

Abstract
Chemokines and their receptors play an important role in the pathogenesis of acute and chronic diabetic nephropathy (DN). However, their expression pattern and function in glomerular podocytes have not been investigated as of yet. In the present study, we investigated whether CXCR3 could protect podocytes from high glucose-induced apoptosis and inflammatory cytokine production and explored the possible mechanism. Cell viability, cell cycle and apoptosis were detected by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and flow cytometry, respectively. The level of intracellular reactive oxygen species (ROS) and mitochondrial membrane potential (∆Ψm) was measured using a dichlorofluorescein diacetate (DCFH-DA) ortetrechloro-tetraethylbenzimidazol carbocyanine iodide (JC-1) fluorescent probe, respectively. Quantitative real-time PCR was used to determine the gene expression of CXCR3. Western blots were carried out for the related protein expression in podocytes, including CXCR3, Nephrin, Podocin, Bcl-2, Bax, and Caspase-3. Firstly, we found that CXCR3 expression was significantly up-regulated and cell viability was decreased in high glucose (HG)-treated mouse podocytes in a dose-dependent manner. Secondly, knockdown of CXCR3 in mouse podocytes significantly suppressed HG-induced viability decrease, cell cycle arrest, ROS generation and ∆Ψm reduction. Moreover, knockdown of CXCR3 reduced the podocytes injury in cell apoptosis and inflammation through increasing the expression of Nephrin, Podocin and Bcl-2, and decreasing the expression of Bax and Caspase-3. In conclusion, CXCR3 knockdown protected podocytes from HG-induced apoptosis and inflammation in vitro, suggesting that inhibition of CXCR3 may have a therapeutic potential in DN treatment.
Keywords: CXCR3, high glucose, podocyte, apoptosis, inflammatory cytokine
Introduction
Diabetic nephropathy (DN) is a serious complication of diabetes and a frequent cause of end-stage renal disease (ESRD). The structural changes in kidney are generally observed in the patients of DN, including thickening of glomerular basement membrane, mesangial expansion, glomerular hypertrophy, fibroblast proliferation, matrix deposition glomerulosclerosis and tubular necrosis [1,2]. The causes of DN have been intensively studied and various mechanisms have been established, such as high blood glucose, polyol pathway activation, advanced glycation end product formation, activation of the protein kinase C pathway and reactive oxygen species (ROS) generation [3,4]. Podocytes are terminally differentiated cells that contribute to maintaining the integrity of the glomerular basement membrane and have been confirmed to play a vital role in the development of the pathological changes that characterize DN [5]. A reduction in podocyte number resulting from apoptosis has been shown to be the strongest predictor of progression of DN, where fewer cells predicted more rapid progression [6,7]. Thus, seeking effective methods for inhibiting podocyte apoptosis may be of great clinical importance in the treatment of DN. However, the consequences, molecular pathways and pathomechanism(s) that underlie the loss of podocytes in DN remain poorly understood.
Mitochondrial dysfunction, triggered by hyperglycemia causes excess production and accumulation of ROS which result in oxidative stress, is increasingly recognized as contributing to glomerular diseases and is an early event in podocyte apoptosis [8,9]. An increased ROS generation and a loss of mitochondrial membrane potential are associated with the pathogenesis of DN [10]. High glucose induces intracellular ROS in mesangial cells and tubular epithelial cells, which can be successfully blocked by the inhibition of NADPH oxidase [11]. Therefore, ameliorating mitochondrial dysfunction is a potential way to improve podocyte apoptosis and DN.
Diabetic nephropathy is increasingly considered as an inflammatory disease characterized by leukocyte infiltration at every stage of renal involvement. Chemokines, a group of small peptides that are subdivided into four families comprising more than 50 ligands with at least 17 different receptors, are important participators in the recruitment of specific subpopulations of inflammatory cells into renal compartments [12,13]. Within the glomerulus, chemokines and their receptors are expressed in infiltrating cells as well as in resident glomerular cells. Glomerular-produced chemokines seem not only to induce recruitment of inflammatory cells, but can also alter functions of resident glomerular cells, such as the formation of extracellular matrix [14]. Previous study showed that downregulation of CXCL9 receptor CXCR3 suppressed the loss of renal function and may be a potential therapeutic target for human immune-mediated nephritis [15]. It is unknown whether CXCR3 expressed in podocyte, and whether CXCR3 involved in podocyte injury and the development of DN.
In this study, we investigate the molecular events of CXCR3-mediated renal injury in DN by in vitro experiment. We demonstrate that CXCR3 is up-regulated in DN, contributing to podocyte apoptosis and inflammatory cytokines release.
Materials and methods
Chemicals and reagents
RPMI 1640 medium and FBS (fetal bovine serum) were obtained from Gibco Invitrogen Corporation (Carlsbad, China). Trypsin, D-glucose and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma (St Louis, MO). Fluorescein isothiocyanate (FITC)-conjugated annexin V and propidium iodide were purchased from KeyGen Biotech (Nanjing, China). siRNA was synthesized by SBS Genetech Co., Ltd (Beijing, China). Lipofectamine 2000 reagent was obtained from Invitrogen (CA, USA). CXCR3, Nephrin, Podocin, Bcl-2, Bax, Caspase-3 and GAPDH antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). Horseradish peroxidase-conjugated secondary antibodies were purchased from Zhongshanjinqiao Biotechnology (Beijing, China).
Cell culture and treatment
Conditionally immortalized mouse podocytes were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 10 U/ml recombinant mouse interferon-γ at 33°C under a humidified atmosphere of 5% CO2 for propagation. Proliferating cells were transferred to an incubator at 37°C in interferon-γ-free medium for 7-14 days to induce differentiation. Cells were incubated with serum-free RPMI 1640 medium for 24 h and then exposed to different doses of D-glucose (5.5, 15, 25, 35 or 50 mM) for the experimental group.
CXCR3 siRNA transfection
For CXCR3 silencing, podocytes (2×105 cells per well) were seeded in 6-well plates and transfected with 30 nM of CXCR3 small interfering RNA (siRNA) by using Lipofectamine 2000 reagent according to the manufacturer’s instructions. Non-specific control siRNA was used as a negative control (NC). Transfected cells were incubated at 37°C for 6 h, and then cells were treated with 35 mM HG for 48 h. Subsequently, the levels of CXCR3, Nephrin, Podocin, Bcl-2, Bax, Caspase-3 and GAPDH were analyzed by western blots.
MTT assay
MTT assay was used to measure cell viability. Podocytes (104 cells per well) were incubated in 96-well plates. Before the experiments, RPMI 1640 medium was removed and replaced with serum-free medium for 24 h incubation. Then cells were treated with HG (35 mM) with or without CXCR3 siRNA transfection for another 12, 24, 48 or 72 h. 100 μl MTT (0.5 mg/ml) was added. After continued incubation at 37°C for an additional 4 h, the medium was carefully removed. Cells were read at 570 nm using a microplate reader.
Flow cytometry analysis
Podocytes were seeded onto 6-well plate at a density of 3×105 per well. After treatment as described above for 48 h, podocytes were collected and washed with PBS. For cell cycle analysis, cell apoptosis, MMP and ROS, podocytes were incubated with propidium iodide, Annexin V-FITC and PI, dichlorofluorescein diacetate (DCFH-DA) or tetrechloro-tetraethylbenzimidazol carbocyanine iodide (JC-1) for 30 minutes, respectively. The cells were then harvested for flow cytometric analysis by using FACSCalibur Flow Cytometer (BD Bioscience).
Measurement of TNF-α, IL-6 and IL-1β release
The podocytes were cultured in 6-well plates. They were kept at 37°C for 48 h at treatment as described above before supernatants were taken. TNF-α, IL-6 and IL-1β concentrations were measured with an ELISA (R&D Systems, Wiesbaden, Germany) following the manufacturer’s instructions.
Quantitative real-time PCR
Total RNA from cultured podocytes (106 cells) was extracted using Trizol reagent (Invitrogen, Carlsbad, CA) and a DNeasy Tissue Kit (Qiagen Sciences, Germantown, MD) according to the manufacturer’s instructions. The sequences of the primers were as follows: CXCR3 5’-GACTCAAAGCCACCTCATTC-3’ and 5’-GCCTTGCCTTCCTAAATACC-3’; GAPDH 5’-ATCACTGCCACCCAGAAG-3’ and 5’-TCCACGACGGACACATTG-3’. Quantitative real-time PCR was used to detect the relative CXCR3 and GAPDH content using an ABI 7300 Real-time PCR Detection System. The values were determined relative to the control sample after normalizing to GAPDH gene control values and calculated by the comparative cycle threshold (ΔΔCt) method.
Western blot
The cells were rinsed with ice-cold phosphate-buffered saline (PBS) and lysed in RIPA buffer mixed with protease inhibitors (P8340, Sigma-Aldrich) for 30 min on ice. The concentration of protein was measured using a BCA protein assay kit (Beyotime Biotechnology, Beijing, China) according to the manufacturer’s instructions. An equal amount of protein (50 μg) from each sample was separated by SDS-PAGE and transferred to PVDF membranes. After blocking in blocking buffer containing 5% skim milk for 30 min, the membranes were incubated with primary antibodies for CXCR3, Nephrin, Podocin, Bcl-2, Bax, Caspase-3 and GAPDH separately at 4°C overnight. The membranes were incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h and signals were detected by using enhanced chemiluminescence (BioRad, Richmond, CA, USA). Densitometric analysis was performed using Image Pro Plus 6.0 software. Values were corrected with GAPDH as a control.
Statistical analysis
All values were presented as mean ± SD and analyzed with SPSS 17.0. Differences in mean values were tested using student’s t test or one-way ANOVA test. Differences are statistically if P<0.05.
Results
Effect of high glucose on cell viability and CXCR3 expression in cultured podocytes
The dose-dependent effect of high glucose (HG) on cell viability of podocytes was investigated firstly. Treatment of podocytes with different doses of HG (15, 25, 35 or 50 mM) for 12, 24, 48 and 72 h significantly inhibited the cell viability of podocytes compared with normal control group (5.5 mM HG) and showed a dose-dependent manner (Figure 1A). The increased expression of CXCR3 was also detected in podocytes exposed to high glucose (HG) in vitro. As shown in Figure 1B-D, the mRNA and protein levels of CXCR3 expression dramatically increased after 48 h in different doses of HG (15, 25, 35 or 50 mM) medium compared with normal control group (5.5 mM HG). The results suggested that the HG-promoted expression and distribution of CXCR3 might be involved in the biological changes in podocytes in diabetes.
Figure 1.
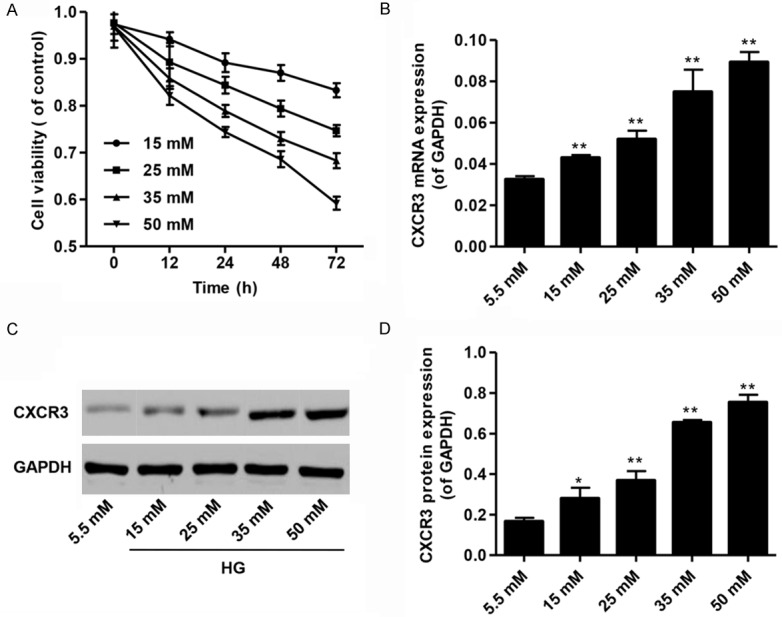
Cell viability and CXCR3 expression in the cultured podocytes under the high glucose treatment. A. The cell viability of podocytes cultured under high glucose (HG; 15, 25, 35 or 50 mM) for 12, 24, 48 or 72 h by MTT assay. B. CXCR3 mRNA expression in cultured podocytes with HG (15, 25, 35 or 50 mM) for 48 h. C, D. Representative western blot and quantification of CXCR3 expression in cultured podocytes with HG (15, 25, 35 or 50 mM) for 48 h. *P<0.05, **P<0.01 compared with normal control (5.5 mM D-glucose).
CXCR3 knockdown increases cell viability in podocytes with HG treatment
To further investigate the role of CXCR3 on podocyte function, CXCR3 siRNA or control shRNA (NC) were used in this study. Our results showed a reduced expression of CXCR3 mRNA and protein in podocytes transduced with CXCR3 siRNA (Figure 2A-C). HG (35 mM) treatment led to a significant decrease in cell viability compared with normal control as measured by MTT, but CXCR3 siRNA significantly inhibited HG-induced decrease in cell viability (Figure 2D). These findings indicated that CXCR3 was involved in the cell viability in podocyte injury promoted by HG.
Figure 2.
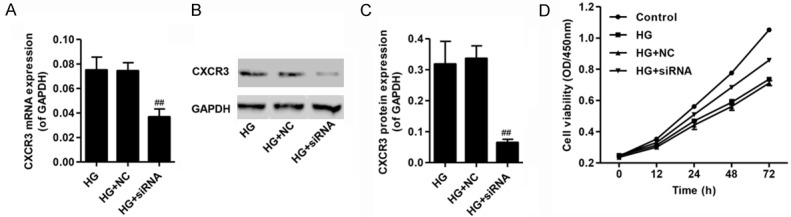
Knockdown of CXCR3 increased cell viability of podocytes with high glucose (35 mM) treatment. A. Quantification of CXCR3 mRNA expression showing the efficiency of CXCR3 knockdown by siRNA transfection. B, C. Representative western blot and quantification showing the efficiency of CXCR3 knockdown by siRNA transfection. D. The cell viability of podocytes cultured under HG (35 mM) for 12, 24, 48 or 72 h with or without CXCR3 siRNA transfection by MTT assay. ##P<0.01 compared with HG.
CXCR3 knockdown inhibits cell cycle arrest and apoptosis in podocytes with HG treatment
Moreover, HG treatment led to a significant cell cycle arrest at S and G2/M phases and increase in cell apoptosis compared with normal control as measured by flow cytometry, but CXCR3 siRNA significantly inhibited HG-induced cell cycle arrest at S and G2/M phases and increase in cell apoptosis (Figure 3A-D). These findings indicated that CXCR3 was involved in the cell apoptosis in podocyte injury promoted by HG.
Figure 3.

Knockdown of CXCR3 suppressed cell cycle arrest and apoptosis in podocytes with high glucose (35 mM) treatment. The effects of CXCR3 knockdown on the cell cycle (A, B) and cell apoptosis (C, D) in podocytes with HG (35 Mm) treatment for 48 h were determined by flow cytometry analysis. **P<0.01 compared with normal control (5.5 mM D-glucose). ##P<0.01 compared with HG.
CXCR3 knockdown increases ∆Ψm level and decreases ROS generation in podocytes with HG treatment
To investigate whether cell apoptosis was associated with mitochondrial dysfunction, we analyzed ∆Ψm and ROS changes in podocytes. As shown in Figure 4A-D, HG treatment significantly reduced the ∆Ψm level and increased the ROS production in podocytes compared with normal control, which was partially rescued by CXCR3 knockdown. These data suggested that HG induces cell apoptosis through the intrinsic pathway.
Figure 4.
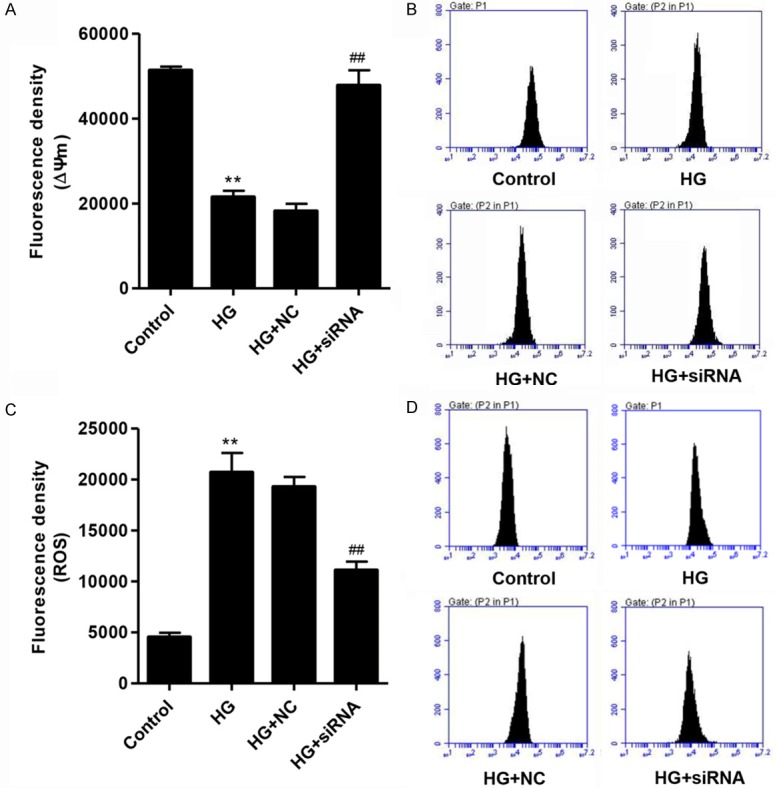
Knockdown of CXCR3 induced ∆Ψm increase and ROS decrease in podocytes with high glucose (35 mM) treatment. The effects of CXCR3 knockdown on the ∆Ψm (A, B) and ROS generation (C, D) in podocytes with HG (35 Mm) treatment for 48 h were determined by flow cytometry analysis. **P<0.01 compared with normal control (5.5 mM D-glucose). ##P<0.01 compared with HG.
CXCR3 knockdown inhibits inflammatory cytokine production in podocytes with HG treatment
To further investigate the role of CXCR3 on podocyte inflammatory response, inflammatory cytokines production was measured by ELISA. As shown in Figure 5A-C, HG treatment increased the production of TNF-α, IL-6 and IL-1β compared with normal control, which was partially rescued by CXCR3 knockdown. These findings indicated that CXCR3 was involved in the inflammatory response in podocyte injury promoted by HG.
Figure 5.

Knockdown of CXCR3 reduced inflammatory response in podocytes with high glucose (35 mM) treatment. Levels of inflammatory cytokines, including TNF-α (A), IL-1β (B) and IL-6 (C), were evaluated by ELISA assay. **P<0.01 compared with normal control (5.5 mM D-glucose). ##P<0.01 compared with HG.
Effect of CXCR3 knockdown on protein expression in podocytes with HG treatment
Nephrin and Podocin are the most important components of slit membrane of podocytes [16]. We verified the expression of Nephrin and Podocin in podocytes by western blot assay. HG significantly reduced the expression of Nephrin and Podocin in podocytes, which were up-regulated by CXCR3 knockdown (Figure 6A and 6B). Meanwhile, HG treatment increased the expression of Bax and Caspase-3, two pro-apoptosis proteins, while decreased level of the anti-apoptosis protein Bcl-2 in podocytes. In addition, when CXCR3 was knockdown by siRNA, the expression of Bax and Caspase-3 was decreased, while expression of Bcl-2 increased dramatically (Figure 6A and 6B). These data suggested that CXCR3 plays a key role on HG-induced podocytes apoptosis.
Figure 6.

Effect of knockdown of CXCR3 on protein expression in podocytes with high glucose (35 mM) treatment. A, B. Representative western blot and quantification of Nephrin, Podocin, Bcl-2, Bax and Caspase-3 expression in cultured podocytes with HG (35 mM) for 48 h. *P<0.01, **P<0.01 compared with normal control (5.5 mM D-glucose). #P<0.05, ##P<0.01 compared with HG.
Discussion
The CXC chemokines, the important chemotactic molecules that control leukocyte trafficking and function, play an important role in several additional biological functions, such as regulation of lymphocyte development, expression of adhesion molecules, cell proliferation, angiogenesis, virus-target cell interactions, and in various aspects of cancer [17,18]. Our study is the first evidence suggesting that CXCR3 contributes to podocyte injury and the development of DN. In the present study, the up-regulation of CXCR3 expression was observed in podocytes with DN induced by HG treatment and associated with cell viability and apoptosis of podocytes, ROS production, mitochondrial membrane potential and inflammatory cytokines release. In addition, our results showed that gene silencing of CXCR3 significantly decreased HG-induced podocytes injury in vitro, and attenuated renal injury through, regulating apoptosis factors, increasing Nephrin and Podocin expression, and reducing proinflammatory cytokines production in podocytes. It is significantly noted that CXCR3 takes a key role in the onset and development of DN and could be a potential target molecule in the regulation of podocyte function.
CXCR3 has been detected in mesangial cells of patients with IgA nephropathy, membranoproliferative glomerulonephritis, and rapidly progressive glomerulonephritis, indicating that CXCR3 might contribute to mesangial cell proliferation in these diseases [12,19]. A clinical study has demonstrated that elevated blood glucose level was the main risk factor in the development of DN. High glucose was often used as a stimulant to induce podocyte apoptosis in previous studies and the phenotype of podocytes cultured in HG environments resembles the phenotype of DN [20,21]. Previous studies conducted by other groups indicate that abnormalities in podocyte structure and function occur early in the course of DN [22]. Based on the results from this study and the importance of podocytes in the pathogenesis of DN, we focused on podocyte injury in this study. In the present study, we treated the mouse podocytes with different doses of HG resulting in up-regulation of CXCR3 expression and a decrease in cell viability of podocytes. With the transfection of CXCR3 siRNA, cell viability and the expression of two components of slit membrane of podocytes, Podocin and Nephrin, which suggested podocyte injury had occurred, were markedly increased, compared with the HG group. Moreover, our results suggested that gene silencing of CXCR3 significantly decreased HG-induced podocytes apoptosis by increasing the protein levels of an anti-apoptosis protein (Bcl-2), and decreasing protein levels of a proapoptosis protein (Bax) and the apoptosis marker (cleaved Caspase-3).
Meanwhile, the pathogenesis of DN involves numerous factors, such as oxidative stress and inflammation [23], which are closely associated with permeability changes in the glomerular filtration barrier and proteinuria in DN [24]. We also evaluated the level of oxidative stress including intracellular ROS production in our study. Mitochondria are not only a major source of ROS, but also the main targets attacked by ROS. ROS-mediated effects of HG have been implicated in podocyte injury and apoptosis [25]. Recent report also emphasized that enhanced ROS formation is mediated by mitochondrial disturbances induced by dyslipidemia, along with a sustained activation of NADDPH oxidase isoforms that play a crucial role in different renal pathophysiological mechanisms [26,27]. In the present study, gene silencing of CXCR3 significantly inhibited HG-induced decrease in mitochondrial membrane potential and increase in ROS production. Cytokines (TNF-α, IL-1β and IL-6) and chemokines (CXCL9 and CXCL10) are relevant to the development of DN [28]. The present results showed that HG induced cytokines production in podocytes, which may be inhibited by the gene silencing of CXCR3. It is indicated that CXCR3 may be involved in the inflammatory events in the development of DN with unclear mechanism. Therefore, further study is needed to investigate the pro-inflammatory effect of CXCR3 on podocytes in DN.
In summary, the present study shows that HG directly promotes podocyte apoptosis and up-regulates CXCR3 expression in podocytes in vitro. CXCR3 contributes to HG-induced podocyte apoptosis via increasing ROS generation and loss of mitochondrial membrane potential. Thus, the CXCR3 may be a new candidate for therapeutic targets to podocyte injury during the progression of DN.
Acknowledgements
This study was supported by the Tianjin Health Industry Key Research Projects (15KG101) and Tianjin Science and Technology Support Project (13ZCDSY01300).
Disclosure of conflict of interest
None.
References
- 1.Thomson SE, McLennan SV, Kirwan PD, Heffernan SJ, Hennessy A, Yue DK, Twigg SM. Renal connective tissue growth factor correlates with glomerular basement membrane thickness and prospective albuminuria in a non-human primate model of diabetes: possible predictive marker for incipient diabetic nephropathy. J Diabetes Complications. 2008;22:284–294. doi: 10.1016/j.jdiacomp.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 2.Xiong J, Wang Y, Zhu Z, Liu J, Wang Y, Zhang C, Hammes HP, Lang F, Feng Y. NG2 proteoglycan increases mesangial cell proliferation and extracellular matrix production. Biochem Biophys Res Commun. 2007;361:960–967. doi: 10.1016/j.bbrc.2007.07.113. [DOI] [PubMed] [Google Scholar]
- 3.Dronavalli S, Duka I, Bakris GL. The pathogenesis of diabetic nephropathy. Nat Clin Pract Endocrinol Metab. 2008;4:444–452. doi: 10.1038/ncpendmet0894. [DOI] [PubMed] [Google Scholar]
- 4.Arora MK, Singh UK. Molecular mechanisms in the pathogenesis of diabetic nephropathy: an update. Vascul Pharmacol. 2013;58:259–271. doi: 10.1016/j.vph.2013.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Liu F, Zong M, Wen X, Li X, Wang J, Wang Y, Jiang W, Li X, Guo Z, Qi H. Silencing of histone deacetylase 9 expression in podocytes attenuates kidney injury in diabetic nephropathy. Sci Rep. 2016;6:33676. doi: 10.1038/srep33676. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Susztak K, Raff AC, Schiffer M, Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–233. [PubMed] [Google Scholar]
- 7.Liu WT, Peng FF, Li HY, Chen XW, Gong WQ, Chen WJ, Chen YH, Li PL, Li ST, Xu ZZ, Long HB. Metadherin facilitates podocyte apoptosis in diabetic nephropathy. Cell Death Dis. 2016;7:e2477. doi: 10.1038/cddis.2016.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan Y, Huang S, Wang W, Wang Y, Zhang P, Zhu C, Ding G, Liu B, Yang T, Zhang A. Activation of peroxisome proliferator-activated receptorgamma coactivator 1alpha ameliorates mitochondrial dysfunction and protects podocytes from aldosterone-induced injury. Kidney Int. 2012;82:771–789. doi: 10.1038/ki.2012.188. [DOI] [PubMed] [Google Scholar]
- 9.Cai X, Bao L, Ren J, Li Y, Zhang Z. Grape seed procyanidin B2 protects podocytes from high glucose-induced mitochondrial dysfunction and apoptosis via the AMPK-SIRT1-PGC-1alpha axis in vitro. Food Funct. 2016;7:805–815. doi: 10.1039/c5fo01062d. [DOI] [PubMed] [Google Scholar]
- 10.Bock F, Shahzad K, Wang H, Stoyanov S, Wolter J, Dong W, Pelicci PG, Kashif M, Ranjan S, Schmidt S, Ritzel R, Schwenger V, Reymann KG, Esmon CT, Madhusudhan T, Nawroth PP, Isermann B. Activated protein C ameliorates diabetic nephropathy by epigenetically inhibiting the redox enzyme p66Shc. Proc Natl Acad Sci U S A. 2013;110:648–653. doi: 10.1073/pnas.1218667110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He T, Guan X, Wang S, Xiao T, Yang K, Xu X, Wang J, Zhao J. Resveratrol prevents high glucose-induced epithelial-mesenchymal transition in renal tubular epithelial cells by inhibiting NADPH oxidase/ROS/ERK pathway. Mol Cell Endocrinol. 2015;402:13–20. doi: 10.1016/j.mce.2014.12.010. [DOI] [PubMed] [Google Scholar]
- 12.Huber TB, Reinhardt HC, Exner M, Burger JA, Kerjaschki D, Saleem MA, Pavenstadt H. Expression of functional CCR and CXCR chemokine receptors in podocytes. J Immunol. 2002;168:6244–6252. doi: 10.4049/jimmunol.168.12.6244. [DOI] [PubMed] [Google Scholar]
- 13.Ruster C, Wolf G. The role of chemokines and chemokine receptors in diabetic nephropathy. Front Biosci. 2008;13:944–955. doi: 10.2741/2734. [DOI] [PubMed] [Google Scholar]
- 14.Wang G, Lai FM, Chow KM, Kwan BC, Pang WF, Luk CC, Leung CB, Li PK, Szeto CC. Urinary mRNA levels of ELR-negative CXC chemokine ligand and extracellular matrix in diabetic nephropathy. Diabetes Metab Res Rev. 2015;31:699–706. doi: 10.1002/dmrr.2654. [DOI] [PubMed] [Google Scholar]
- 15.Menke J, Zeller GC, Kikawada E, Means TK, Huang XR, Lan HY, Lu B, Farber J, Luster AD, Kelley VR. CXCL9, but not CXCL10, promotes CXCR3-dependent immune-mediated kidney disease. J Am Soc Nephrol. 2008;19:1177–1189. doi: 10.1681/ASN.2007111179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarz K, Simons M, Reiser J, Saleem MA, Faul C, Kriz W, Shaw AS, Holzman LB, Mundel P. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108:1621–1629. doi: 10.1172/JCI12849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vielhauer V, Anders HJ. Chemokines and chemokine receptors as therapeutic targets in chronic kidney disease. Front Biosci (Schol Ed) 2009;1:1–12. doi: 10.2741/s1. [DOI] [PubMed] [Google Scholar]
- 18.Segerer S, Nelson PJ, Schlondorff D. Chemokines, chemokine receptors, and renal disease: from basic science to pathophysiologic and therapeutic studies. J Am Soc Nephrol. 2000;11:152–176. doi: 10.1681/ASN.V111152. [DOI] [PubMed] [Google Scholar]
- 19.Segerer S, Banas B, Wornle M, Schmid H, Cohen CD, Kretzler M, Mack M, Kiss E, Nelson PJ, Schlondorff D, Grone HJ. CXCR3 is involved in tubulointerstitial injury in human glomerulonephritis. Am J Pathol. 2004;164:635–649. doi: 10.1016/S0002-9440(10)63152-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma Y, Yang Q, Chen X, Liang W, Ren Z, Ding G. c-Abl contributes to glucose-promoted apoptosis via p53 signaling pathway in podocytes. Diabetes Res Clin Pract. 2016;113:171–178. doi: 10.1016/j.diabres.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 21.Kim D, Lim S, Park M, Choi J, Kim J, Han H, Yoon K, Kim K, Lim J, Park S. Ubiquitinationdependent CARM1 degradation facilitates Notch1-mediated podocyte apoptosis in diabetic nephropathy. Cell Signal. 2014;26:1774–1782. doi: 10.1016/j.cellsig.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 22.Weil EJ, Lemley KV, Mason CC, Yee B, Jones LI, Blouch K, Lovato T, Richardson M, Myers BD, Nelson RG. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int. 2012;82:1010–1017. doi: 10.1038/ki.2012.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–340. doi: 10.1038/nrneph.2011.51. [DOI] [PubMed] [Google Scholar]
- 24.Navarro-Gonzalez JF, Mora-Fernandez C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol. 2008;19:433–442. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 25.Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, Abboud HE. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes. 2009;58:1201–1211. doi: 10.2337/db08-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mima A. Inflammation and oxidative stress in diabetic nephropathy: new insights on its inhibition as new therapeutic targets. J Diabetes Res. 2013;2013:248563. doi: 10.1155/2013/248563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montero RM, Covic A, Gnudi L, Goldsmith D. Diabetic nephropathy: what does the future hold? Int Urol Nephrol. 2016;48:99–113. doi: 10.1007/s11255-015-1121-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wong CK, Ho AW, Tong PC, Yeung CY, Kong AP, Lun SW, Chan JC, Lam CW. Aberrant activation profile of cytokines and mitogen-activated protein kinases in type 2 diabetic patients with nephropathy. Clin Exp Immunol. 2007;149:123–131. doi: 10.1111/j.1365-2249.2007.03389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]