

Abstract
Src associated in mitosis, 68 kDa (Sam68) is a KH domain RNA-binding protein that regulates a broad scope of biological events, including RNA metabolism, transcription and signal transduction. Herein, we aimed to explore the expression, clinical significance and biological function of Sam68 in human non-small cell lung cancer (NSCLC). By applying quantitative real-time PCR (qRT-PCR), western blotting and immunohistochemistry (IHC) methods, we found that nucleic localized Sam68 was markedly overexpressed in NSCLC tissues and cell lines. By X2 analysis and Kaplan-Meier survivial analysis between Sam68 expression and various clinicopathological features, Sam68 was found to be significantly associated with clinical T stage, advanced tumor grade, and short overall survival. Finally, in vitro loss-of-function studies showed that knockdown of Sam68 inhibited cell proliferation, colony formation and cell cycle progression in NSCLC cells. Moreover, our results clarified that knockdown of Sam68 could suppress NSCLC cell proliferation via the inhibition of Wnt/β-catenin pathway. To conclude, our results demonstrated that upregulation of Sam68 in NSCLC resulted in poor prognosis, and it promoted cell proliferation via activating Wnt/β-catenin signaling pathway, which could serve as a novel biomarker for the prognosis and therapy of NSCLC.
Keywords: Sam68, NSCLC, cell proliferation, Wnt/β-catenin signaling
Introduction
Sam68 (SRC associated in mitosis of 68 kDa), also known as KHDRBS1 (KH domain-containing, RNA-binding, signal-transduction associated 1), belongs to the STAR (signal transduction and activation of RNA metabolism) family of RNA binding proteins (RBPs). It is initially identified by two groups as a protein that physically interacts with and is phosphorylated by the tyrosine kinase c-SRC during mitosis [1,2]. Sam68 contains an evolutionarily conserved hnRNP K homology domain (KH domain) for RNA binding, and it modulates several steps of RNA processing, including nuclear export, cytoplasmic RNA translation and alternative splicing by binding to the AU-rich RNA structures [3-5]. Besides, this protein has been described as an adaptor recruited in various signal transduction pathways via its proline- and tyrosine-rich sequence motifs, linking cellular signaling to RNA processing [6]. All these published reports demonstrate that Sam68 is a multifunctional factor that plays important roles in various cellular processes, including proliferation and differentiation.
Although ubiquitously expressed, Sam68 has been proved to be upregulated and play specialized roles in different cellular environments in a variety of human cancers [7]. Indeed, it is known that disruption of Sam68 in DT40 cell line results in markedly growth retardation due to G2/M arrest [8]. Increasing studies suggest that Sam68 may act as an oncogene to promote cell proliferation, chemoresistance and metastasis [7,9]. Moreover, Richard and colleagues have documented that haploinsufficiency of Sam68 delays mammary tumorigenesis and metastasis in the PyMT transgenic mice model [10]. Nevertheless, whether the deregulation of Sam68 is a prevalent event in human cancer needs further investigation.
NSCLC (non-small cell lung cancer) is one of the leading cause of cancer-related mortality in the world, with high incidence and an approximately 16% of 5-year survival rate [11-13]. With air pollution becoming seriously and disgusting, NSCLC has become one of the most common newly diagnosed cancers and leading causes of cancer-related death in the developing countries, especially in China [14-17]. The mutations, polymorphisms and overexpressions of p53, K-ras and EGFR (epidermal growth factor receptor) gene are all closely associated with NSCLC [18-21]. The elevated expression of Sam68 has also been observed in NSCLC, and high Sam68 expression predicts poor prognosis of NSCLC patients [22]. To our best knowledge, the role of Sam68 in NSCLC tumorigenesis is still lacking from the present studies. Given that Sam68 is increased and correlated with lymph node metastasis and tumor TNM stage in NSCLC [22], and it drives mammary tumourigenesis [7,10], we suppose that it may also exert proto-oncogenic functions in NSCLC.
In an effort to investigate the role of Sam68 in NSCLC, we designed a series of experiments to clarify its expression pattern, clinical significance, biological functions and underlined mechanisms in NSCLC cells. Our study indicated that knockdown of Sam68 suppressed cell proliferation, colony formation and cell cycle progression via inhibiting Wnt/β-catenin pathway, and the upregulation of Sam68 could serve as a novel biomarker of prognosis and therapy target for NSCLC.
Materials and methods
Patients and tissues
Sixty fresh NSCLC tissues and their matched non-cancerous normal lung tissues were collected by Dr. Hua Feng (Department of Thoracic Surgery, Shandong Cancer Hospital and Institute, Jinan, P. R. China) from NSCLC patients. All patients were diagnosed with histologically confirmed NSCLC and clinicopathological data were available. All patients had provided the written informed consent use of their tissues. For total RNA and total protein extraction, tissues were immediately frozen by liquid nitrogen and stored at -80°C until used.
Cell culture and reagents
Human bronchial epithelial Beas-2B cells and eight different lung cancer cells were cultured in 1640 or DMEM medium supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 units/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified 5% CO2 atmosphere. All these cell lines were from the cell bank of Tianjin Lung Cancer Institute (Tianjin, P. R. China). The antibody against Sam68, cyclin D1 and c-Myc was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibody against β-actin was from Sigma (St. Louis, MO, USA).
Short interference RNAs and transfections
For knocking down of endogenous Sam68 gene (NCBI Reference Sequence: NM_006559.2), two independent short interference RNA (siRNA) oligos targeting Sam68 (siSam68-1 and siSam68-2) and control siRNA oligos (Control) were from Ribobio (Guangzhou, China). The sequences of these oligos are: Control: 5’-UUCUCCGAACGUGUCACGUdTdT-3’; siSam68-1: 5’-AGAGCUGUCCUACUUGAAUdTdT-3’; siSam68-2: 5’-AAGGCUACGAAGGCUAUUAdTdT-3’.
Transient transfections of cells were performed using Lipofectamine® 2000 (Invitrogen, Carlsbad, CA, USA) as per the manufacture’s instructions.
Quantitative real-time PCR (qRT-PCR)
Relative gene expressions were determined by qRT-PCR as described previously [23]. In brief, total RNA was extracted from the tissues or cells by TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. qRT-PCR was performed on Applied Biosystems Step Two Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using the comparative threshold cycle (Ct) quantization method. SYBR Premix Ex Taq (Takara, Tokyo, Japan) was used to detect and quantify the expression level of target gene. β-actin was used as an internal control. The fold changes of mRNA levels were calculated by the 2-ΔΔCt method. The primer sequences are as follows: β-actin F: 5’-GATCATTGCTCCTCCTGAGC-3’; β-actin R: 5’-ACTCCTGCTTGCTGATCCAC-3’; Sam68 F: 5’-GCGAGTGCTGATACCTGTCAAG-3’; Sam68 R: 5’-TCATTGAGCCCTTTCCCAAT-3’; cyclin D1 F: 5’-CTGGAGGTCTGCGAGGAACA-3’; cyclin D1 R: 5’-CTGCAGGCGGCTCTTTTTC-3’; c-Myc F: 5’-GCTCCTGGCAAAAGGTCAGA-3’; c-Myc R: 5’-CGCTGCGTAGTTGTGCTGAT-3’.
Western blotting
Relative protein expressions were determined by western blotting as described previously [23]. In brief, tissues or cells were lysed on ice for 30 min in RIPA buffer (Beyotime Biotechnology, Shanghai, China) supplemented with 1 mM phenylmethylsulfonyl fluoride (Beyotime Biotechnology, Shanghai, China). The supernatant was collected after centrifugation at 4°C, 13000 rpm for 30 min. Equal amounts of protein were resolved on SDS-PAGE and transferred to a nitro-cellulose membrane. Proteins of interest were detected by western blotting using specific antibodies.
Immunohistochemistry staining
Immunohistochemistry staining of tissues was performed as described [24]. Tissues were formaldehyde-fixed and paraffin-embedded. The sections (5 μm thickness) were heat-immobilized, deparaffinized and rehydrated. Endogenous peroxidases were blocked by 75% H2O2 in phosphate-buffered saline (PBS) for 30 min. Antigen retrieval was finished by incubation in 10 mM citrate buffer (pH6.0) for 10 min, followed by incubation in 5% BSA blocking buffer for 1 h. The sections were incubated with primary anti-Sam68 antibody (1:200) at 4°C overnight. After washes, the sections were incubated with secondary antibody for 1 h, and detected by incubation with streptavidin-horseradish peroxidase complex. The sections were finally visualized by 3,3-diaminobenzidine (DAB) and subsequently photographed under a microscope.
Cell proliferation assays
A549 and H1299 cells (1.2×103/well) were seeded into 96-well plates after transfection and incubated for another 4 d. At each day, WST-8 (10 μl/well) from Cell Counting Kit-8 (Dojindo, Tokyo, Japan) was added and the absorbance at 450 nm was measured with a SpectraMax® M5 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA, USA).
Colony formation assay
Colony formation assay was performed as described before [24]. After transfection, H1299 cells (2.0×103/well) were seeded in 12-well plates with the medium containing 10% FBS. Medium was changed every other day. 12 days later, after removing the medium, the cells were fixed with 4% formalin and stained with 0.5% crystal violet solution for 0.5 h. After that, cells were washed twice with PBS, aspirated, allowed to air dry and photographed. Cells were resolved with 1% SDS solution. The absorbance was measured using a SpectraMax® M5 Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) at 600 nm.
Cell cycle analysis
Cell cycle analysis was determined by propidium iodide (PI) staining and flow cytometry method as described previously [23]. Briefly, transfected A549 cells were fixed by 70% ice-cold ethanol and stained with freshly prepared nuclei staining buffer (0.1% Triton X-100 in PBS, 50 μg/ml of PI and 200 μg/ml of RNaseA) for 20 min at 37°C. Cell-cycle histograms were generated and analyzed with a FACS-CaliburTM Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ, USA). The percentage of cells in the G0/G1, S and G2/M phases were counted and compared.
Luciferase reporter assay
A549 and H1299 cells were seeded in triplicate in 24-well plates and allowed to settle overnight. For basal level of Wnt/β-catenin reporter assay: cells were co-transfected with 0.1 μg of the TOP flash or FOP flash reporter plasmid (containing three copies of wild type or mutant TCF binding motif), 50 pmol siRNA oligos and 0.05 μg of β-galactosidase expressing plasmid as an internal control for transfection efficiency by using Lipofectamine® 2000. For activated level of Wnt/β-catenin reporter assay: cells were co-transfected with 0.1 μg of the TOP flash, 50 pmol siRNA oligos, 0.05 μg of β-galactosidase expressing plasmid and flag-β-catenin expression plasmid by using Lipofectamine® 2000. 72 h after transfection, cells were harvested and analyzed for luciferase activities using Luciferase Reporter Assay Kit (Promega Corporation, Fitchburg, WI, USA). β-galactosidase (β-gal) activity was measured in assay buffer (100 mM pH7.5 phosphate, 2 mM MgCl2, 100 mM β-mercaptoethanol, 1.33 mg/mL o-nitrophenyl β-D-galactopyranoside) at 420 nm. Relative Luc activity was calculated as the ratio of Luc/β-gal activity.
Statistical analyses
Statistical analyses were performed by two-tailed Student t-test using GraphPad Prism 5 software (La Jolla, CA, USA). Protein quantification was performed by ImageJ 1.46r software (NIH, Bethesda, MD, USA). Correlation between Sam68 expression and clinicopathological features was performed by X2 test using SAS 8.02 software (SAS Institute Inc., Cary, NC, USA). Kaplan-Meier survival analysis was performed by logrank test using original data from KM plotter (http://www.kmplot.com/) or TCGA database (https://cancergenome.nih.gov/). Differences with P<0.05 were considered statistically significant. Data were represented as mean ± standard deviation (SD).
Results
Upregulation of Sam68 in primary NSCLC tissues
We firstly applied qRT-PCR, western blotting and IHC methods to determine the expression levels of Sam68 in 60 pairs of primary NSCLC tissues. The results displayed that both mRNA and protein levels of Sam68 were much higher in NSCLC tissues than those in matched non-cancerous lung tissues (Figure 1A and 1B). Next, IHC analysis revealed that the major immunoreactivity of Sam68 located in the nucleus. In agreement with the above results, the immunoreactivity of Sam68 was more obviously stained in the NSCLC tissues, compared with the paired adjacent lung tissues. Moreover, the immunoreactivity was even absent in the normal lung tissues (Figure 1C). Collectively, these results confirmed that Sam68 is upregulated in NSCLC tissues at both transcriptional and translational levels, keeping in line with the previous report [22].
Figure 1.

The expression of Sam68 is upregulated in NSCLC tissues. A. Determination of Sam68 mRNA level by qRT-PCR. ΔCtN: Ct value of β-actin was subtracted from Ct value of Sam68 of paired normal tissue. ΔCtT: Ct value of β-actin was subtracted from that of Sam68 of NSCLC tissue. Bar value (ΔCtN-ΔCtT) represented the difference between Sam68 mRNA level of NSCLC tissues and paired normal tissues. Bar value ≤-1 indicated that the expression of Sam68 was decreased in NSCLC tissues. Bar value ≥1 indicated that the expression of Sam68 was increased in NSCLC tissues. B. Determination of protein level of Sam68 by western blotting in 18 pairs of NSCLC tissues. The lower panel showed the quantitative results. N: normal; T: tumor. C. Representative images of Sam68 expression in alveolar epithelium (a), bronchial epithelial (b), adjacent lung adenocarcinoma (c), lung adenocarcinoma (d), adjacent squamous cell lung carcinoma (e) and squamous cell lung carcinoma (f). Scale bar: 50 μm.
The prognostic significance of Sam68 in NSCLC
Basing on the abovementioned results of Sam68 expression in NSCLC tissues (Figure 1), we re-grouped these samples according to mRNA level as non-increased Sam68 group (n=28) and increased Sam68 group (n=32). Then statistic X2 analysis was conducted to evaluate the association between Sam68 expression and each clinicopathological feature. As shown in Table 1, higher Sam68 expression was strongly associated with higher T stage (P=0.0221) and advanced tumor grade (P=0.0392) in this cohort of 60 NSCLC patients. However, we found no difference between Sam68 expression and other clinical variables, such as age, sex, smoking history, tumor type or N stage (P>0.05). In addition, Kaplan-Meier survival analysis was further performed by logrank test using original data from KM plotter or TCGA database to elucidate the prognosis value of Sam68 in NSCLC. As shown in Figure 2A and 2B, patients with higher levels of Sam68 harbored significantly shorter overall survival rates than patients with lower Sam68 expression (P<0.0001), as calculated from two independent databases. Combining with the above results, we strengthened the conclusion that Sam68 is overexpressed in NSCLC tissues, and its higher expression predicts worse outcomes.
Table 1.
Relationship between expression level of Sam68 and clinical and pathological features of the NSCLC individuals
| Clinical Characteristics | Non-increased (ΔΔCt≤1) n=28 | Increased (ΔΔCt>1) n=32 | Test of Significance |
|---|---|---|---|
| Sex | |||
| Female | 10 | 11 | X2=0.0118 |
| Male | 18 | 21 | P=0.9136 |
| Age | |||
| <60 | 16 | 20 | X2=0.1786 |
| ≥60 | 12 | 12 | P=0.6726 |
| Smoking History | |||
| Non-smoking | 11 | 12 | X2=0.0201 |
| Smoking | 17 | 20 | P=0.8871 |
| Tumor Type | |||
| Squamous cell lung carcinoma | 12 | 13 | |
| Lung adenocarcinoma | 15 | 17 | X2=0.2327 |
| Others | 1 | 2 | P=0.8902 |
| T Stage | |||
| T1 | 17 | 10 | X2=5.2381 |
| T2+T3 | 11 | 22 | P=0.0221 |
| N Stage | |||
| N0 | 15 | 16 | X2=0.0763 |
| N1+N2 | 13 | 16 | P=0.7824 |
| Tumor Grade | |||
| I+II | 22 | 17 | X2=4.2504 |
| III+IV | 6 | 15 | P=0.0392 |
Figure 2.
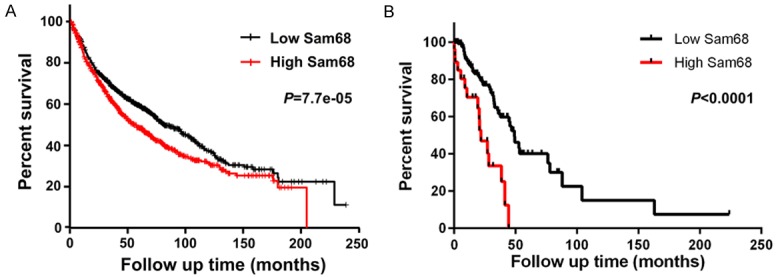
Higher Sam68 expression predicts worse overall survival. A. Kaplan-Meier analysis of the correlation between Sam68 and overall survival of the LC patients using the data from KM plotter (n=1926, http://www.kmplot.com). B. Kaplan-Meier analysis of the correlation between Sam68 and overall survival of the LC patients using the data from TCGA database (n=230, https://cancergenome.nih.gov/). Patients were stratified into high and low expression subgroups using the median of Sam68 mRNA level.
Upregulation of Sam68 in primary NSCLC cell lines
Furthermore, the expression of Sam68 in a panel of NSCLC cell lines and normal bronchial epithelial cell line was also examined. As shown in Figure 3A, we observed that Sam68 was markedly overexpressed in most of the NSCLC cell lines. Among them, 4 cell lines exhibited over 2-folds upregulation than the normal bronchial epithelial Beas-2B cells. Moreover, Sam68 protein was also confirmed to be increased in six lung cancer cell lines (Figure 3B). Taken together, Sam68 is significantly upregulated in both NSCLC tissues and cell lines, indicating its tumor promoting roles in NSCLC.
Figure 3.
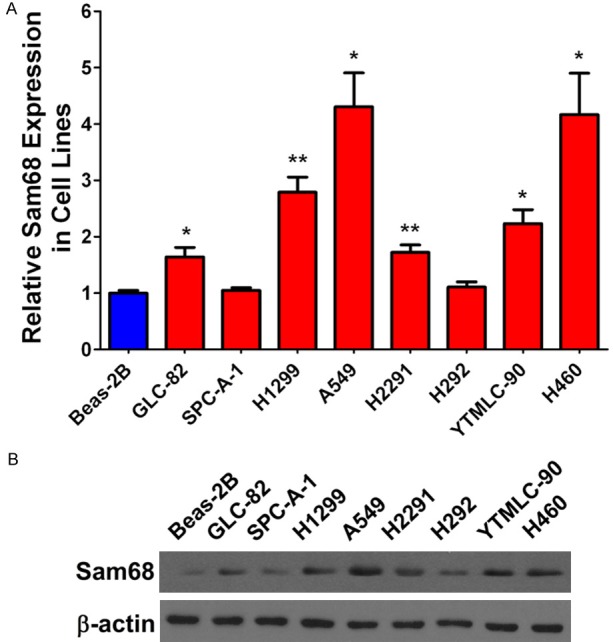
Expression of Sam68 is upregulated in lung cancer cells. A. Determination of Sam68 mRNA by qRT-PCR in human bronchial epithelial Beas-2B cells and eight lung cancer cell lines (GLC-82, SPC-A-1, H1299, A549, H2291, H292, YTMLC-90 and H460). B. Determination of protein level of Sam68 by western blotting in human bronchial epithelial Beas-2B cells and eight lung cancer cell lines. *: P<0.05, **: P<0.01 versus Beas-2B cells.
Knockdown of Sam68 inhibits cell proliferation and cell cycle progression of NSCLC cells
To investigate the biological function of Sam68 in NSCLC, Sam68-silenced A549 and H1299 cells were constructed using two independent siRNAs targeting Sam68. Efficient reduction of Sam68 expression was obtained in these cell lines (Figure 4A and 4B). Next, CCK-8 assay revealed that knockdown of Sam68 resulted in drastic inhibition of cell proliferation in both A549 and H1299 cells (Figure 4C). Subsequent detection of colony formation ability among H1299 cells also showed that knockdown of Sam68 significantly suppressed clonogenic survival, by forming fewer colonies with smaller size (Figure 4D). To examine if cell cycle progression was impaired by knockdown of Sam68, PI staining followed by flow cytometry analysis was performed in A549 cells. The result demonstrated that knockdown of Sam68 caused G0/G1 arrest (Figure 4E), indicating that Sam68 may promote NSCLC cell proliferation in a cell cycle-dependent manner.
Figure 4.

Knockdown of Sam68 inhibits cell proliferation of lung cancer cell. (A) A549 or H1299 cells were transfected with either control siRNA oligos or two Sam68 siRNA oligos. 72 h later, mRNA levels of Sam68 were detected by qRT-PCR. (B) A549 or H1299 cells were transfected as described in (A). 72 h later, protein levels of Sam68 were detected by western blotting. (C) A549 or H1299 cells were transfected as described in (A). In 24 h, cells were trypsinzed and seeded into 96-well plate. Cell proliferation assay was performed as described in Materials and Methods. (D) A549 or H1299 cells were transfected as described in (A). In 24 h, the cells were trypsinzed and seeded into 12-well plate. Colony formation assay was performed as described in Materials and Methods. (E) A549 or H1299 cells were transfected as described in (A). In 72 h, the cells were harvested for cell cycle assay as described in Materials and Methods. The left panel depicted DNA content and the right panel showed the quantitative results. *: P<0.05, **: P<0.01, ***: P<0.001 versus control group.
Knockdown of Sam68 suppresses cell proliferation via inhibiting Wnt/β-catenin signaling
Available data indicate that Wnt/β-catenin signaling substantially impacts on NSCLC tumorigenesis, prognosis, and resistance to therapy [25]. Meanwhile, we realized from one report that Sam68 could promote the nuclear accumulation of β-catenin, facilitie Wnt/β-catenin signaling activation and upregulate TCF/LEF transcription activity in breast cancer cells [26]. To investigate whether knockdown of Sam68 altered Wnt/β-catenin signaling activity, luciferase reporter assay was utilized using its signaling reporter TOP Flash or FOP Flash which contains three copies of wild type or mutant TCF binding motif [27]. We found that knockdown of Sam68 suppressed the TOP Flash activity, whereas no significant change could be observed on TOP Flash activity (Figure 5A). Next, qRT-PCR and western blotting were performed to examine the levels of β-catenin-TCF/LEF-activated genes, such as cyclin D1 and c-Myc. We found that expression of cyclin D1 and c-Myc displayed downregulation upon knockdown of Sam68 at both mRNA and protein levels in A549 or H1299 cells (Figure 5B and 5C). Moreover, we overexpressed flag-β-catenin in H1299 cells to investigate whether loss of Sam68 could also alter the activated level of Wnt/β-catenin signaling. We found that knockdown of Sam68 effectively inhibited flag-β-catenin-activated Wnt luciferase reporter (P<0.001, Figure 5D). Finally, we investigated whether Sam68 knockdown could also inhibit colony formation when Wnt/β-catenin signaling was activated. The results showed that after β-catenin was overexpressed, the clonogenic formation ability was significantly impaired by loss of Sam68 (~70% inhibition), compared with that at basal level (~50% inhibition) (Figure 5E). Taken together, we demonstrated that knockdown of Sam68 suppressed NSCLC cell proliferation via inhibiting Wnt/β-catenin signaling.
Figure 5.

Knockdown of Sam68 suppresses cell proliferation by inhibiting Wnt/β-catenin signaling pathway. A. A549 cells were co-transfected with 0.1 μg of the TOP flash or FOP flash reporter plasmid, 50 pmol siRNA oligos and 0.05 μg of β-galactosidase expressing plasmid. 72 h later, cells were lysed, luciferase and β-galactosidase activities were examined. Fold induction (Luc/β-gal) were calculated and expressed as means ± SD. B. Knockdown of Sam68 reduced the mRNA levels of downstream cyclin D1 and c-Myc genes as determined by qRT-PCR in A549 cells. C. Knockdown of Sam68 decreased the protein levels of downstream cyclin D1 and c-Myc as determined by western blotting in A549 and H1299 cells. D. H1299 cells were co-transfected with 0.1 μg of the TOP flash, 50 pmol siRNA oligos, 0.05 μg of β-galactosidase expressing plasmid and 0.25 μg flag-β-catenin expression plasmid. 72 h later, cells were lysed, luciferase and β-galactosidase activities were examined. Fold induction (Luc/β-gal) were calculated and expressed as means ± SD. E. H1299 cells were co-transfected with Sam68 siRNA oligos and flag-β-catenin expression plasmid. In 24 h, the cells were trypsinzed and seeded into 12-well plate. Colony formation assay was performed as described in Materials and Methods. **: P<0.01, ***: P<0.001, NS: no significance versus control group.
Discussion
As is known, lung cancer is the leading cause of cancer-related death in the developed world, and NSCLC accounts for approximately 80% of all lung cancer subtypes, which contains adenocarcinoma, squamous cell carcinoma and large cell carcinoma [12,28]. The 5-year survival rate for lung cancer is only 16%, because a high proportion of patients are already with metastatic and advanced disease at diagnosis [13]. High mortalities of lung cancer patients can be majorly attributed to: (a) absence of accurate early diagnostic technologies, (b) resistance to currently available therapeutic methods. Short of understanding the precise molecular mechanism governing lung cancer carcinogenesis makes the actuality worse.
Gene regulation in eukaryotes is a complex, multi-step process, including RNA transcription, alternative splicing, transport, localization and translation [29], in which RBPs partner the nascent RNA throughout its journey in the cell. The multi-functionality and the vast scope of targets regulated by RBPs make them important post transcriptional regulators. Now increasing evidence shows that the RBPs are dysregulated and make contributions to cancer development [30].
In the present study, we sought to investigate the expression, clinical significance and biological roles of Sam68, a well-studied RBP, in human NSCLC cells. The first mentioned two aspects have been disclosed by Zhang et al previously, that is, Sam68 is overexpressed in NSCLC tissues and high Sam68 expression predicts poor prognosis [22]. Through systematic investigation, we also conclude that Sam68 is upregulated in both NSCLC tissues and cell lines, and its higher expression is significantly associated with clinical T stage, advanced tumor grade, and short overall survival. Besides, our cellular functional studies showed that knockdown of Sam68 inhibited cell proliferation, colony formation and cell cycle progression. Therefore, our findings add to the understanding of the mechanism of NSCLC tumorigenesis in association with Sam68. Furthermore, our mechanistic studies revealed that knockdown of Sam68 suppresses cell proliferation via inhibiting Wnt/β-catenin signaling, which is prominently activated in NSCLC and plays a critical role in lung tumorgenesis and metastasis [31]. However, the detailed mechanisms of Sam68 in promoting cell proliferation through activating Wnt/β-catenin signaling remain unclear. Its RNA binding and scaffolding features may be used as the central breakthroughs. In addition, whether other mechanisms exist which underlies Sam68-mediated growth promotion in NSCLC needs to be further explored.
In summary, our study provides the first comprehensive view of aberrant upregulation of Sam68 in NSCLC cell proliferation in vitro via a mechanism linked to activation of the Wnt/β-catenin signaling pathway. We believe that our findings provide new insights into the molecular pathogenesis of NSCLC and implicate Sam68 as a potential prognostic biomarker and therapeutic target for NSCLC.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (No. 81572288, to Qinghua Zhou; No. 81302002, to Xuebing Li; No. 81502166, to Xuexia Zhou), the Key Project of International Cooperation of Science and Technology Innovation between Governments, the National Key Research and Development Plan of China (No. 2016YEE0103400, to Qinghua Zhou), the Tianjin Natural Science Foundation (No. 14JCQNJC12300, to Xuebing Li; No. 17JCYBJC27100, to Xuexia Zhou; No. 17JCQNJC11700, to Hongli Pan; No. 17JCYBJC25400, to Yaguang Fan), and the “New Century” Talent Training Project of Tianjin Medical University General Hospital (2014, to Xuebing Li; 2016, to Xuexia Zhou).
Disclosure of conflict of interest
None.
References
- 1.Fumagalli S, Totty NF, Hsuan JJ, Courtneidge SA. A target for Src in mitosis. Nature. 1994;368:871–874. doi: 10.1038/368871a0. [DOI] [PubMed] [Google Scholar]
- 2.Taylor SJ, Shalloway D. An RNA-binding protein associated with Src through its SH2 and SH3 domains in mitosis. Nature. 1994;368:867–871. doi: 10.1038/368867a0. [DOI] [PubMed] [Google Scholar]
- 3.Lin Q, Taylor SJ, Shalloway D. Specificity and determinants of Sam68 RNA binding. Implications for the biological function of K homology domains. J Biol Chem. 1997;272:27274–27280. doi: 10.1074/jbc.272.43.27274. [DOI] [PubMed] [Google Scholar]
- 4.Paronetto MP, Messina V, Bianchi E, Barchi M, Vogel G, Moretti C, Palombi F, Stefanini M, Geremia R, Richard S, Sette C. Sam68 regulates translation of target mRNAs in male germ cells, necessary for mouse spermatogenesis. J Cell Biol. 2009;185:235–249. doi: 10.1083/jcb.200811138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoss O, Olbrich M, Hartmann AM, Konig H, Memmott J, Andreadis A, Stamm S. The STAR/GSG family protein rSLM-2 regulates the selection of alternative splice sites. J Biol Chem. 2001;276:8665–8673. doi: 10.1074/jbc.M006851200. [DOI] [PubMed] [Google Scholar]
- 6.Lukong KE, Richard S. Sam68, the KH domain-containing superSTAR. Biochim Biophys Acta. 2003;1653:73–86. doi: 10.1016/j.bbcan.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Bielli P, Busa R, Paronetto MP, Sette C. The RNA-binding protein Sam68 is a multifunctional player in human cancer. Endocr Relat Cancer. 2011;18:R91–R102. doi: 10.1530/ERC-11-0041. [DOI] [PubMed] [Google Scholar]
- 8.Li QH, Haga I, Shimizu T, Itoh M, Kurosaki T, Fujisawa J. Retardation of the G2-M phase progression on gene disruption of RNA binding protein Sam68 in the DT40 cell line. FEBS Lett. 2002;525:145–150. doi: 10.1016/s0014-5793(02)03103-4. [DOI] [PubMed] [Google Scholar]
- 9.Valacca C, Bonomi S, Buratti E, Pedrotti S, Baralle FE, Sette C, Ghigna C, Biamonti G. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J Cell Biol. 2010;191:87–99. doi: 10.1083/jcb.201001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richard S, Vogel G, Huot ME, Guo T, Muller WJ, Lukong KE. Sam68 haploinsufficiency delays onset of mammary tumorigenesis and metastasis. Oncogene. 2008;27:548–556. doi: 10.1038/sj.onc.1210652. [DOI] [PubMed] [Google Scholar]
- 11.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 12.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 13.Torre LA, Siegel RL, Jemal A. Lung cancer statistics. Adv Exp Med Biol. 2016;893:1–19. doi: 10.1007/978-3-319-24223-1_1. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Zheng R, Baade PD, Zhang S, Zeng H, Bray F, Jemal A, Yu XQ, He J. Cancer statistics in China, 2015. CA Cancer J Clin. 2016;66:115–132. doi: 10.3322/caac.21338. [DOI] [PubMed] [Google Scholar]
- 15.Dehghani M, Keshtgar L, Javaheri MR, Derakhshan Z, Oliveri Conti G, Zuccarello P, Ferrante M. The effects of air pollutants on the mortality rate of lung cancer and leukemia. Mol Med Rep. 2017;15:3390–3397. doi: 10.3892/mmr.2017.6387. [DOI] [PubMed] [Google Scholar]
- 16.Downward GS, Hu W, Rothman N, Reiss B, Tromp P, Wu G, Wei F, Xu J, Seow WJ, Chapman RS, Lan Q, Vermeulen R. Quartz in ash, and air in a high lung cancer incidence area in China. Environ Pollut. 2017;221:318–325. doi: 10.1016/j.envpol.2016.11.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng R, Zeng H, Zuo T, Zhang S, Qiao Y, Zhou Q, Chen W. Lung cancer incidence and mortality in China, 2011. Thorac Cancer. 2016;7:94–99. doi: 10.1111/1759-7714.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aviel-Ronen S, Blackhall FH, Shepherd FA, Tsao MS. K-ras mutations in non-small-cell lung carcinoma: a review. Clin Lung Cancer. 2006;8:30–38. doi: 10.3816/CLC.2006.n.030. [DOI] [PubMed] [Google Scholar]
- 19.Liu C, Xu X, Zhou Y. Association between EGFR polymorphisms and the risk of lung cancer. Int J Clin Exp Pathol. 2015;8:15245–15249. [PMC free article] [PubMed] [Google Scholar]
- 20.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37(Suppl 4):S9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 21.Rodin SN, Rodin AS. Human lung cancer and p53: the interplay between mutagenesis and selection. Proc Natl Acad Sci U S A. 2000;97:12244–12249. doi: 10.1073/pnas.180320897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z, Xu Y, Sun N, Zhang M, Xie J, Jiang Z. High Sam68 expression predicts poor prognosis in non-small cell lung cancer. Clin Transl Oncol. 2014;16:886–891. doi: 10.1007/s12094-014-1160-3. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Zhou X, Li Y, Zu L, Pan H, Liu B, Shen W, Fan Y, Zhou Q. Activating transcription factor 3 promotes malignance of lung cancer cells in vitro. Thorac Cancer. 2017;8:181–191. doi: 10.1111/1759-7714.12421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Zhou X, Fan Y, Zhang Y, Zu L, Yao F, Zhou Q. WW45, a Gli1 binding protein, negatively regulated hedgehog signaling in lung cancer. Oncotarget. 2016;7:68966–68975. doi: 10.18632/oncotarget.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart DJ. Wnt signaling pathway in nonsmall cell lung cancer. J Natl Cancer Inst. 2014;106:djt356. doi: 10.1093/jnci/djt356. [DOI] [PubMed] [Google Scholar]
- 26.Wang L, Tian H, Yuan J, Wu H, Wu J, Zhu X. CONSORT: Sam68 is directly regulated by MiR-204 and promotes the self-renewal potential of breast cancer cells by activating the Wnt/Beta-catenin signaling pathway. Medicine (Baltimore) 2015;94:e2228. doi: 10.1097/MD.0000000000002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N, Waterman M, Bowerman B, Clevers H, Shibuya H, Matsumoto K. The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta-catenin and transcription factor TCF. Nature. 1999;399:798–802. doi: 10.1038/21674. [DOI] [PubMed] [Google Scholar]
- 28.Stella GM, Luisetti M, Pozzi E, Comoglio PM. Oncogenes in non-small-cell lung cancer: emerging connections and novel therapeutic dynamics. Lancet Respir Med. 2013;1:251–261. doi: 10.1016/S2213-2600(13)70009-2. [DOI] [PubMed] [Google Scholar]
- 29.Aparicio LA, Abella V, Valladares M, Figueroa A. Posttranscriptional regulation by RNA-binding proteins during epithelial-to-mesenchymal transition. Cell Mol Life Sci. 2013;70:4463–4477. doi: 10.1007/s00018-013-1379-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wurth L, Gebauer F. RNA-binding proteins, multifaceted translational regulators in cancer. Biochim Biophys Acta. 2015;1849:881–886. doi: 10.1016/j.bbagrm.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Stewart DJ, Chang DW, Ye Y, Spitz M, Lu C, Shu X, Wampfler JA, Marks RS, Garces YI, Yang P, Wu X. Wnt signaling pathway pharmacogenetics in non-small cell lung cancer. Pharmacogenomics J. 2014;14:509–522. doi: 10.1038/tpj.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]