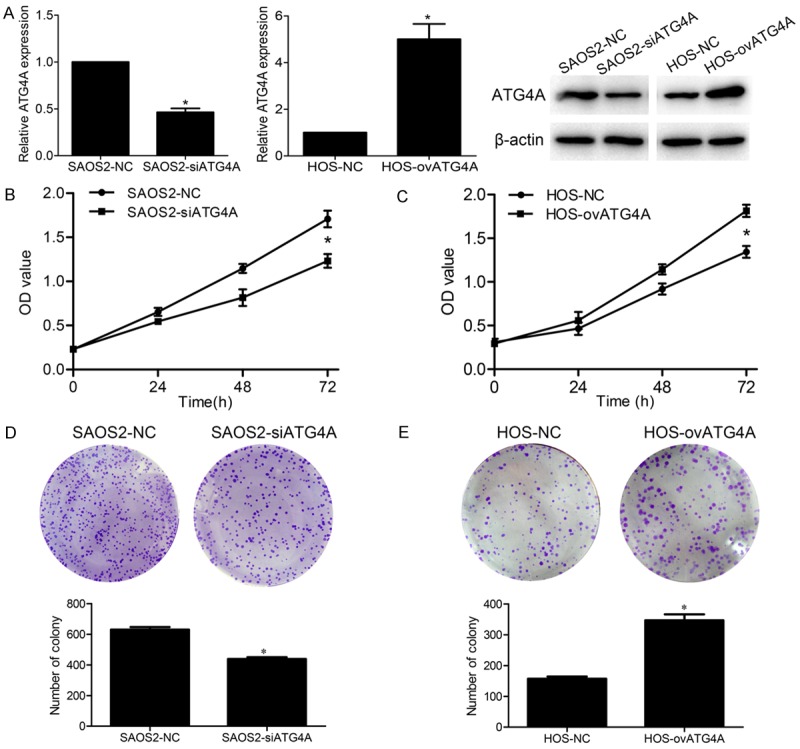
Effects of ATG4A on osteosarcoma cell proliferation. (A) The mRNA and protein level of ATG4A after knockdown and overexpression in SAOS2 and HOS cell lines. Knockdown of ATG4A inhibited SAOS2 cell proliferation (B) and clone formation ability (D). Overexpression of ATG4A promotes HOS cell proliferation (C) and clone formation ability (E). *P<0.05. NC, negative control.
ATG4A regulates the migration and invasion processes of osteosarcoma cells
The effects of ATG4A on osteosarcoma cell migration and invasion were investigated. In the wound healing assay, down-regulation of ATG4A slowed the wound closure rate of SAOS2 cells, while ectopic expression of ATG4A accelerated the wound closure rate in HOS cells (Figure 3A and 3B). In the transwell assay, the invasive activity of SAOS2-siATG4A cells was reduced compared to SAOS2-NC cells (Figure 3C). On the contrary, HOS-ovATG4A cells exhibited an enhanced invasive ability (Figure 3D).
Figure 3.
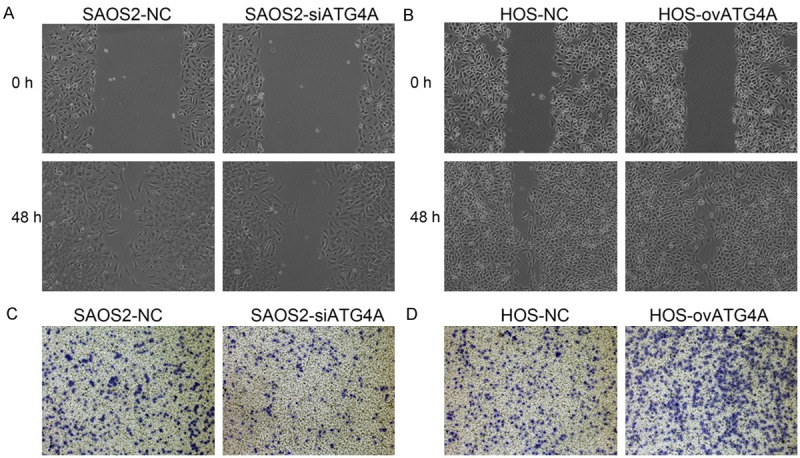
Effects of ATG4A on osteosarcoma cell migration and invasion. Knockdown of ATG4A inhibited SAOS2 cell migration (A) and invasion (C) ability. Overexpression of ATG4A promotes HOS cell migration (B) and invasion (D). NC, negative control.
ATG4A regulated the EMT process of osteosarcoma cell partially through the Notch signaling pathway
Having shown that ATG4A induce behavior changes of osteosarcoma cell, so we further investigate whether ATG4A participate the EMT process. The expression of EMT markers E-cadherin and vimentin were detected. Compared with SAOS2-NC cell, the level of E-cadherin was increased and vimentin was decreased in SAOS2-siATG4A cell. In HOS cells, overexpressed ATG4A down-regulated the expression of E-cadherin and up-regulated vimentin (Figure 4A). Previous study has revealed that ATG4A regulates EMT of gastric cancer cells via the Notch signaling pathway [11], so we examined the expression of Notch1 and Hes1. The results showed Notch1 and Hes1 were down-regulated in ATG4A-silencing SAOS2 cells and up-regulated in ATG4A-overexpressing HOS cells (Figure 4A). Next, HOS-ovATG4A cells were treated with DAPT (the Notch signaling inhibitor) or saline for 24 h and subjected to migration and invasion assays. The data indicated DAPT treatment inhibited the migration and invasion of HOS-ovATG4A cells (Figure 4B and 4C).
Figure 4.
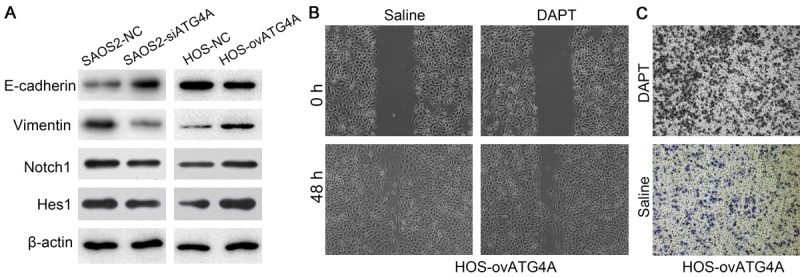
Overexpression of ATG4A regulated the EMT process of osteosarcoma cell partially through the Notch signaling pathway. (A) The protein level of E-cadherin, vimentin Notch1 and Hes1 in osteosarcoma cell lines with knocked down and overexpressed ATG4A. Treatment of DAPT on ATG4A over-expressed HOS cell inhibited cell migration (B) and invasion (C).
Discussion
Tumor metastasis is a major cause of cancer-related death, while EMT is a critical process in metastasis [12,13]. During the EMT process, epithelial cells undergo multiple biochemical changes to acquire mesenchymal cells phenotype and gene expression patterns [14]. After that, cancer cells gain increased mobility and invasiveness, and could detach from the basement membrane, migrate and invade to surrounding tissues [15]. A number of molecules and pathways have engaged in the EMT process, such as the transcription factors Snail and Slug, the ERK/MAPK and PI3K/Akt pathways [16-18]. In this study, we analyzed the role and mechanism of ATG4A in the EMT process of osteosarcoma cell lines.
ATG4A is an isoform of ATG4, which has been shown to play a key role in autophagy. Aberrant autophagy process has been reported to contribute to cancer progression [19]. In ovarian cancer patients, hypomethylated ATG4A correlated with clinical stage and poor progression-free survival. While in ovarian cancer cells, high expression of ATG4A increased the stem properties, and promoted cisplatin resistance, cell migration and anchorage-independent growth [20]. In gastric cancer, ATG4A was up-regulated in tumor tissues, and promoted gastric cancer cell stem-like properties and the EMT [9]. Banzhou Pan et al found down-regulation of ATG4A by the target miR-24-3p suppressed autophagy in small-cell lung cancer cells and allow cells to be re-sensitized to VP16-DDP [8]. In breast cancer cells, ATG4A regulates the stem-cell phenotype and tumourigenicity [10].
In the current study, we analyzed the function of ATG4A in osteosarcoma, and found ATG4A was up-regulated in osteosarcoma tissues. The results suggested that ATG4A participated in the tumorigenesis of osteosarcoma. However, given the small sample size, we will collect more samples to verify the present results. Then, we investigated the biological function of ATG4A in osteosarcoma cell lines. ATG4A was knockdown or overexpressed in osteosarcoma cell lines, and we found knockdown of ATG4A inhibited osteosarcoma cell proliferation, clone formation, migration and invasion. On the contrary, overexpression of ATG4A promoted osteosarcoma cell proliferation, clone formation, migration and invasion. Since ATG4A promoted the aggressive behavior of osteosarcoma cells, we examined the EMT markers to investigate whether ATG4A participate in regulating the EMT process. E-cadherin is the epithelial cell marker and vimentin is the mesenchymal cell marker. We found E-cadherin was up-regulated after silencing of ATG4A and down-regulated after overexpressing of ATG4A. By comparison, vimentin was decreased following transfection with the ATG4A siRNAs and increased following transfection with the ATG4A expression vector. These data confirmed that ATG4A participated in the EMT process. Numerous studies have reported that the Notch signaling pathway plays a significant role in tumor metastasis [21,22]. The present data showed that ATG4A knockdown inhibited the expression of Notch1 and Hes-1, and ATG4A overexpression promoted the expression of Notch1 and Hes-1. Further research demonstrated that Notch signaling inhibitor DAPT abrogated ATG4A-induced cell migration and invasion. Notch1 has been reported to repress E-cadherin expression and up-regulate Snail and Slug expression, so we presumed that ATG4A regulates the EMT process of osteosarcoma cells partly though Notch signaling pathway.
In conclusion, we demonstrated that ATG4A is up-regulated in osteosarcoma tissues. ATG4A promotes the EMT process of osteosarcoma cells partly though Notch signaling pathway.
Disclosure of conflict of interest
None.
References
- 1.Baroy T, Chilamakuri CS, Lorenz S, Sun J, Bruland OS, Myklebost O, Meza-Zepeda LA. Genome analysis of osteosarcoma progression samples identifies FGFR1 overexpression as a potential treatment target and CHM as a candidate tumor suppressor gene. PLoS One. 2016;11:e0163859. doi: 10.1371/journal.pone.0163859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jo VY, Fletcher CD. WHO classification of soft tissue tumours: an update based on the 2013 (4th) edition. Pathology. 2014;46:95–104. doi: 10.1097/PAT.0000000000000050. [DOI] [PubMed] [Google Scholar]
- 3.Hu YY, Du XY, Zhan AL, Zhou L, Jiang Q, Niu YM, Shen M. Vascular endothelial growth factor polymorphisms are associated with osteosarcoma susceptibility. Oncotarget. 2016;7:47711–47719. doi: 10.18632/oncotarget.10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang J, Liu K, Yu Y, Xie M, Kang R, Vernon P, Cao L, Tang D, Ni J. Targeting HMGB1-mediated autophagy as a novel therapeutic strategy for osteosarcoma. Autophagy. 2012;8:275–277. doi: 10.4161/auto.8.2.18940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Z, Li B, Ren Y, Ye Z. T-cell-based immunotherapy for osteosarcoma: challenges and opportunities. Front Immunol. 2016;7:353. doi: 10.3389/fimmu.2016.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts SS, Chou AJ, Cheung NK. Immunotherapy of childhood sarcomas. Front Oncol. 2015;5:181. doi: 10.3389/fonc.2015.00181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan B, Chen Y, Song H, Xu Y, Wang R, Chen L. Mir-24-3p downregulation contributes to VP16-DDP resistance in small-cell lung cancer by targeting ATG4A. Oncotarget. 2015;6:317–331. doi: 10.18632/oncotarget.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang SW, Ping YF, Jiang YX, Luo X, Zhang X, Bian XW, Yu PW. ATG4A promotes tumor metastasis by inducing the epithelial-mesenchymal transition and stem-like properties in gastric cells. Oncotarget. 2016;7:39279–39292. doi: 10.18632/oncotarget.9827. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Wolf J, Dewi DL, Fredebohm J, Muller-Decker K, Flechtenmacher C, Hoheisel JD, Boettcher M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013;15:R109. doi: 10.1186/bcr3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clemo NK, Collard TJ, Southern SL, Edwards KD, Moorghen M, Packham G, Hague A, Paraskeva C, Williams AC. BAG-1 is up-regulated in colorectal tumour progression and promotes colorectal tumour cell survival through increased NF-kappaB activity. Carcinogenesis. 2008;29:849–857. doi: 10.1093/carcin/bgn004. [DOI] [PubMed] [Google Scholar]
- 12.Liu TY, Chen J, Shang CL, Shen HW, Huang JM, Liang YC, Wang W, Zhao YH, Liu D, Shu M, Guo LY, Hu Z, Yao SZ. Tripartite motif containing 62 is a novel prognostic marker and suppresses tumor metastasis via c-Jun/Slug signalingmediated epithelial-mesenchymal transition in cervical cancer. J Exp Clin Cancer Res. 2016;35:170. doi: 10.1186/s13046-016-0445-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei X, Xu JF, Chang RM, Fang F, Zuo CH, Yang LY. JARID2 promotes invasion and metastasis of hepatocellular carcinoma by facilitating epithelial-mesenchymal transition through PTEN/AKT signaling. Oncotarget. 2016;7:40266–40284. doi: 10.18632/oncotarget.9733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tseng CH, Murray KD, Jou MF, Hsu SM, Cheng HJ, Huang PH. Sema3E/plexin-D1 mediated epithelial-to-mesenchymal transition in ovarian endometrioid cancer. PLoS One. 2011;6:e19396. doi: 10.1371/journal.pone.0019396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rumman M, Jung KH, Fang Z, Yan HH, Son MK, Kim SJ, Kim J, Park JH, Lim JH, Hong S, Hong SS. HS-173, a novel PI3K inhibitor suppresses EMT and metastasis in pancreatic cancer. Oncotarget. 2016;7:78029–78047. doi: 10.18632/oncotarget.12871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ha JH, Ward JD, Radhakrishnan R, Jayaraman M, Song YS, Dhanasekaran DN. Lysophosphatidic acid stimulates epithelial to mesenchymal transition marker Slug/Snail2 in ovarian cancer cells via Galphai2, Src, and HIF1alpha signaling nexus. Oncotarget. 2016;7:37664–37679. doi: 10.18632/oncotarget.9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang L, Li J, Ouyang L, Liu B, Cheng Y. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016;373:19–26. doi: 10.1016/j.canlet.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 20.Liao YP, Chen LY, Huang RL, Su PH, Chan MW, Chang CC, Yu MH, Wang PH, Yen MS, Nephew KP, Lai HC. Hypomethylation signature of tumor-initiating cells predicts poor prognosis of ovarian cancer patients. Hum Mol Genet. 2014;23:1894–1906. doi: 10.1093/hmg/ddt583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brzozowa-Zasada M, Piecuch A, Dittfeld A, Mielanczyk L, Michalski M, Wyrobiec G, Harabin-Slowinska M, Kurek J, Wojnicz R. Notch signalling pathway as an oncogenic factor involved in cancer development. Contemp Oncol (Pozn) 2016;20:267–272. doi: 10.5114/wo.2016.61845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Chang H, Peng X, Gu Y, Yi L, Zhang Q, Zhu J, Mi M. 3,6-dihydroxyflavone suppresses the epithelial-mesenchymal transition in breast cancer cells by inhibiting the Notch signaling pathway. Sci Rep. 2016;6:28858. doi: 10.1038/srep28858. [DOI] [PMC free article] [PubMed] [Google Scholar]