

Abstract
The present study aimed to elucidate the roles and possible molecular mechanisms of long noncoding RNA (lncRNA) urothelial carcinoma associated 1 (UCA1) in neuronal pheochromocytoma (PC)-12 cells under hypoxic conditions. The neuronal PC-12 cells were exposed to hypoxic and normoxic conditions followed by the measurement of the expression of lncRNA UCA1. In addition, the cells were transfected with short hairpin RNAs (sh-RNAs) against UCA1 (sh-UCA1), SOX6 (sh-SOX6), negative control (sh-NC), pEX-SOX6, pEX, miR-18a mimic, mimic NC, miR-18a inhibitor, and inhibitor NC. Under different treatments of transfection, cell viability and migration and invasion potential were analyzed. In addition, the induction of apoptosis was investigated by studying the expression profiles of apoptosis-related proteins. Hypoxia treatment significantly enhanced the expression of UCA1, which in turn induced injury in PC-12 cells characterized by the inhibition of cell viability, the reduction in migration and invasion potential, and the promotion of cell apoptosis. Moreover, the suppression of UCA1 alleviated the hypoxia injury. In addition, the relationship between UCA1 and miR-18a and between miR-18a and SRY-box containing gene 6 (SOX6) were explored. MiR-18a was found to be a direct target of UCA1, an upregulation of which mediated the effects of suppression of UCA1 (alleviated hypoxic injury). Besides, SOX6 was found to be a target of miR-18a whose expression could be negatively regulated by miR-18a. An overexpression of SOX6 could also aggravate hypoxia injury in PC-12 cells, whereas a knockdown of SOX6 exhibited contrary results. Our findings indicated that the down-regulation of UCA1 promoted the expression of miR-18a that led to a reduction in the expression of its target protein, SOX6, thereby contributing to the hypoxia injury following cerebral ischemia.
Keywords: Cerebral ischemia, long non-coding RNA urothelial carcinoma associated 1, miR-18a, SOX6, hypoxia injury
Introduction
Cerebral or brain ischemia is a condition characterized by an insufficient flow of blood to the brain, leading to cerebral hypoxia and subsequent neuronal injury [1]. It is considered to be one of the main causes of cerebral infarction and ischemic stroke, which constitute major public health problems [2]. Thus, an elucidation of the key mechanism of ischemic cerebral injury would provide a new insight into the development of effective targets for therapeutic intervention of cerebral infarction and ischemic stroke.
Long non-coding RNAs (lncRNAs) are a kind of RNA molecules with non-protein transcripts that are longer than 200 nucleotides with a molecular weight of up to approximately 100 kb [3,4]. Interestingly, transcriptomic profiles of lncRNAs in brain microvascular endothelium are reported to be altered after cerebral ischemia, suggesting the potential pathological roles of these transcripts in mediating endothelial responses to ischemic stimuli [5]. In addition, a better understanding of the expression and functions of lncRNAs would pave the way for the utilization of these molecules as promising neuroprotectants against ischemic brain injury [6]. Following cerebral ischemia, the neuroprotective roles of several lncRNAs against ischemic brain injury have been reported. For instance, the non-coding RNA, lncRNA H19, has been demonstrated to be upregulated by hypoxia and subsequently activate autophagy to induce cerebral ischemia-reperfusion injury and therefore may act as a potential target for the treatment of ischemic stroke [7]. The downregulation of lncRNA, Meg3, could regulate the Notch signal transduction pathway to promote angiogenesis after ischemic brain injury [8]. Similarly, lncRNA, C2dat1, was demonstrated to modulate Ca2+/calmodulin-dependent protein kinase II δ-isoform (CaMKIIδ) expression leading to the neuronal survival following cerebral ischemia, and therefore, may serve as a novel target for the prevention and therapy of cerebral ischemia [9]. Recently, human urothelial carcinoma associated 1 (UCA1), an lncRNA that was first identified in human bladder carcinoma [10], has been shown to play an oncogenic role in the initiation and progression of a variety of prevalent cancers, such as osteosarcoma [11], glioma [12], gastric cancer [13], lung cancer [14], and breast cancer [15]. However, the roles of UCA1 in preventing brain injury following cerebral ischemia have not been completely illustrated and therefore remain elusive.
In the present study, with an aim to investigate the role of UCA1 in cerebral ischemia, we studied the expression of UCA1 under hypoxia and the effects of its suppression on preventing hypoxia injury in neuronal PC-12 cells. In addition, the study involved an investigation of the relationship between UCA and miR-18a and between miR-18a and SRY-box containing gene 6 (SOX6), a protein actively involved in neuronal differentiation. Moreover, the roles of the UCA1-miR-18a-SOX6 axis in countering hypoxia-induced injury in PC-12 cells were further explored. Therefore, our study aimed to elucidate the roles and regulatory mechanisms of UCA1 in preventing hypoxia-induced neuronal injury to provide new insights into its therapeutic potential for the treatment of ischemic brain injury following cerebral ischemia.
Materials and methods
Cell culture and treatment
The neuronal pheochromocytoma (PC)-12 cells (Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming, China) were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. The cells were maintained in a humidified incubator containing 5% CO2 at 37°C. For the hypoxic and normoxic conditions, the cells were exposed to 3% and 10% O2 culture conditions, respectively.
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from PC-12 cells using Trizol reagent (Life Technologies Corporation, Carlsbad, CA, USA) following the manufacturer’s instructions. After determining the quality of total RNA extracted using ultraviolet spectrophotometer (SMA 400 UV-VIS, Merinton, Shanghai, China), it was reverse-transcribed into complementary DNA (cDNA) using iScript™ cDNA Synthesis Kit (Bio-Rad Laboratories, USA). To check the expression levels of UCA1, SOX6, and miR-18a, the quantitative real-time polymerase chain reaction (qRT-PCR) analysis was conducted using One Step SYBR® PrimeScript®PLUS RT-PCR Kit (Takara, Dalian, China), RNA PCR Kit (AMV) Ver.3.0 (Takara Dalian, China), and TaqMan Universal Master Mix II with the TaqMan MicroRNA Assay (Applied Biosystems, Foster City, CA, USA), respectively. The relative expressions of targets were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) that served as the internal control and calculated by the relative quantification (2-ΔΔCt) method.
Cell transfection
Short-hairpin RNAs (shRNAs) against human lncRNA UCA1 (sh-UCA1) and SOX6 (sh-SOX6) were ligated into the U6/GFP/Neo plasmid (GenePharma, Shanghai, China) for the suppression of UCA1 and SOX6, respectively. The full-length SOX6 sequence was cloned into the pEX-2 vector (GenePharma, pEX-SOX6) to overexpress SOX6. The vector carrying a non-targeting sequence was used as a negative control (NC). MiR-18a mimic, miR-18a inhibitor, and their respective NCs were also synthesized (Life Technologies Corporation, MD, USA). For cell transfection, the cells were seeded into a 24-well plate, followed by the transfection with sh-UCA1, sh-SOX6, sh-NC, pEX-SOX6, pEX, miR-18a mimic, mimic NC, miR-18a inhibitor, and inhibitor NC using Lipofectamine 2000 (Life Technologies Corporation, Carlsbad, CA, USA) according to the manufacturer’s instructions. The cells were harvested at 48 h post-transfection for subsequent experiments and analyses.
Cell viability assay
For assessing the cell viability, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed. PC-12 cells at a density of 5 × 104 cells/mL were cultured into a 96-well plate in triplicate. After 48 h of transfection, 20 μL of MTT solution was added to each well and the cells were incubated for 4 h. To dissolve the formazan crystals (MTT reduced by living cells), 200 μL of dimethyl sulfoxide (DMSO) was added to each well. The absorbance of each well was then measured with a microplate reader at 492 nm (BioTek, USA). The cell viability rate (%) was calculated as the absorbance of formazan in the treatment group/the absorbance of formazan in the control group × 100%. The measured formazan is presumed to be directly proportional to the number of living cells.
Migration assay
Cell migration acts as a major mechanism for the progression of many diseases including cancer and contributes to metastasis [16]. The cell migration was determined by Transwell culture chamber with a pore size of 8 mm. Briefly, the cells were suspended in serum-free medium and seeded into the upper compartment of a 24-well Transwell culture chamber. Complete medium was then added to the lower compartment of the chamber, which served as a chemoattractant. After 12 h of incubation at 37°C (to allow the cells to settle down), non-migrated cells in the upper compartment of the chamber were carefully removed with a cotton swab. The traversed cells across the membrane that were considered to have migrated into the lower chamber were washed, fixed with methanol, stained with leucocrystal violet, and counted under a microscope spanning a total of five fields.
Invasion assay
Cell invasion was determined using a modified invasion chamber with a pore size of 8 mm. Briefly, after 48 h of transfection, cells at a density of 5.0 × 104 were cultured in serum-free DMEM medium and seeded into the upper chamber of a BD BioCoatTM MatrigelTM Invasion Chamber (BD Biosciences). The cells were allowed to invade into the lower chamber filled with complete medium containing 10% FBS. After incubation for 48 h at 37°C, a cotton swab was used to remove the non-invaded cells in the upper chamber carefully. The cells that invaded into the bottom of the Matrigel matrix-coated chamber were washed, fixed in methanol, stained with 0.1% crystal violet, and were counted in a total of five fields under a microscope (Olympus, Tokyo, Japan).
Apoptosis assay
Cell apoptosis was analyzed by double staining with fluorescein isothiocyanate (FITC)-conjugated Annexin V and propidium iodide (PI). Briefly, cells in the different groups were harvested after 48 h of transfection, fixed in 70% ethanol, and then stained with FITC-Annexin V and PI in the presence of 50 μg/mL RNase A (Sigma-Aldrich, St Louis, MO, USA). After incubation for 1 h at room temperature in dark, the apoptotic cells were detected by flow cytometry using a FACS machine (Beckman Coulter, Fullerton, CA, USA). The flow cytometric data were analyzed using CellQuest 3.0 software (Becton Dickinson, NJ, USA).
Reporter vector construction and luciferase reporter assay
To construct the reporter vector, UCA1-wild-type (UCA1-WT), a fragment from UCA1 containing the putative binding site of miR-18a was amplified using polymerase chain reaction (PCR). The amplified fragment was cloned into a pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Madison, WI, USA). The above reporter vector and miR-18a mimics were then co-transfected into human embryonic kidney (HEK) 293T cells. At 48 h of post-transfection, the luciferase activity was then checked using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA). The Renilla luciferase gene was used as an internal control.
Western blot assay
The PC-12 cells in different groups were harvested and lysed in radioimmunoprecipitation (RIPA) buffer (Beyotime Biotechnology, Shanghai, China) containing protease inhibitors (Roche, Guangzhou, China) to extract the total protein. The protein extracts were then quantified with the bicinchoninic acid (BCA)™ Protein Assay Kit (Pierce, Appleton, WI, USA). An equal amount of protein extracts were separated on a 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto the polyvinylidene difluoride (PVDF) membrane (Millipore, MA, USA). The membranes were incubated overnight at 4°C in primary antibodies prepared in 5% blocking buffer. Primary antibodies to Bax, Bcl-2, pro-caspase-3, cleaved-caspase-3, pro-caspase-9, cleaved-caspase-9, SOX6, and GAPDH (1:1000 dilution, Santa Cruz County, Delaware Avenue, CA, USA) were used in this study. GAPDH served as the internal control. Following incubation, the membranes were probed with appropriate secondary antibodies conjugated to horseradish peroxidase (HRP) for 1 h. After rinsing, the membranes were transferred into the Bio-Rad ChemiDoc™ XRS system and visualized by incubation in Immobilon Western Chemiluminescent HRP Substrate (Millipore, MA, USA).
Statistical analysis
Each experiment was conducted in triplicate and the data obtained from multiple experiments were presented as the mean ± standard deviation (SD). The statistical analyses of these data were performed using GraphPad 6.0 statistical software (GraphPad Prism, San Diego, CA, USA). The statistically significant difference was calculated using a one-way analysis of variance (ANOVA) with the threshold value of P < 0.05.
Results
Hypoxia induces injury in PC-12 cells
In the current study, we first analyzed the effects of hypoxia on PC-12 cells. In comparison to the control group, hypoxic conditions significantly inhibited the viability (Figure 1A), migration ability (Figure 1B), and invasion potential of PC-12 cells (Figure 1C). The treatment also induced massive apoptosis (Figure 1D, P < 0.05) as evident by an increase in the expression of apoptosis-related proteins. As shown in Figure 1E, the expression of Bcl-2, an anti-apoptotic protein, was considerably reduced after hypoxia treatment compared with the control groups, whereas the expression levels of pro-apoptotic proteins, such as Bax, cleaved-caspase-3, and cleaved-caspase-9 were markedly increased.
Figure 1.
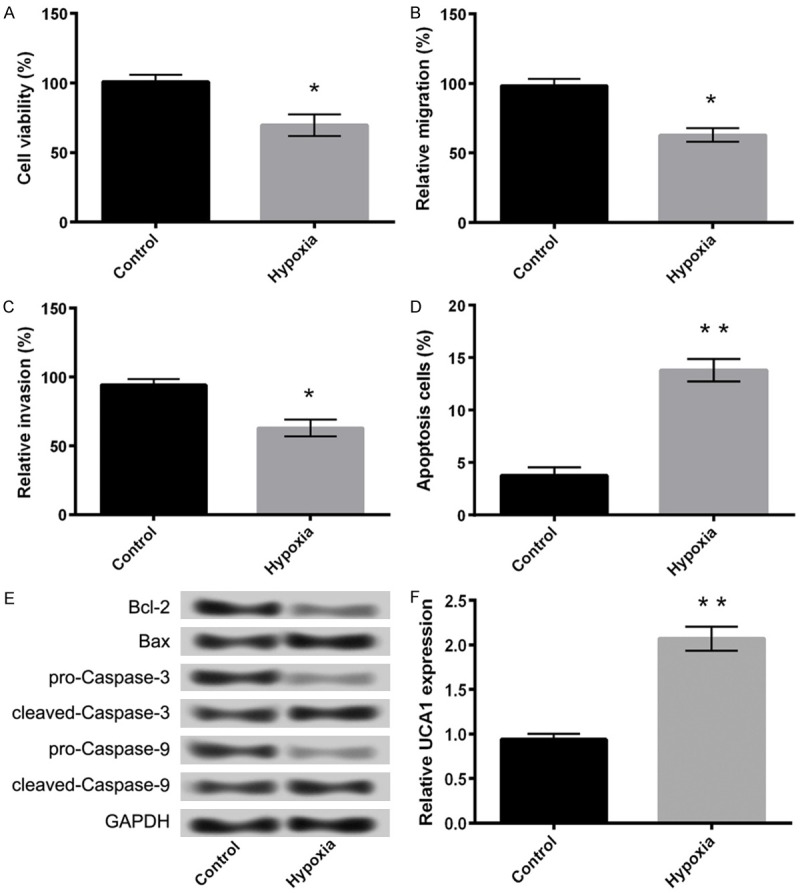
Hypoxia induces injury in PC-12 cells. A: Cell viability; B: Cell migration; C: Cell invasion; D: Cell apoptosis; E: The expression of apoptosis-related proteins; F: Hypoxia promotes the expression of UCA1 in PC-12 cells. Data are expressed as mean ± SD. *, P < 0.05, **, P < 0.01.
Hypoxia promotes expression of UCA1 in PC-12 cells
Figure 1F depicts the effect of hypoxia on the expression of UCA1 in PC-12 cells. The results demonstrated a significant upregulation in the expression of UCA1 after hypoxia insult of cells as compared with control (P < 0.05).
Suppression of UCA1 alleviates hypoxia injury in PC-12 cells
To investigate the effects of UCA1 expression on hypoxic PC-12 cells, we conducted sh-RNA-mediated suppression of UCA1 in PC-12 cells. As presented in Figure 2A, sh-UCA1 could effectively reduce the expression of UCA1 in sh-UCA1 group than in the control or si-NC group (P < 0.05), indicating a successful inhibition of UCA1 in PC-12 cells. In addition, the results demonstrated that the suppression of UCA1 substantially alleviated the hypoxia-induced decline in the cell viability (Figure 2B), migration ability (Figure 2C), and invasion potential (Figure 2D). It also inhibited hypoxia-induced cell apoptosis (Figure 2E, P < 0.05) as shown by a reversal in the expression of the apoptosis-related proteins after the suppression of UCA1 (P < 0.05, Figure 2F). These data indicated that the suppression of UCA1 could efficiently mitigate the injurious effects of hypoxia in PC-12 cells.
Figure 2.
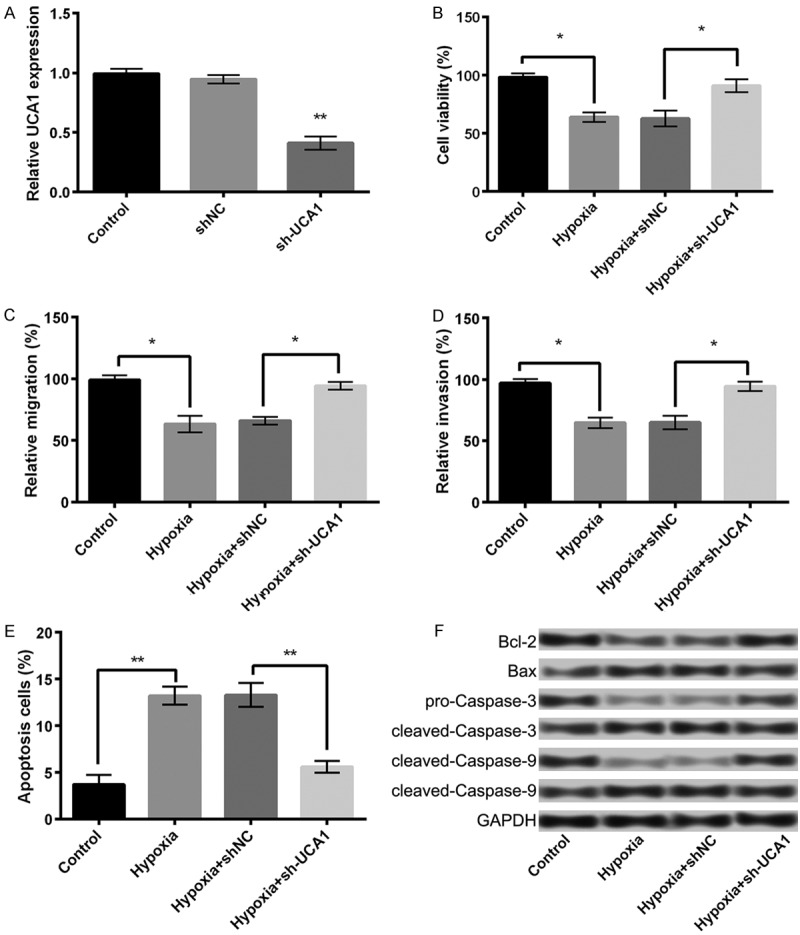
The suppression of UCA1 alleviates hypoxia injury in PC-12 cells. A: The expression of UCA1 in different treatment groups; B: Cell viability; C: Cell migration; D: Cell invasion; E: Cell apoptosis; F: The expression of apoptosis-related proteins. Data are expressed as mean ± SD. *, P < 0.05, **, P < 0.01.
MiR-18a is a direct target of UCA1
We further investigated the relationship between miR-18a and UCA1. The results demonstrated a significantly higher expression of miR-18a in sh-UCA1 group as compared to the control or sh-NC group (P < 0.05, Figure 3A), suggesting a negative regulatory relationship between miR-18a and UCA1. In addition, the results of luciferase reporter assay displayed that miR-18a mimic remarkably inhibited the luciferase reporter activity of UCA1-WT (P < 0.05) rather than UCA1-MUT (Figure 3B). These data are indicative of miR-18a to be a direct target of UCA1.
Figure 3.
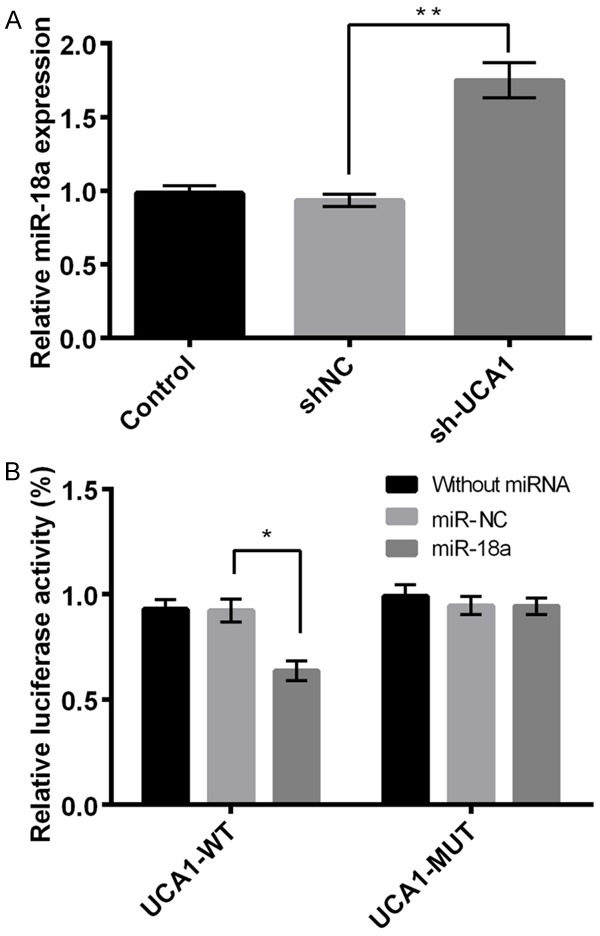
miR-18a as the direct target of UCA1. A: The expression of miR-18a in different treatment groups; B: The luciferase report activity of UCA1-WT and UCA1-MUT. Data are expressed as mean ± SD. *, P < 0.05, **, P < 0.01.
Suppression of miR-18a aggravates hypoxia injury in PC-12 cells
As shown in Figure 4A, an appreciable increase in the expression of miR-18a after transfection of PC-12 cells by its mimic was observed as compared with the control or mimic NC groups. However, its expression in miR-18a inhibitor group was markedly decreased (P < 0.05), indicating that the transfection of PC-12 cells with miR-18a mimic and miR-18a inhibitor could efficiently overexpress and suppress miR-18a, respectively. The combined effect of sh-UCA1 and miR-18a inhibitor was further explored. We found that in comparison with hypoxia + sh-UCA1 + inhibitor NC group, the suppression of miR-18a aggravated the hypoxia-induced ischemic injury in hypoxia + sh-UCA1 + miR-18a inhibitor transfected cells. The effect was clearly visible in terms of a notable reduction in cell viability (Figure 4B), migration potential (Figure 4C), and invasion property (Figure 4D). We also reported an induction of apoptosis (Figure 4E, P < 0.05) since the expression of Bcl-2 in hypoxia + sh-UCA1 + miR-18a inhibitor group was significantly decreased compared with hypoxia + sh-UCA1 + inhibitor NC. Contrary to this, the expressions of Bax, cleaved-caspase-3, and cleaved-caspase-9 were markedly elevated (Figure 4F). These data corroborate the finding that the suppression of UCA1 alleviates hypoxia injury in PC-12 cells through an upregulation of expression of miR-18a.
Figure 4.

The suppression of miR-18a aggravates hypoxia injury in PC-12 cells. A: The expression of miR-18a in different treatment groups; B: Cell viability; C: Cell migration; D: Cell invasion; E: Cell apoptosis; F: The expression of apoptosis-related proteins. Data are expressed as mean ± SD. *, P < 0.05, **, P < 0.01.
SOX6 is a direct target of miR-18a
In the present study, based on the information of TargetScanHuman, we predicted SOX6 to be a potential target of miR-18a (Figure 5A). The results of luciferase reporter assay further confirmed the above possibility, as miR-18a mimic significantly inhibited the luciferase reporter activity of SOX6-WT (P < 0.05) and not of SOX6-MUT (Figure 5B). Moreover, the mRNA and protein expression levels of SOX6 were significantly reduced in the miR-18a mimic group compared with the mimic NC group. On the other hand, the expression levels were markedly increased in miR-18a inhibitor group compared with inhibitor NC group (P < 0.05, Figure 5C and 5D). The above findings indicated that SOX6 was a direct target of miR-18a and negatively regulated by it.
Figure 5.

SOX6 was a direct target of miR-18a. A: The predicted information using TargetScanHuman; B: The luciferase reporter activity of SOX6-WT and SOX6-MUT; C: The mRNA expression of SOX6 in different treatment groups; D: The protein expression of SOX6 in different treatment groups. Data are expressed as mean ± SD. *, P < 0.05, **, P < 0.01.
Overexpression of SOX6 aggravates hypoxia injury in PC-12 cells
To detect whether SOX6 is a downstream target of miR-18a, it was simultaneously overexpressed and knocked down in PC-12 cells by transfection with pEX-SOX6 and sh-SOX6, respectively. As shown in Figure 6A, SOX6 expression was considerably increased in the pEX-SOX6 group, whereas it decreased in the sh-SOX6 group compared with control or their respective NC groups (P < 0.05). In comparison with hypoxia or hypoxia + pEX group, the overexpression of SOX6 aggravated the hypoxia-induced ischemic injury in PC-12 cells by significantly inhibiting cell viability (Figure 6C), migration potential (Figure 6D), and invasive property (Figure 6E), and inducing apoptosis (Figure 6F, P < 0.05). However, the knockdown of SOX6 exhibited contrary results.
Figure 6.
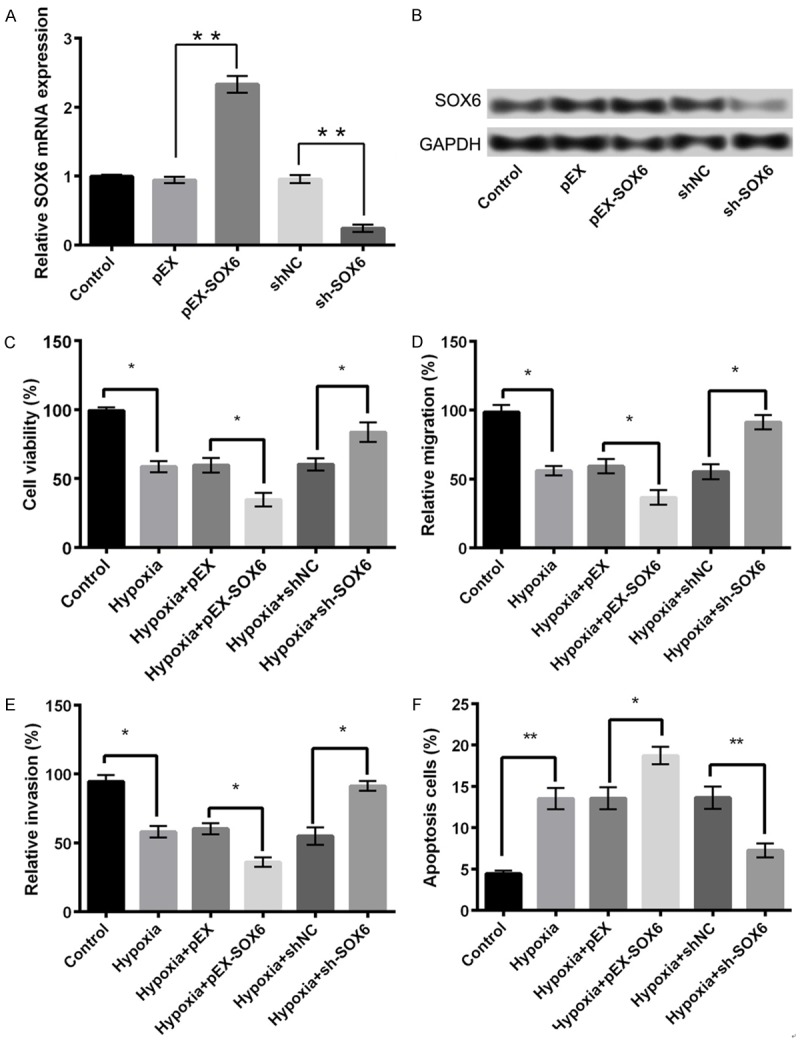
An overexpression of SOX6 aggravates hypoxia injury in PC-12 cells. A: The mRNA expression of SOX6 in different treatment groups; B: The protein expression of SOX6 in different treatment groups; C: Cell viability; D: Cell migration; E: Cell invasion; F: Cell apoptosis. Data are expressed as mean ± SD. *, P < 0.05, **, P < 0.01.
Discussion
In the present study, the effects and mechanisms of lncRNA UCA1 on PC-12 cells exposed to hypoxia-induced stress were studied. The results demonstrated some interesting findings. The results revealed that an exposure to hypoxia inhibited cell viability, reduced migration, and invasion potential, and promoted cell apoptosis in PC-12 cells, collectively called “hypoxia-induced injury”. Moreover, hypoxia treatment enhanced the expression of UCA1 in PC-12 cells. We also found miR-18a to be a direct target of UCA1. The up-regulation of miR-18a significantly decreased UCA1 expression, thereby mitigating the effects of UCA1-mediated hypoxia injury. Besides, miR-18a was shown to target SOX6 whose expression was negatively regulated by miR-18a. In addition, overexpressed SOX6 could aggravate the hypoxia injury in PC-12 cells, whereas a knockdown of it exhibited contrary results. Taken together, our study provides a novel and potential strategy for the treatment of neuronal injury induced by hypoxia.
In the previous studies, UCA1 has been implicated in several cancers, where its pathological roles were found to be mediated by miRNAs. For instance, Wei demonstrated that UCA1 contributed to melanoma cell proliferation, invasion, and G0/G1 cell cycle arrest by targeting miR-507 [17]. Bian reported that UCA1 enhanced colorectal cancer cell proliferation and mediated resistance to 5-fluorouracil by inhibiting the expression of miR-204-5p [18]. Similarly, Nie confirmed UCA1 to play an oncogenic role in non-small cell lung cancer through targeting miR-193a-3 [19]. On the same lines, in our study, we found miR-18a to be a direct target of UCA1. MiR-18a, a reported brain-specific miRNA, has been shown to be upregulated in frontal lobe and hippocampus after treatment with duloxetine [20]. However, it has been shown to be downregulated under hypoxic conditions in PC12 cells, which in turn alleviates the cerebral ischemic injury through targeting ATXN1 [21]. In addition, miR-18a can regulate the activity or expression of hypoxia inducible factor-1α (HIF-1α), thereby mediating the progression of several diseases [22-24]. HIF-1α is shown to have both neuroprotective and neurotoxic effects in hypoxic ischemia and may serve as a target for the therapy of hypoxic ischemia [25]. In our study, UCA1 was upregulated under hypoxic conditions and a suppression of it alleviated hypoxia injury in PC-12 cells. Upregulation of miR-18a mediated the effects of suppression of UCA1 on hypoxia injury. Although the role of UCA1 after hypoxic ischemia has not been completely investigated, considering the protective effects of miR-18a against the cerebral ischemic injury, we speculate that UCA1 may contribute to the cerebral hypoxia injury by targeting miR-18a. Another important finding of our study was that SOX6 served as a target of miR-18a, and its expression could be negatively regulated by miR-18a. SOX6 belongs to the D-sub family of SOXs and has been shown to participate in the development of the central nervous system by mediating neuronal differentiation and insulin resistance [26,27]. SOX6 is also shown to be implicated in the determination of cell fate, proliferation, and differentiation [28]. During neocortical development, it can function as a central regulator of dorsal progenitor identity and interneuron diversity [29]. The pathological roles of SOX6 are found in several disease conditions, such as endometriosis [30], sepsis-induced cardiac apoptosis [31], and various cancers [32,33]. In the present study, an overexpression of SOX6 could aggravate the injurious effects of hypoxic stress in PC-12 cells, whereas a knockdown of SOX6 completely exhibited contrary results. Although the role of SOX6 in hypoxic ischemia is not characterized, we speculate that it may be acting downstream of UCA1-miR-18a pathway to control the process of cerebral hypoxia injury.
In conclusion, the present study provides a comprehensive analysis of UCA1-miR-18a-SOX6 axis in the hypoxia injury following cerebral ischemia. The results revealed that the upregulation of UCA1 decreases the expression of miR-18a, consequently leading to an increase in the expression of SOX6, thereby contributing to the hypoxia injury following cerebral ischemia. We believe that our findings may serve as a platform for the synthesis of novel and potential therapeutic strategies for the treatment of cerebral infarction and ischemic stroke.
Acknowledgements
This work was supported by National Natural Science Foundation of China, Grant No: 8160231, 81160338, 31360238, 81560209 and 81660527, National University Student Innovation Program, Grant No: 201410752002, Key projects of universities in Ningxia, Grant No: NGY2011039, and West China First-Class Discipline Construction Project Funded by Ningxia Medical University, Grant No: 20130181601.
Disclosure of conflict of interest
None.
References
- 1.Hori M, Nakamachi T, Rakwal R, Shibato J, Nakamura K, Wada Y, Tsuchikawa D, Yoshikawa A, Tamaki K, Shioda S. Unraveling the ischemic brain transcriptome in a permanent middle cerebral artery occlusion mouse model by DNA microarray analysis. Dis Model Mech. 2012;5:270–283. doi: 10.1242/dmm.008276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thompson B. Hypoxia/Reoxygenation stress modulates atorvastatin transport at the bloodbrain barrier: a role for organic anion transporting polypeptide. 2014 [Google Scholar]
- 3.Vemuganti R. All’s well that transcribes well: Non-coding RNAs and post-stroke brain damage. Neurochem Int. 2013;63:438. doi: 10.1016/j.neuint.2013.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yin KJ, Hamblin M, Chen YE. Non-coding RNAs in cerebral endothelial pathophysiology: emergingroles in stroke. Neurochem Int. 2014;77:9–16. doi: 10.1016/j.neuint.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang J, Yuan L, Zhang X, Hamblin MH, Zhu T, Meng F, Li Y, Chen YE, Yin KJ. Altered long non-coding RNA transcriptomic profiles in brain microvascular endothelium after cerebral ischemia. Exp Neurol. 2015;277:162–170. doi: 10.1016/j.expneurol.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kaur P, Liu F, Tan JR, Lim KY, Sepramaniam S, Karolina DS, Armugam A, Jeyaseelan K. Non-coding RNAs as potential neuroprotectants against ischemic brain injury. Brain Sci. 2013;3:360. doi: 10.3390/brainsci3010360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Cao B, Han D, Sun M, Feng J. Long non-coding RNA H19 induces cerebral ischemia reperfusion injury via activation of autophagy. Aging Dis. 2017;8:71. doi: 10.14336/AD.2016.0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Li Q, Zhang KS, Hu B, Niu X, Zhou SM, Li SG, Luo YP, Wang Y, Deng ZF. Downregulation of the long non-coding RNA Meg3 promotes angiogenesis after ischemic brain injury by activating notch signaling. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-0270-z. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu Q, Deng F, Xing Z, Wu Z, Cen B, Xu S, Zhao Z, Nepomuceno R, Bhuiyan MI, Sun D. Long non-coding RNA C2dat1 regulates CaMKIIδ expression to promote neuronal survival through the NF-κB signaling pathway following cerebral ischemia. Cell Death Dis. 2016;7:e2173. doi: 10.1038/cddis.2016.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang XS, Zhang Z, Wang HC, Cai JL, Xu QW, Li MQ, Chen YC, Qian XP, Lu TJ, Yu LZ. Rapid identification of UCA1 as a very sensitive and specific unique marker for human bladder carcinoma. Clin Cancer Res. 2006;12:4851–4858. doi: 10.1158/1078-0432.CCR-06-0134. [DOI] [PubMed] [Google Scholar]
- 11.Wei L, Peng X, Ruan W. Overexpression of lncRNA UCA1 promotes osteosarcoma progression and correlates with poor prognosis. J Bone Oncol. 2016;5:80–85. doi: 10.1016/j.jbo.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao W, Sun C, Cui Z. A long noncoding RNA UCA1 promotes proliferation and predicts poor prognosis in glioma. Clin Transl Oncol. 2017;19:735–741. doi: 10.1007/s12094-016-1597-7. [DOI] [PubMed] [Google Scholar]
- 13.Zuo ZK, Gong Y, Chen XH, Ye F, Yin ZM, Gong QN, Huang JS. TGFβ1-Induced LncRNA UCA1 upregulation promotes gastric cancer invasion and migration. DNA Cell Biol. 2017;36:159. doi: 10.1089/dna.2016.3553. [DOI] [PubMed] [Google Scholar]
- 14.Wang HM, Lu JH, Chen WY, Gu AQ. Upregulated lncRNA-UCA1 contributes to progression of lung cancer and is closely related to clinical diagnosis as a predictive biomarker in plasma. Int J Clin Exp Med. 2015;8:11824. [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao C, Wu CH, Hu HZ. LncRNA UCA1 promotes epithelial-mesenchymal transition (EMT) of breast cancer cells via enhancing Wnt/beta-catenin signaling pathway. Eur Rev Med Pharmacol Sci. 2016;20:2819. [PubMed] [Google Scholar]
- 16.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 17.Wei Y, Sun Q, Zhao L, Wu J, Chen X, Wang Y, Zang W, Zhao G. LncRNA UCA1-miR-507-FOXM1 axis is involved in cell proliferation, invasion and G0/G1 cell cycle arrest in melanoma. Med Oncol. 2016;33:1–9. doi: 10.1007/s12032-016-0804-2. [DOI] [PubMed] [Google Scholar]
- 18.Bian Z, Jin L, Zhang J, Yuan Y, Quan C, Hu Y, Feng Y, Liu H, Fei B, Mao Y. LncRNA-UCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer by inhibiting miR-204-5p. Sci Rep. 2016;6:23892. doi: 10.1038/srep23892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nie W, Ge HJ, Yang XQ, Sun X, Huang H, Tao X, Chen WS, Li B. LncRNA-UCA1 exerts oncogenic functions in non-small cell lung cancer by targeting miR-193a-3p. Cancer Lett. 2015;371:99–106. doi: 10.1016/j.canlet.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Pan B, Liu Y. Effects of duloxetine on microRNA expression profile in frontal lobe and hippocampus in a mouse model of depression. Int J Clin Exp Pathol. 2015;8:15454. [PMC free article] [PubMed] [Google Scholar]
- 21.Ke W, Lu ZN, Deng XR, Li WL, Du M, Yang H, Liu YM. MicroRNA-18a targets ATXN1 to alleviate injury induced by permanent middle cerebral artery occlusion in mice. Int J Clin Exp Med. 2017;10:965–971. [Google Scholar]
- 22.Li P, Gao Y, Li F, Pan Q, Liu Z, Lu X, Song C, Diao X. MicroRNA-18a regulates invasive meningiomas via hypoxia-inducible factor-1α. Exp Ther Med. 2015;10:1165. doi: 10.3892/etm.2015.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu F, Huang W, Wang X. microRNA-18a regulates gastric carcinoma cell apoptosis and invasion by suppressing hypoxia-inducible factor-1α expression. Exp Ther Med. 2015;10:717. doi: 10.3892/etm.2015.2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han F, Wu Y, Jiang W. MicroRNA-18a decreases choroidal endothelial cell proliferation and migration by inhibiting HIF1A expression. Med Sci Monit. 2015;21:1642. doi: 10.12659/MSM.893068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan X, Heijnen CJ, van der Kooij MA, Groenendaal F, Bel FV. The role and regulation of hypoxia-inducible factor-1α expression in brain development and neonatal hypoxic-ischemic brain injury. Brain Res Rev. 2009;62:99–108. doi: 10.1016/j.brainresrev.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Hamadakanazawa M, Ishikawa K, Ogawa D, Kanai M, Kawai Y, Narahara M, Miyake M. Suppression of Sox6 in P19 cells leads to failure of neuronal differentiation by retinoic acid and induces retinoic acid-dependent apoptosis. FEBS Lett. 2004;577:60–66. doi: 10.1016/j.febslet.2004.09.063. [DOI] [PubMed] [Google Scholar]
- 27.Iguchi H, Urashima Y, Inagaki Y, Ikeda Y, Okamura M, Tanaka T, Uchida A, Yamamoto TT, Kodama T, Sakai J. SOX6 suppresses cyclin D1 promoter activity by interacting with beta-catenin and histone deacetylase 1, and its down-regulation induces pancreatic betacell proliferation. J Biol Chem. 2007;282:19052–19061. doi: 10.1074/jbc.M700460200. [DOI] [PubMed] [Google Scholar]
- 28.Snyder M, Huang XY, Zhang JJ. Stat3 is essential for neuronal differentiation through direct transcriptional regulation of the Sox6 gene. FEBS Lett. 2011;585:148–152. doi: 10.1016/j.febslet.2010.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Azim E, Jabaudon D, Fame RM, Macklis JD. SOX6 controls dorsal progenitor identity and interneuron diversity during neocortical development. Nat Neurosci. 2009;12:1238–1247. doi: 10.1038/nn.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang D, Li Y, Tian J, Zhang H, Wang S. MiR-202 promotes endometriosis by regulating SOX6 expression. Int J Clin Exp Med. 2015;8:17757. [PMC free article] [PubMed] [Google Scholar]
- 31.Wu HL, Li SM, Hu J, Yu X, Xu H, Chen Z, Ye ZQ. Demystifying the mechanistic and functional aspects of p21 gene activation with double-stranded RNAs in human cancer cells. J Exp Clin Cancer Res. 2016;35:145. doi: 10.1186/s13046-016-0423-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Zheng D, Zhang B, Liu L, Ou J, Chen W, Xiong S, Gu Y, Yang J. Mir-208 promotes cell proliferation by repressing SOX6 expression in human esophageal squamous cell carcinoma. J Transl Med. 2014;12:1–9. doi: 10.1186/1479-5876-12-196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong G, Yun W, Ding Q, Yang L. Hsamir-1269 genetic variant contributes to hepatocellular carcinoma susceptibility through affecting SOX6. Am J Transl Res. 2015;7:2091. [PMC free article] [PubMed] [Google Scholar]