

Abstract
Gorlin syndrome, a rare autosomal dominant disease, is characterized by numerous basal cell carcinomas, multiple jaw cysts, palmar and plantar pits and embryological deformities. Mutations in the PTCH1 gene are the most common molecular defects associated with Gorlin syndrome. We detected a duplication of thymine after nucleotide position 2927 in exon 18 of the PTCH1 gene (c.2927 dupT) in a fifty-year-old male proband with peri-anal basal cell carcinoma and his brother. The mutation creates a frameshift and leads to a premature stop codon (p.Tyr977 Leufs* 16) lacking 5 of the 12 transmembrane-spanning domains. However, the functional significance of truncation of the N terminal regions remains currently unknown and to be further investigated. The current findings indicate that genetic testing of PTCH1 gene mutational status may aid in the early diagnosis of Gorlin syndrome in which multiple complex abnormalities are present, hampering prompt diagnosis and treatment.
Keywords: Nevoid basic cell carcinoma syndrome, Gorlin syndrome, PTCH1, mutation, truncation
Introduction
The nevoid basal cell carcinoma syndrome, also known as Gorlin syndrome, is a rare autosomal dominant disease with a prevalence rate of 1/150000 to 1/570000 [1]. The syndrome is characterized by numerous basal cell carcinomas, multiple jaw cysts, palmar and plantar pits and embryological deformities. Patients with Gorlin syndrome suffer from developmental defeats and a high susceptibility oflow-frequency tumors, such as basal cell carcinoma, medulloblastoma, rhabdomyosarcoma, and cardiac fibromas [2]. Mutations in the PTCH1 gene (patched homolog 1, located on chromosome 9q 22) are the most common molecular defects associated with Gorlin syndrome. To date, a total of 348 different mutations in the PTCH1 gene have been reported in the human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/all.php). Missense, nonsense and small deletions remain the most common mutations in the PTCH1 gene while gross deletions, insertions or duplications account for less than 10% of all PTCH1 gene mutations.
In this article, we report the identification of a novel duplication mutation in the PTCH1 gene, leading to a truncated PTCH1, in a 50-year-old male patient with familiar Gorlin syndrome and we further describe the clinicopathologic features of Gorlin syndrome in the proband.
Subjects and methods
Ethical considerations
All procedures were reviewed and approved by the Ethical Review Board of Zhongnan Hospital of Wuhan University. Informed consent was obtained from the patients or their family members for all procedures in this study and permission for use of patient photographs was obtained.
Patient evaluation
The nevoid basal cell carcinoma syndrome in the proband was diagnosed based on the result of multinational analysis of the PTCH1 gene and according to the criteria as described by Larsen et al. [3]. The patient was examined by an experienced dermatologist and then evaluated radiologically for skeletal deformities and with head CT scan. The anatomic distribution of BCCS was recorded according to six body sites including head and neck, face, scalp, upper extremities, lower extremities, and trunk. The peri-anal tumor tissue was biopsied and sectioned and stained with H&E for histological examination as previously described [4]. In addition, a detailed family history was obtained of the proband.
Mutation analysis
Peripheral blood samples were taken from the proband and his family members. Genomic DNA was extracted from blood samples using the TIANamp Blood DNA kit as instructed by the manufacturer (Tiangen, Beijing, China). Primer covering all coding regions and flanking introns of the PTCH1 gene (OMIM: 603673) was designed and synthesized (Qingwei, Wuhan, China) according to the PTCH1 sequence from Ensembl (http://asia.ensembl.org/index.html) via NCBI Primer-BLAST (http://frodo.wimit.edu/primer3). Primer sequences are available upon request. All the sequences of the PTCH1 gene were amplified by polymerase chain reaction (PCR). The purified PCR products were sent to Grandomics Biosciences Co. (Wuhan, China) for sequencing analysis. Nucleotide numbering is based on GenBank sequence NM_000264.3, where the A of the ATG translation initiation site represents the nucleotide+1. The affected transcript and protein were denoted using the Human Genome Variation Society (HGVS) nomenclature version 2.0 (Mutalyzer 2.0.beta-32, https://mutalyzer.nl/).
Results
Clinicopathologic characteristics of the proband
The proband was a 50-year-old male who presented to the Department of Dermatology of our hospital because of perianal neoplasia for 3 years. The clinical features of the nevoid basal cell carcinoma syndrome in the proband are shown in Table 1. The patient exhibited double-facial asymmetry, ocular hypertelorism and a broad base of the nose (Figure 1A and 1B). Palmar and plantar pits without scales or keratinization were present (Figure 1C). Skin examination showed an irregular and rugged soft ecru neoplasia around the anus, which was about 1.5 cm in diameter, with a clear boundary; the tumor had rough surface and exhibited mild erosions (Figure 1D). Furthermore, head CT scans demonstrated a broadened septum pellucidum and multiple calcifications in the bilateral tentorium cerebella and cerebral falx (Figure 2A and 2B), a widened subdural cavity in the bilateral cerebellar hemisphere and right maxillary sinusitis. Pathological examination of a biopsy specimen showed basal cell carcinoma characterized by proliferation of basaloid cells with palisading nuclei at the periphery of the nests (Figure 2C and 2D).
Table 1.
Clinical features of the nevoid basal cell carcinoma syndrome in the proband and his first degree relatives
| Clinical features | The proband | Mother | Brother | Sister |
|---|---|---|---|---|
| Major manifestations | Healthy | |||
| Basal cell carcinoma | Basal cell carcinoma | Basal cell carcinoma | ||
| Palmar or plantar pitting | Palmar or plantar pitting | Palmar or plantar pitting | ||
| Calcifications of the falx cerebri | Head CT scan was not performed. | Head CT scan was not performed | ||
| First degree relatives (mother and brother) with basal cell carcinoma | ||||
| Minor manifestations | Ocular hypertelorism | Ocular hypertelorism | Ocular hypertelorism |
Figure 1.

Clinical manifestations of Gorlin syndrome in the proband. A and B. Double-facial asymmetry, ocular hypertelorism and broad base of the nose; C. Palmar pits. D. An irregular and rugged soft ecru tumor is present near the anus, which is about 1.5 cm in diameter, with a clear boundary. The surface is rough and mild erosions are present.
Figure 2.
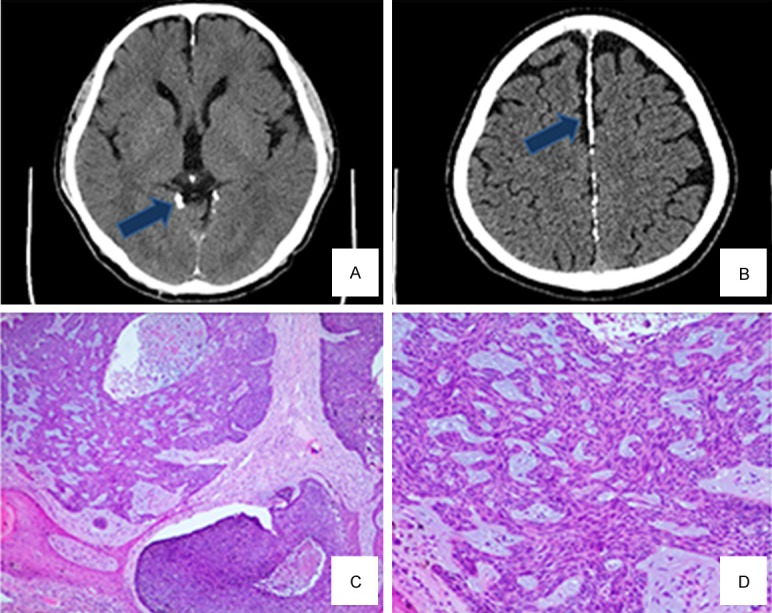
Head CT scan shows calcification of the tentorium cerebella (A) and calcification of the cerebral falx (B) Pathological biopsy of the perianal neoplasm reveals proliferation of basaloid cells with palisading nuclei at the periphery of the nests and contraction gaps between tumors and the surrounding tissues (C and D: H&E, magnification × 40).
Pedigree characteristics of the proband
The first degree relatives of the proband were interviewed. The mother and brother of the proband had a history of basal cell carcinoma, and they also complained of other relevant manifestations (Table 1). His sister did not have basal cell carcinoma or any features indicative of Gorlin syndrome. The father of the proband was not examined because he was deceased. The family pedigree of the proband is shown in Figure 3.
Figure 3.

A. The family pedigree of the proband (black arrow) co-segregating the PTCH1 germline mutation (c.2927 dupT). B. Structural modeling of the PTCH1 protein showing that the protein is truncated at amino acid position 977.
PTCH1 gene mutational characteristics of the proband
We performed a mutation analysis to detect possible mutations responsible for Gorlin syndrome in the proband and his family. All available family members were analyzed through direct sequencing of all the exons and introns of the PTCH1 gene. We detected a duplication of thymine after nucleotide position 2927 in exon 18 of the PTCH1 gene (c.2927 dupT), resulting in a frameshift mutation in PTCH1. This mutation was found to be heterozygous in his brother and no such mutation was detected in his sister (Figure 4A and 4B). The mutational status of PTCH1 was unknown in his mother because she was not available for providing blood samples for gene sequencing. Compared with the published sequence at the Emsembl (http://www.ensembl.org), this frameshift mutation eventually introduces a premature stop-codon in translation (p.Tyr977 Leufs* 16). This mutation has not previously been reported in any patients with Gorlin syndrome according to the PTCH1 mutation database (NM_000264.3, Human Gene Mutation Database professional 2017.2, http://www.hgmd.org/).
Figure 4.

DNA sequencing diagram of the PTCH1 exon 18 sequence showing the heterozygous germline duplication of a thymine (T) in the DNA of the proband and his brother. (A) The DNA and corresponding amino-acid sequences of wild-type and mutant PTCH1 alleles are also shown. The affected transcript and protein were labeled NM_000264.3 (PTCH1_v001):c.2927 dupT and NM_000264.3 (PTCH1_i001):p.(Tyr977Leufs* 16), respectively, using the Human Genome Variation Society (HGVS) nomenclature version 2.0 (Mutalyzer 2.0.beta-32, https://mutalyzer.nl/). The arrow indicates the duplication point in the proband and his brother and the normal nucleotide in the sister (B).
Discussion
The proband in the present paper exhibited clinical features typical of Gorlin syndrome including basal cell carcinoma, palmar and plantar pits, lamellar calcification of the falx cerebli and ocular hypertelorism [3]. The diagnosis of Gorlin syndrome is further supported by molecular evidence showing a truncation mutation of PTCH1. The familiar nature of this disease in the proband was corroborated by the presence of probable Gorlin syndrome and detection of the identical mutation in the brother of the proband.
Aberrant activation of the hedgehog signaling pathway has been well documented in multiple cancers. Several effectors, such as n-Myc, EGF, and EMP, are downstream targets of this important pathway. The hedgehog signaling network consists of secreted ligands (sonic hedgehog, Shh), the Patched family hedgehog receptors (PTCH1 and PTCH2) and the Smoothened (SMO) transmembrane effector protein, and its downstream transcription factors (the GLI family). It functions in cell-cell communication and regulates pattern formation, proliferation, cell fate and self-renewal in many organs. Compared with Shh and SMO, PTCH1 is expressed widely as one of the regulators of the sonic hedgehog pathway. The PCTH1 gene is also reported as a cancer suppressor gene, which encodes the patched protein that binds to SMO and maintains SMO in the inactive state in order to inhibit signaling to downstream genes. Mutations of PCTH1 may incapacitate the patched protein; thus, the SMO remains activated without the inhibitor and transmits the stimulating signal to the downstream factors [5].
Gorlin syndrome is an autosomal dominant disorder with mutations in the PTCH1 gene being the most commonly detected. The PTCH1 gene consists of 23 exons encoding a 1447-amino-acid protein which contains 12 transmembrane-spanning domains, two large extracellular loops and a large intracellular loop [6]. The PTCH1 protein is the ligand binding component of the sonic hedgehog receptor complex, whose absence may inactivate the Patched protein and overstimulate its downstream the hedgehog signaling pathway [7,8]. Among totally 348 different mutations in the PTCH1 gene, most, like the mutation in the current report, are frameshift or nonsense mutations that lead to the synthesis of a truncated protein. In the current paper, we described a novel germline duplication (insertion) mutation of PTCH1, c.2927 dupT, in a Chinese patient with familiar Gorlin syndrome. This mutation creates a premature termination codon in the mutant allele that results in the truncation of the PTCH1 protein. The mutation in the PTCH1 occurs in the extracellular loop and the truncated PTCH1 lacks 5 of the 12 transmembrane-spanning domains. Lindstrom et al. [9] reported that the germline mutations in Gorlin syndrome were mainly truncating mutations concentrated in the large extracellular loops, the large intracellular loop, and the N-terminal region. The truncating mutation in our case, on the other hand, occurs adjacent to the C terminus. Okamoto et al. reported one of the shortest forms of truncated PTCH1 missing lacks 11 of the 12 transmembrane-spanning domains [10]. However, the functional significance of truncation of the N terminal regions remains currently unknown and to be further investigated.
In most cases of Gorlin syndrome, patients present with basal cell carcinoma, which is one of the crucial standards for diagnosis of Gorlin syndrome. Patients with Gorlin syndrome have a high burden of basal cell carcinoma. In the United Kingdom, 75% of the patients over 20 years old with Gorlin syndrome had basal cell carcinoma, and the incidence increased to 90% in patients over 40 years old. Korean patients seem to have a reduced frequency of basal cell carcinoma (reduced by 15% comparing with UK) [11]. In Japan, 38% of the patients over 20 years old had basal cell carcinoma and 53% of the patients over 40 years old suffered from basal cell carcinoma [12]. The discrepant prevalence suggested the incidence of basal cell carcinoma may be influenced by genetic and environmental factors. But there is no doubt that with the growth of age, the higher the incidence of the tumor will grow. Molecular genetic testing will be helpful not only to confirm the diagnosis in patients with atypical phenotype or possibly for prenatal diagnosis, but also to searching novel targeting treatment of genetic disease.
In conclusion, we detected a truncation mutation of PTCH1 in a familiar case of Gorlin syndrome and this novel mutation may be the cause of Gorlin syndrome in the patient. The current findings indicate that genetic testing of PTCH1 gene mutational status may aid in the early diagnosis of Gorlin syndrome in which multiple complex abnormalities are present, hampering prompt diagnosis and treatment.
Acknowledgements
We are grateful to the families and patients whose generosity has made this study possible. We thank the Dermatological Department of Zhongnan Hospital of Wuhan University for financial support.
Disclosure of conflict of interest
None.
References
- 1.Acocella A, Sacco R, Bertolai R, Sacco N. Genetic and clinicopathologic aspects of Gorlin-Goltz syndrome (NBCCS): presentation of two case reports and literature review. Minerva Stomatol. 2009;58:43–53. [PubMed] [Google Scholar]
- 2.Bholah Z, Smith MJ, Byers HJ, Miles EK, Evans DG, Newman WG. Intronic splicing mutations in PTCH1 cause Gorlin syndrome. Fam Cancer. 2014;13:477–480. doi: 10.1007/s10689-014-9712-9. [DOI] [PubMed] [Google Scholar]
- 3.Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, Bale AE, Bale SJ. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69:299–308. [PubMed] [Google Scholar]
- 4.Go JW, Kim SH, Yi SY, Cho HK. Basal cell nevus syndrome showing several histologic types of Basal cell carcinoma. Ann Dermatol. 2011;23(Suppl 1):S36–40. doi: 10.5021/ad.2011.23.S1.S36. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Ruiz i Altaba A. Therapeutic inhibition of Hedgehog-GLI signaling in cancer: epithelial, stromal, or stem cell targets? Cancer Cell. 2008;14:281–283. doi: 10.1016/j.ccr.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 6.Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, Noll M, Hooper JE, de Sauvage F, Rosenthal A. The tumour-suppressor gene patched encodes a candidate receptor for sonic hedgehog. Nature. 1996;384:129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- 7.Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by hedgehog. Genes Dev. 1996;10:301–312. doi: 10.1101/gad.10.3.301. [DOI] [PubMed] [Google Scholar]
- 8.Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- 9.Lindstrom E, Shimokawa T, Toftgard R, Zaphiropoulos PG. PTCH mutations: distribution and analyses. Hum Mutat. 2006;27:215–219. doi: 10.1002/humu.20296. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto N, Naruto T, Kohmoto T, Komori T, Imoto I. A novel PTCH1 mutation in a patient with Gorlin syndrome. Hum Genome Var. 2014;1:14022. doi: 10.1038/hgv.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasaki R, Saito K, Watanabe Y, Takayama Y, Fujii K, Agawa K, Miyashita T, Ando T, Akizuki T. Nevoid basal cell carcinoma syndrome with cleft lip and palate associated with the novel PTCH gene mutations. J Hum Genet. 2009;54:398–402. doi: 10.1038/jhg.2009.51. [DOI] [PubMed] [Google Scholar]
- 12.Fujii M, Noguchi K, Urade M, Muraki Y, Moridera K, Kishimoto H, Hashimoto-Tamaoki T, Nakano Y. Novel PTCH1 mutations in Japanese Nevoid basal cell carcinoma syndrome patients: two familial and three sporadic cases including the first Japanese patient with medulloblastoma. J Hum Genet. 2011;56:277–283. doi: 10.1038/jhg.2011.2. [DOI] [PubMed] [Google Scholar]