

Abstract
Esophageal cancer (EC) is one of common digestive tract malignant tumors which morbidity and mortality were increased year by year. This study was aimed to investigate the role of microRNA (miR)-200a in EC. Human esophageal squamous cell carcinoma (ESCC) cells TE3 was transfected with miR-200a mimic or scramble control. Cell viability and invasion were assessed by MTT and Transwell assay, respectively. Binding effect of miR-200a on 3’UTR of RKIP was verified by luciferase activity assay. RKIP expression in miR-200a mimic transfected cells was measured. RKIP was overexpressed in miR-200a transfected cells and cell viability and invasion were measured. The expressions of Raf1, ERK, MMP-14, LIN28 and GRK-2 were also measured by qRT-PCR and Western blot analysis, respectively. Results showed that miR-200a mimic transfection increased cell viability and invasion of TE3 cells in vitro. miR-200a binding with 3’UTR of RKIP negatively regulated RKIP expression. RKIP overexpression inhibited effects of miR-200a on cell viability and invasion, as well as the increased phosphorylation levels of Raf1 and ERK. miR-200a increased expressions of MMP-14, LIN28 and GRK-2 in TE3 cells, and the up-regulations were inhibited by RKIP overexpression. In conclusion, the up-regulation of miR-200a in TE3 cells promoted cell viability and invasion via negatively regulating RKIP expression. RKIP was a direct target of miR-200a. miR-200a might be involved in activation of MAPK/ERK signaling pathway and expression of MMP-14, LIN28 and GRK-2 which were important factors of intracellular information transduction. Our findings demonstrated that miR-200a regulated ESCC cells via regulating RKIP expression.
Keywords: microRNA-200a, Raf kinase inhibitor protein, MAPK/ERK signal pathway, esophageal cancer
Introduction
Esophageal cancer (EC) is one of the most aggressive digestive tract tumors [1]. In recent years, incidence of human EC was continually increased with the improvement of life standard and impact of eating habit and environment [2,3]. The esophageal squamous cell carcinoma (ESCC) accounts for more than 90% of all EC cases. ESCC in early stage is usually asymptomatic, most ESCC are diagnosed at advanced stages, following local invasion and lymph node metastasis [4]. In spite of advances in surgical techniques and perioperative management in recent years, mortality of ESCC has not been decreased significantly. The discovery of oncogenes, tumor suppressor or proteins specifically related to esophageal carcinogenesis might reveal new diagnostic biomarkers and targets for EC therapy.
MicroRNAs (miRNAs) as a large class of short RNAs (20-24 nucleotides in length) have been proved to be involved in the complex life process, including growth, development, organ formation, cell proliferation and apoptosis, and etc. [5]. miRNA regulates gene expression, especially at the post-transcriptional level. It has key roles in cell development and differentiation by mediating the post-transcriptional regulation of protein-coding genes [6]. Abnormal expression of miRNA was found in many kinds of tumors including EC and was closely related with tumor development and metastasis [7]. Researchers suggested that the deregulation of miRNAs not only affected cancer progression, but also directly promoted tumor initiation [8]. The development of EC is a complex process involving in many pathogenic factors, multiple stages, and accumulation of multiple gene mutations and interactions. These factors cause dysregulation of oncogenes, tumor-suppressor genes and signaling protein molecules at the molecular level [9]. Previous studies found that miRNAs were frequently deregulated in EC, indicating that miRNAs were important in tumorigenesis of EC [10]. Nowadays, several miRNAs were supported to be potential therapeutic targets for the treatment of ESCC, such as miRNA (miR)-21, miR-375, miR-218 and etc. [10-12]. But there is still little known about the role of miRNAs in EC. As a member of miR-200 family which is becoming a new hotspot in researches of human cancer, miR-200a was also believed to be a tumor suppressor in cancers, but the exactly role of miR-200a in ESCC is still not clear.
In this study, we explored the effects of miR-200a on human ESCC cells by using miR-200a mimic transfection in vitro. The probable target gene of miR-200a in ESCC cells was also verified, as well as the potential mechanisms of miR-200a in regulating ESCC cells. Our findings might provide potential treatment targets and novel strategies for ESCC clinical therapy.
Materials and methods
Cell culture and transfection
Human ESCC TE3 cells were previously established from primary EC and maintained in 90% RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 100 U/ml penicillin and 100 U/ml streptomycin (GIBCO, Grand Island, NY, USA) at 37°C, in a humidified incubator under 5% CO2 condition. Cells were passaged at a ratio of 1:3 when 80% confluence was reached [13].
For miRNA transfection, synthetic miR-200a mimic and scramble miRNAs as negative control were purchased from Genepharma (Shanghai, China). For overexpression of Raf kinase inhibitor protein (RKIP), full length of RKIP sequence was cloned into the pBABE retroviral vector (Cell Biolabs, Inc., San Diego, CA, USA) and empty vector was used as negative control. All the transfections were performed by using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. After 48 h of transfection, cells were selected and collected for the next analysis.
RNA extraction and quantitative real-time PCR (qPCR) analysis
Total RNA of cells was extracted by using TRIzol reagent (Invitrogen) and was quantified by using agarose gel (1%) electrophoresis and spectrophotometric analysis (A260/A280 ratio). The RNA (500 ng) was reversely transcribed to cDNA by using NCode miRNA First-Strand cDNA synthesis kit (Invitrogen) according to the manufacturer’s instructions. The cDNA was used as the template for real-time PCR FastStart Universal SYBR green Master (Roche, Branchburg, NJ, USA) supplemented with the universal reverse primer in the kit. The qPCR analysis was performed on Applied Biosystems real-time detection system (Applied Biosystems, Foster City, CA, USA), and the thermocycling parameters were 95°C for 3 min and 40 cycles of 95°C for 15 s followed by 60°C for 30 s. Each sample was run in triplicate and was normalized to U6 snRNA levels (for miR-200a) or GAPDH levels (for mRNAs) [14]. Melting curve analysis was performed to confirm the specificity of the PCR products. The replicates were then averaged, and fold induction was determined by a ΔΔCT-based fold change calculation [15].
Cell viability assay
After corresponding treatments, cells were seeded in triplicate in 96-well plates at a density of 2 × 103 cells/well and were cultured at 37°C, in a humidified incubator with 5% CO2. The cell viability was assessed by using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay according to the standard methods. Briefly, on each day of the MTT assay, 50 µg of MTT (Sigma, St. Louis, MO, USA) were added into each well and the mix was incubated for another 4 h at 37°C. At the end of incubation, 100 μl of 0.04 N HCl in 2-propanol (Sigma) was mixed thoroughly into each well by shaking for 10 min. The absorbance of each well of plates was read on a Molecular Devices microplate reader (Sunnyvale, CA, USA) at a wavelength of 570 nm, with a background reading at 650 nm subtracted. Triplicate reading for each sample was averaged.
Cell invasion assay
Cell invasion was assessed by using 24-well Transwells and Matrigel (BD Biosciences, Franklin Lakes, NJ, USA). In brief, a total of 50 µl dissolved Matrigel was evenly coated onto the bottom of upper chamber of each Transwell. After 48 h of corresponding administration, cells were digested and centrifuged. Then cells were re-suspended in culture medium without FBS, adjusting the concentration of cells to 5 × 105/ml. Then, 20 µl of cell suspension was added onto the upper chamber, and 600 µl RPMI-1640 medium containing 10% FBS was added into the lower chamber. Transwells were incubated at 37°C with 5% CO2 for 48 h. The non-invasive cells on the upper chamber were carefully removed by using cotton swabs. Then Transwell was inverted and 600 µl anhydrous ethanol was added. After been air-dried and rinsed, cells were stained with crystal violet, counted and photographed under microscope (Leica Microsystems, Wetzlar, Germany).
Luciferase activity assay
The 3’ untranslated region (UTR) segment of RKIP gene containing the miR-200a binding sites was amplified by PCR and was inserted into the pmirGLO Dual-Luciferase miRNA target Expression vector (Promega, Madison, WI, USA) as RKIP 3’UTR wild type (WT). Cells were co-transfected with RKIP 3’UTR WT and miR-200a mimic or miRNA scramble control by using Lipofectamine 2000 reagent (Invitrogen). The binding site of miR-200a in RKIP 3’UTR WT was mutated via point mutation, which was referred to as negative control (RKIP 3’UTR Mt). The Firefly and Renilla luciferase activity were analyzed at 48 h post-transfection by using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) according to the manufacturer’s instructions. For each transfection, the luciferase activity was averaged from three replicates.
Western blot analysis
The proteins were extracted by RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) supplemented with protease inhibitors (Roche). The samples were quantified by using BCA™ Protein Assay Kit (Pierce, Appleton, WI, USA). Equal amount of samples were lysed with 1 × SDS-loading buffer (50 mM Tris-Cl pH 6.8, 100 mM DTT, 2% SDS, 10% glycerol and 0.1% bromophenol blue) as the whole-cell sample and were subjected to SDS-PAGE. All blots were transferred on to the poluvinylidene difluoride (PVDF) membranes. Immunoblots were carried out with primary antibodies at the dilution of 1:1000 including anti-RKIP (sc-101504), anti-Beta-Tubulin (sc-101527), anti-Raf1 (sc-52827), anti-phosphorylated (p)-Raf1 (sc-81513), anti-ERK1/2 (sc-94), anti-p-ERK1/2 (sc-101760), anti-matrix metalloproteinase (MMP)-14 (sc-377097), anti-LIN-28 (sc-293120) and anti-G-protein-coupled receptor kinase (GRK)-2 (sc-13143) (Santa Cruz, USA), respectively. Then the membranes were incubated with secondary antibody marked by horseradish peroxidase (HRP) (sc-516102, Santa Cruz), at the dilution of 1:5000. The intensity of blots were detected by enhanced chemiluminescence (ECL-plus, Amersham Pharmacia Biotech, Piscataway, New Jersey, USA) and quantified by using Image LabTM Software (Bio-Rad, Shanghai, China).
Statistical analysis
The results of multiple experiments were presented as mean + standard deviation (SD) except for the results of cell viability, which were presented as mean ± SD. Statistical analysis was performed by using GraphPad Prism 6 software (GraphPad Software, San Diego, CA, USA). The P-values were calculated by using a one-way analysis of variance (ANOVA) for multiple groups, and two-tailed Student’s t-test between two groups. P value of < 0.05 was considered to indicate statistically significant.
Results
miR-200a promoted cell viability and invasion of TE3 cells
To verify the effect of miR-200a on human ESCC, TE3 cells were transfected with miR-200a mimic or scramble miRNAs as negative control. The transfection efficiency was measured by qPCR. Results in Figure 1A showed that miR-200a mimic transfection significantly increased miR-200a expression in TE3 cells compared with control group (P < 0.01), suggesting that miR-200a expression level in TE3 cells was effectively increased after miR-200a mimic transfection.
Figure 1.
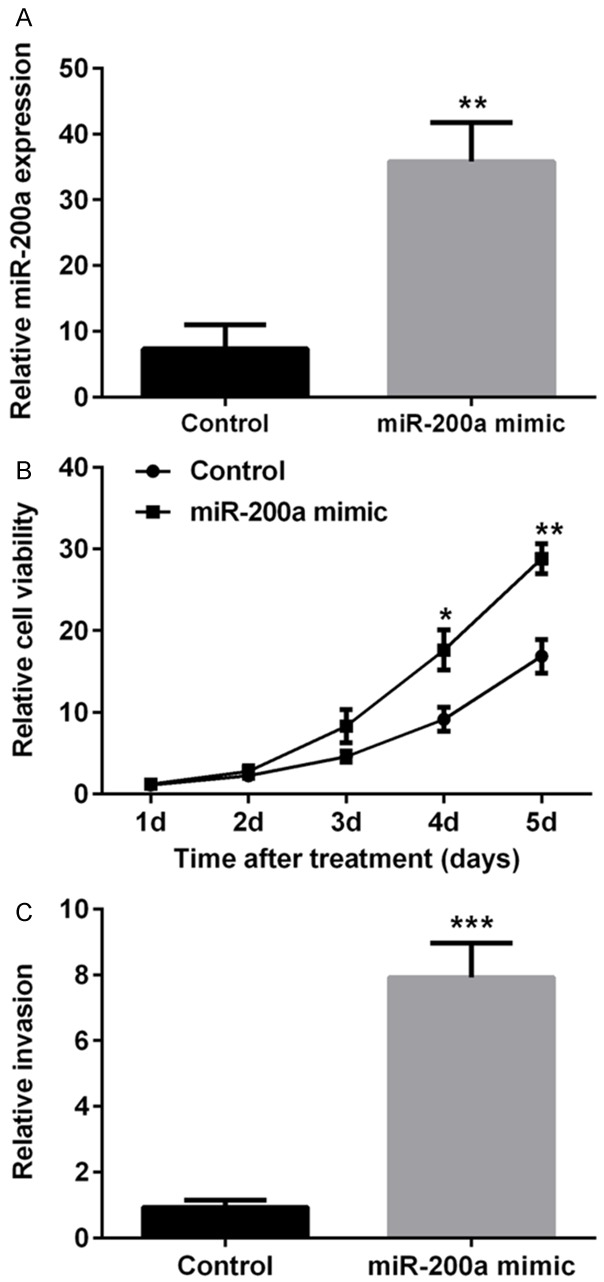
miR-200a mimic transfection promoted cell viability and invasion of human esophageal squamous cell carcinoma TE3 cells. miR-200a mimics or scramble control (control) were transfected into TE3 cells. Real-time PCR was performed to measure the expression level of miR-200a (A). Cell viability (B) and invasiveness (C) were measured by MTT and Transwell assays, respectively. *, P < 0.05; **, P < 0.01; ***, P < 0.001 compared to control group or that in corresponding time point of control group.
The cell viability and invasion of TE3 cells were also assessed after miRNA transfection. Results of MTT assay were shown in Figure 1B, cell viability was increased compared with control group at the corresponding test time (P < 0.05 or P < 0.01). The results of invasion assay in Figure 1C showed that miR-200a mimic transfection significantly increased cell invasion compared with that in control group (P < 0.001). These results suggested that miR-200a mimic transfection promoted human ESCC TE3 cell viability and invasion in vitro.
miR-200a regulated RKIP expression in TE3 cells via binding with RKIP 3’UTR
According to the predication analysis, the segment of RKIP 3’UTR had the potential binding sits of miR-200a as shown in Figure 2A. We assessed the binding effect of miR-200a on RKIP 3’UTR, luciferase assay results in Figure 2B showed that the fluorescence signal intensity was significantly inhibited in RKIP 3’UTR WT + miR-200a mimic transfection group compared with RKIP 3’UTR WT + miRNA scramble control transfection group (P < 0.001), while in RKIP 3’UTR Mt groups, there were no significant differences. These results suggested that RKIP might be a direct target of miR-200a in TE3 cells.
Figure 2.
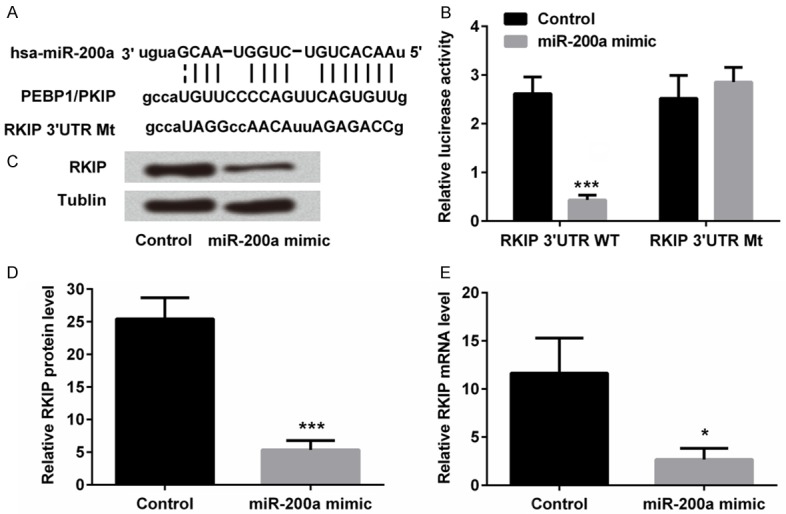
miR-200a directly regulated the expression of RKIP in human esophageal squamous cell carcinoma TE3 cells. The potential miR-200a binding sits on RKIP 3’UTR wild type (WT) and mutant (Mt) as shown (A). miR-200a and 3’UTR of RKIP were co-transfected into TE3 cells and luciferase activities were performed to assess binding effect of miR-200a on 3’UTR of RKIP (B). miR-200a mimic or scramble control (control) were transfected into TE3 cells. Western blot analysis and real-time PCR were performed to determine the protein (C and D) and mRNA (E) expressions of RKIP. *, P < 0.05; ***, P < 0.001 compared to control group.
Meanwhile, expression of RKIP in TE3 cells after miR-200a transfection was also measured. RKIP protein (Figure 2C and 2D) and mRNA (Figure 2E) expression levels in TE3 cells after transfection were measured, respectively. RKIP expression was decreased after miR-200a mimic transfection in both mRNA level (P < 0.05) and protein level (P < 0.001), suggesting that miR-200a negatively regulated expression of RKIP in TE3 cells.
RKIP overexpression inhibited effects of miR-200a on cell viability and invasion as well as activation of MAPK/ERK pathway in TE3 cells
RKIP was overexpressed in TE3 cells combined with miR-200a mimic transfection to further verify the effect of miR-200a. In Figure 3A, MTT assay results showed that miR-200a mimic transfection increased cell viability compared with control (P < 0.05 or P < 0.01 at corresponding time point), while combined with RKIP overexpression, cell viability was decreased with no significant difference with control group. Meanwhile, the invasion analysis results in Figure 3B showed that even miR-200a mimic transfection increased cell invasion compared with control group (P < 0.001), overexpression of RKIP in miR-200a transfected TE3 cells also significantly decreased cell invasion compared with miR-200a + empty vector group (P < 0.001). It suggested that as the direct target of miR-200a, overexpression of RKIP in TE3 cells inhibited the effects of miR-200a mimic transfection on cell viability and invasion.
Figure 3.
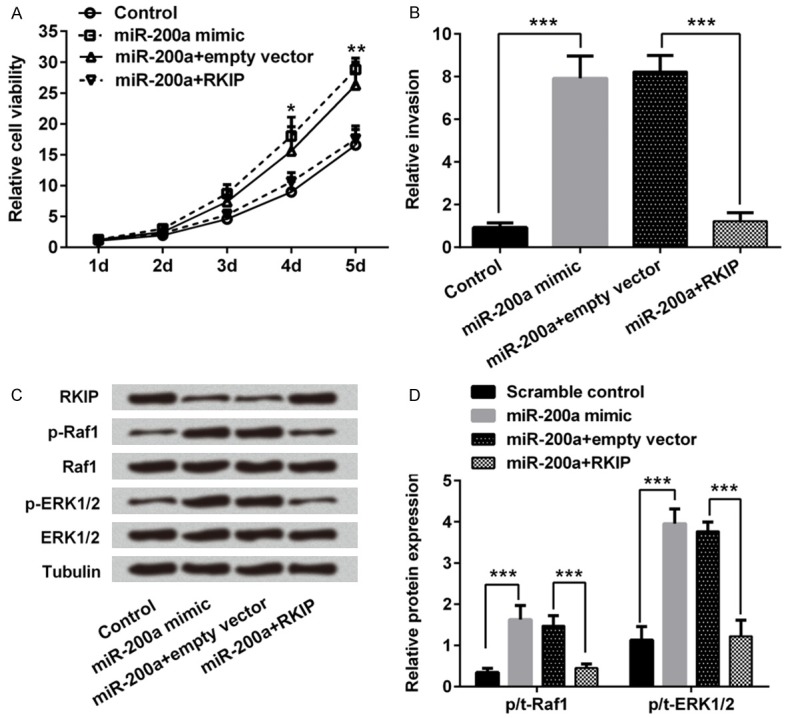
RKIP overexpression inhibited miR-200a induced increases of cell viability and invasion in human esophageal squamous cell carcinoma TE3 cells via inactivating MAP/ERK pathway. Cells were transfected with RKIP overexpression vector (RKIP) and/or miR-200a mimic. MTT and Transwells assays was performed to determine cell viability at corresponding time (A) and invasiveness (B). Protein expressions of RKIP, Raf1, phosphorylated (p)-Raf1, ERK-1/2 and p-ERK1/2 were measured by western blotting (C). Statistical results of phosphorylation levels about Raf1 and ERK1/2 to assess the activation of MAPK/ERK pathway in TE3 cells (D). *, P < 0.05; **, P < 0.01; compared to that in corresponding time point of control group. ***, P < 0.001 compared to the corresponding negative control.
RKIP was identified as an endogenous inhibitor of MAPK/ERK signaling, we then verified the potential mechanism of miR-200a-associated modulation of TE3 cells. The expression of Raf1 and EKR1/2 was determined in miR-200a mimic and/or RKIP transfection cells. Western blot assay results in Figure 3C showed that expression of RKIP was decreased in miR-200a mimic transfected groups, while phosphorylated Raf1 and ERK 1/2 expressions were increased after miR-200a mimic transfection. According to the statistical analysis results as shown in Figure 3D, phosphorylation levels of Raf1 and ERK1/2 were significantly increased in miR-200a transfected cells compared with control group (P < 0.001), while overexpression of RKIP in miR-200a mimic transfected cells significantly decreased phosphorylation levels of Raf1 and ERK1/2 compared with that in miR-200a mimic + empty vector group (P < 0.001). All these results suggested that miR-200a might be related with activity of MAPK/ERK pathway via RKIP in human EC TE3 cells.
miR-200a induced the expression of MMPs in TE3 cells
To further explore the role of miR-200a in human EC, expressions of some related factors were also analyzed. Considering that metalloproteinases are related to extracellular matrix (ECM) degradation which is closely associated with tumor cell migration and metastasis, we measured the expression of MMP-14. Meanwhile, LIN28 and GRK-2 which have been proved to play important roles in tumor development were also assessed. The qPCR analysis results in Figure 4A showed that miR-200a mimic transfection significantly increased mRNA expressions of MMP-14, LIN28 and GRK-2 compared with scramble control group (P < 0.001); while overexpression of RKIP in miR-200a mimic transfected cells significantly decreased these factors mRNA expression level compared to the miR-200a mimic + empty vector group (P < 0.001). Western blot assay results shown in Figure 4B and 4C suggested the inducible effect of miR-200a mimic transfection on these factors expression (P < 0.001), as well as that RKIP overexpression inhibited protein expressions compared with miR-200a + empty vector group (P < 0.001). According to these results, we speculated that miR-200a could elevate expressions of MMP-14, LIN28 and GRK-2 in TE3 cells and might regulate cell bioactivity via its target.
Figure 4.
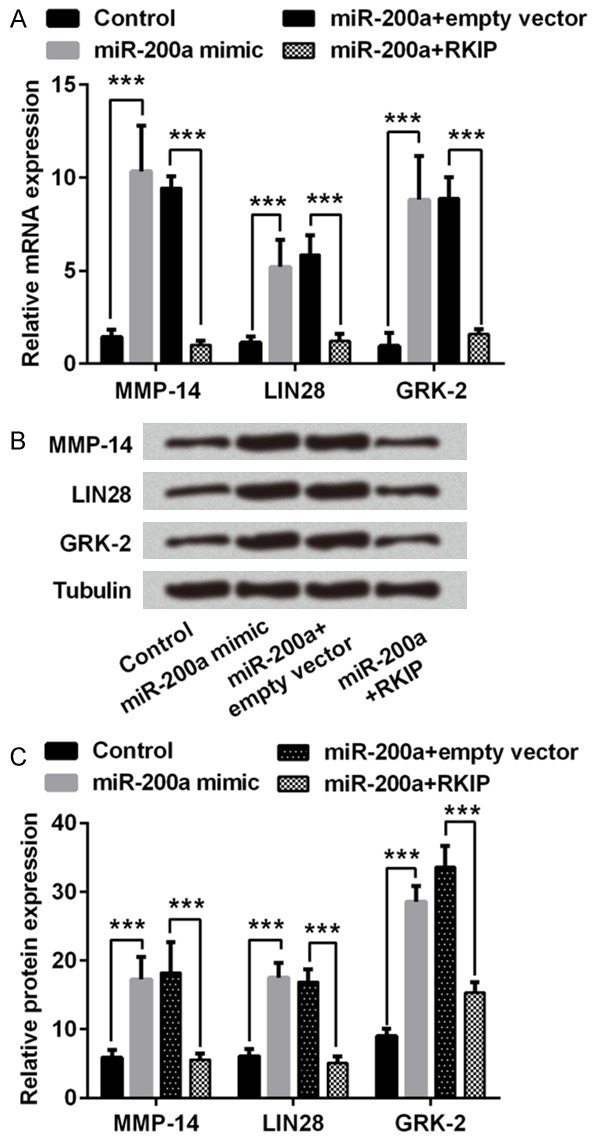
miR-200a increased the expressions of matrix metalloproteinase (MMP)-14, LIN28 and G protein-coupled receptor (GPCR) kinase 2 (GRK-2) in TE3 cells. Cells were transfected with RKIP overexpression vector (RKIP) and/or miR-200a mimic. The mRNA expression level was measured by real-time PCR (A) and protein expression level was assessed by Western blotting (B). Statistical results of immunoblot analysis (C). ***, P < 0.001 compared to the corresponding negative control.
Discussion
EC, especially ESCC, is one of the most common malignant tumors. Most patients with ESCC were diagnosed at an advanced stage [16]. Despite recent improvements in treatment of ECSS, the clinical outcome of ESCC patients remains unsatisfactory [17]. The carcinogenesis of ECSS and its development process is involved in a variety of oncogenes and tumor suppressor genes involved in the abnormal mutation and mutual regulation of complex process. It is urgently needed to investigate the underlying molecular markers to improve the outcome of patients with ESCC [18]. In this study, we made primary researches about the effects of miR-200a on ESCC cells in vitro, and also verified that RKIP was the direct target of miR-200a in TE3 cells. The up-regulation of miR-200a in ESCC TE3 cells promoted cell viability and invasion, as well was suppressed its target RKIP expression via binding with the 3’UTR of RKIP, which was also increased the activation of MAPK/ERK signal pathway. Abnormal expression of miR-200a was probably related with TE3 cell bioactivity.
Previous studies found that miRNAs were frequently deregulated in ESCC, indicating that the deregulation of miRNAs played important roles in the tumorigenesis of ESCC [19]. As a member of miR-200 family which has been proved to act as tumor suppressors, miR-200a is significantly involved in inhibition of epithelial-to-mesenchymal transition (EMT), repression of cancer stem cells (CSCs) self-renewal and differentiation, modulation of cell division and apoptosis, and reversal of chemoresistance [20-22]. Experimental evidence revealed that the majority of human miRNA genes were located at fragile sites and genomic regions involved in cancer, functioning as oncogenes or tumor suppressor genes [23]. In this study, we found that upregulated miR-200a could prompt cell viability and invasion in vitro, suggesting that up-regulation of miR-200a might be related with metastasis of ESCC.
As known, in miRNA biology, seed sequences, in terms of the complementary sequences to binding sites of miRNA within the 3’UTRs of the target genes, play a crucial role in regulatory effects of miRNA on gene expression [24]. In our study, we found that RKIP was a direct target of miR-200a in TE3 cells, miR-200a binding with 3’UTR of RKIP, negatively regulated expression of RKIP in vitro. RKIP is a member of the phosphatidylethanolamine-binding protein (PEBP) family and plays a pivotal modulatory role in several protein kinase signaling cascades [25]. RKIP inhibits Raf-1-mediated phosphorylation of MEK through binding to Raf-1. It is a negative regulator of the Raf/MEK/ERK signaling pathway [26]. Research had proved that ectopic expression of RKIP antagonized the kinase activity of B-Raf, and expression levels of RKIP in melanoma cancer cell lines were low relative to primary melanocytes [27]. The regulatory role of RKIP in cell signaling is reflected in its role in physiology and pathophysiology and thus also be related with regulation of cancer cells [28]. Our results suggested that miR-200a regulated cell viability and invasion of ESCC cells in vitro via regulating its target gene RKIP expression. This regulatory effect was also involved in the MAPK/ERK signaling pathway, that the upregulation of miR-200a in TE3 cells, which inhibited the expression of RKIP as an inhibitor of Raf/MEK/ERK pathway, could effectively increase the phosphorylation levels of Raf and ERK, and thus increased activity of MAPK/ERK signaling pathway.
In the present study, we also found that miR-200a could increase the expression of MMP-14, the RNA-binding protein LIN28 and GRK-2 in both mRNA and protein levels which were inhibited by RKIP in EC TE-3 cells. RKIP was proved to prevent the invasion of cancer cells via controlling the expression of MMPs [29]. MMPs overexpression might indicate a higher risk of poor prognosis in cancers [30]. MMP-1 or MMP-2 in RKIP knockout cancer cells was reverted invasiveness and metastatic potential to normal levels [31]. In the researches about RKIP effect on esophageal TE-1 cancer cells, overexpression of RKIP inhibited MMP-14 expression and reduced transcription of LIN28 which is posttranscriptional repressor of let-7 miRNA biogenesis, and thus reduced the invasiveness of esophageal TE-1 cancer cells [29]. The Lin28-mediated downregulation of let-7 might also play a key role in development, stem cell programming, and tumorigenesis [32]. Meanwhile, GRK-2 is a ubiquitous member of the GRK family that appears to play a central, integrative role in signal transduction cascades. GRK-2 is a negative feedback inhibitory protein for G protein-coupled receptors (GPCRs) which has been shown to phosphorylate GPCRs, and separate G protein from GPCRs [33,34]. In addition, emerging evidence indicates that alterations in GRK-2 levels or functionality participated in the development of several tumor or inflammatory diseases [34]. The binding conversion from Raf-1 to GRK-2 suggests that RKIP serves as a signal regulator in ESCC TE-3 cells, and miR-200a was involved in regulation of the signaling pathways via RKIP.
In conclusion, our findings demonstrated that miR-200a promoted cell viability and invasion of human EC cells via downregulating RKIP, which could inactivate MAP/ERK signaling pathway, and regulated expressions of MMP-14, LIN28 and GRK-2. However, further studies about the expression of miR-200a in ESCC tissues, as well as the usage of animal models simulated in vivo conditions will help to understand the exact role of miR-200a, and provide a potential clinical therapeutic strategy for treatment of ESCC.
Acknowledgements
This work was supported by the Key Laboratory Project of Tumor Molecular Biology of Ningbo City (2015A22011).
Disclosure of conflict of interest
None.
References
- 1.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2004;349:2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 2.Whiteman DC. Esophageal cancer: priorities for prevention. Curr Epidemiol Rep. 2014;1:138–148. [Google Scholar]
- 3.Mao WM, Zheng WH, Ling ZQ. Epidemiologic risk factors for esophageal cancer development. Asian Pac J Cancer Prev. 2011;12:2461–2466. [PubMed] [Google Scholar]
- 4.Zhu ML, He J, Wang M, Sun MH, Jin L, Wang X, Yang YJ, Wang JC, Zheng L, Xiang JQ, Wei QY. Potentially functional polymorphisms in the ERCC2 gene and risk of esophageal squamous cell carcinoma in Chinese populations. Sci Rep. 2014;4:6281. doi: 10.1038/srep06281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jose AM. Movement of regulatory RNA between animal cells. Genesis. 2015;53:395–416. doi: 10.1002/dvg.22871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–433. doi: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- 7.Xie Z, Chen G, Zhang X, Li D, Huang J, Yang C, Zhang P, Qin Y, Duan Y, Gong B. Salivary microRNAs as promising biomarkers for detection of esophageal cancer. PLoS One. 2013;8:e57502. doi: 10.1371/journal.pone.0057502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferracin M, Veronese A, Negrini M. Micromarkers: miRNAs in cancer diagnosis and prognosis. Expert Rev Mol Diagn. 2010;10:297–308. doi: 10.1586/erm.10.11. [DOI] [PubMed] [Google Scholar]
- 9.Qian J, Fang JY. Genetic variations in esophageal cancer. Gastrointestinal Tumors. 2015;2:262–268. [Google Scholar]
- 10.He B, Yin B, Wang B, Xia Z, Chen C, Tang J. MicroRNAs in esophageal cancer. Mol Med Rep. 2012;6:459–465. doi: 10.3892/mmr.2012.975. [DOI] [PubMed] [Google Scholar]
- 11.Fu W, Pang L, Chen Y, Yang L, Zhu J, Wei Y. The microRNAs as prognostic biomarkers for survival in esophageal cancer: a metaanalysis. ScientificWorldJournal. 2014;2014:523979. doi: 10.1155/2014/523979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiang Z, Song Q, Yang S, Zeng R, Li X, Jiang C, Ding W, Zhang J, Zheng Y. Serum microRNA-218 is a potential biomarker for esophageal cancer. Cancer Biomarkers. 2015;15:381. doi: 10.3233/CBM-150480. [DOI] [PubMed] [Google Scholar]
- 13.Chen XY, He QY, Guo MZ. XAF1 is frequently methylated in human esophageal cancer. World J Gastroenterol. 2012;18:2844–2849. doi: 10.3748/wjg.v18.i22.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Motiño O, Francés DE, Mayoral R, Castro-Sánchez L, Fernández-Velasco M, Boscá L, García-Monzón C, Brea R, Casado M, Agra N. Regulation of microRNA 183 by cyclooxygenase 2 in liver is DEAD-Box helicase p68 (DDX5) dependent: role in insulin signaling. Mol Cell Biol. 2015;35:2554–2567. doi: 10.1128/MCB.00198-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 16.Qiu Y, Shi Z. Progress on genomic aberrations in esophageal squamous-cell carcinoma. China J Pathophysiol. 2016;32:952–955. [Google Scholar]
- 17.Hao XW, Zhu ST, He YL, Li P, Wang YJ, Zhang ST. Epigenetic inactivation of secreted frizzled-related protein 2 in esophageal squamous cell carcinoma. World J Gastroenterol. 2012;18:532–540. doi: 10.3748/wjg.v18.i6.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallböhmer D, Brabender J, Metzger R, Hölscher AH. Genetics in the pathogenesis of esophageal cancer: possible predictive and prognostic factors. J Gastrointest Surg. 2010;14(Suppl 1):S75–80. doi: 10.1007/s11605-009-1021-5. [DOI] [PubMed] [Google Scholar]
- 19.Wu BL, Xu LY, Du ZP, Liao LD, Zhang HF, Huang Q, Fang GQ, Li EM. MiRNA profile in esophageal squamous cell carcinoma: downregulation of miR-143 and miR-145. World J Gastroenterol. 2011;17:79–88. doi: 10.3748/wjg.v17.i1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haraguchi T, Kondo M, Uchikawa R, Kobayashi K, Hiramatsu H, Kobayashi K, Chit UW, Shimizu T, Iba H. Dynamics and plasticity of the epithelial to mesenchymal transition induced by miR-200 family inhibition. Sci Rep. 2016;6:21117. doi: 10.1038/srep21117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bao B, Azmi AS, Ahmad A, Li Y, Banerjee S, Kong D, Ali S, Sarkar FH. The biological roles of microRNAs in cancer stem cells. Springer International Publishing; 2014. [Google Scholar]
- 22.Magenta A, Cencioni C, Fasanaro P, Zaccagnini G, Greco S, Sarraferraris G, Antonini A, Martelli F, Capogrossi MC. miR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via ZEB1 inhibition. Cell Death Differ. 2011;18:1628–1639. doi: 10.1038/cdd.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferdin J, Kunej T, Calin GA. Non-coding RNAs: identification of cancer-associated microRNAs by gene profiling. Technol Cancer Res Treat. 2010;9:123–138. doi: 10.1177/153303461000900202. [DOI] [PubMed] [Google Scholar]
- 24.Song CQ, Zhang JH, Shi JC, Cao XQ, Song CH, Hassan A, Wang P, Dai LP, Zhang JY, Wang KJ. Bioinformatic prediction of SNPs within miRNA binding sites of inflammatory genes associated with gastric cancer. Asian Pac J Cancer Prev. 2014;15:937. doi: 10.7314/apjcp.2014.15.2.937. [DOI] [PubMed] [Google Scholar]
- 25.Yousuf S, Duan ML, Moen EL, Crossknorr S, Brilliant K, Bonavida B, Lavalle T, Yeung KC, Almulla F, Chin E. Raf kinase inhibitor protein (RKIP) blocks signal transducer and activator of transcription 3 (STAT3) activation in breast and prostate cancer. PLoS One. 2014;9:e92478. doi: 10.1371/journal.pone.0092478. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Chatterjee D, Sabo E, Tavares R, Resnick MB. Inverse association between Raf kinase inhibitory protein and signal transducers and activators of transcription 3 expression in gastric adenocarcinoma patients: implications for clinical outcome. Clin Cancer Res. 2008;14:2994–3001. doi: 10.1158/1078-0432.CCR-07-4496. [DOI] [PubMed] [Google Scholar]
- 27.Park S, Yeung ML, Beach S, Shields JM, Yeung KC. RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene. 2005;24:3535–3540. doi: 10.1038/sj.onc.1208435. [DOI] [PubMed] [Google Scholar]
- 28.Xinzhou H, Ning Y, Ou W, Xiaodan L, Fumin Y, Huitu L, Wei Z. RKIp inhibits the migration and invasion of human prostate cancer PC-3M cells through regulation of extracellular matrix. Mol Biol. 2011;45:921–928. [PubMed] [Google Scholar]
- 29.Zhao D, Ma J, Shi J, Cheng L, Li F, Jiang X, Jiang H. Raf kinase inhibitor protein inhibits esophageal cancer cell invasion through downregulation of matrix metalloproteinase expression. Oncol Rep. 2013;30:304–312. doi: 10.3892/or.2013.2464. [DOI] [PubMed] [Google Scholar]
- 30.Ren F, Tang R, Zhang X, Luo D, Dang Y, Li Z, Wei K, Chen G. Overexpression of MMP family members functions as prognostic biomarker for breast cancer patients: a systematic review and meta-analysis. PLoS One. 2015;10:e0135544. doi: 10.1371/journal.pone.0135544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beshir AB, Ren G, Magpusao AN, Barone LM, Yeung KC, Fenteany G. Raf kinase inhibitor protein suppresses nuclear factor-κBdependent cancer cell invasion through negative regulation of matrix metalloproteinase expression. Cancer Lett. 2010;299:137–149. doi: 10.1016/j.canlet.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heo I, Joo C, Cho J, Ha M, Han J, Kim VN. Lin28 mediates the terminal uridylation of let-7 precursor microRNA. Mol Cell. 2008;32:276. doi: 10.1016/j.molcel.2008.09.014. [DOI] [PubMed] [Google Scholar]
- 33.Wang L, Gestypalmer D, Fields TA, Spurney RF. Inhibition of WNT signaling by G proteincoupled receptor (GPCR) kinase 2 (GRK2) Mol Endocrinol. 2009;23:1455–1465. doi: 10.1210/me.2009-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Penela P, Murga C, Ribas C, Lafarga V, Mayor F Jr. The complex G protein-coupled receptor kinase 2 (GRK2) interactome unveils new physiopathological targets. Br J Pharmacol. 2010;160:821. doi: 10.1111/j.1476-5381.2010.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]