

Abstract
Small ubiquitin-related modifiers (SUMO) represent a class of ubiquitin-like proteins that are conjugated, like ubiquitin, by a set of enzymes to form cellular regulatory proteins, and play key roles in the control of cell proliferation, differentiation, and apoptosis. We found that ginkgolic acid (GA) can significantly reduce cell vitality in a dose- and time-dependent manner and can also accelerate cyto-apoptosis in both Tca8113 and Cal-27 cells. Migration and wound-healing assays were executed to determine the anti-migration effect of GA in oral squamous cell carcinoma (OSCC) cell lines. GA represses transforming growth factor-β1 (TGF-β1)-induced epithelial-mesenchymal transition (EMT) markers in OSCC cell lines. This investigation is the first evidence that GA suppresses TGF-β1-induced SUMOylation of SMAD4. We show that GA affects the phosphorylation of SMAD2/3 protein and the release of SMAD4. In the xenograft mouse model, the OSCC progression was reduced by GA, effectively suppressing the growth of tumors. In addition, siSMAD4 improved cell migration and viability, which was inhibited by GA in Tca8113 cells. GA suppresses tumorigenicity and tumor progression of OSCC through inhibition of TGF-β1-induced enhancement of SUMOylation of SMAD4. Thus, GA could be a promising therapeutic for OSCC.
Keywords: small ubiquitin-like modifiers, oral squamous cell carcinoma, ginkgolic acid, proliferation, migration
Graphical Abstract
Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common cancer worldwide.1,2 Of all subtypes of oral malignancies, 90% constitute the oral squamous cell carcinoma (OSCC), which is a locally aggressive tumor, and whose invasion and metastasis results from the adaptation to a particular microenvironment.3 Moreover, it has been shown that the high rate of morbidity is due to both locoregional recurrence and distant metastasis. For this reason, the 5-year survival rate remains steady at approximately 50%–55%.2,4 A remarkable phenotype plasticity of epithelial cells underlies morphogenesis, epithelial repair, and tumor invasiveness. Members of the transforming growth factor (TGF) family can initiate and maintain an epithelial-mesenchymal transition in a variety of biological systems and pathophysiological contexts by activating major signaling pathways and transcriptional regulators that are integrated into extensive signaling networks.5 The purpose of this investigation was to review the distinct physiological contexts of EMT and the underlying molecular signaling networks controlled by TGF-β1.6 TGF-β1 regulates its pleiotropic biological activities through the activation of the downstream SMAD signaling pathways.7, 8, 9 Crucially, regulation of the SMAD4 tumor suppressor, the central mediator of the TGF-β/SMAD signaling pathway, is common in malignant HNSCC. The importance of SMAD4 in HNSCC was recently established by a study in which SMAD4 loss caused defects in the Fanconi anemia/BRCA (FANC/BRCA) pathway, leading to genomic instability in mice and spontaneous HNSCC.10
Protein modification by small ubiquitin-related modifiers (SUMOs) controls a diverse array of cellular functions. In particular, the dysregulation of SUMOylation or deSUMOylation processes has been implicated in the development of cancer.11 SUMOylation is mediated by an enzymatic cascade reaction similar to ubiquitination. These enzymes include the E1 SUMO-activating enzyme, the E2-conjugating enzyme Ubc9, and an E3 ligase, which promotes the transfer of SUMO from Ubc9 to specific proteins.12, 13, 14 Post-translational conjugation of small ubiquitin-related modifier proteins has been shown as one of the major modifications that regulate various biological systems. In recent years, it was shown that TGF-β1-induced SUMOylated PML/Pin1 activation in response to TGF-β1/SMAD signaling and contributes significantly to myocardial fibrosis.15 Additionally, SMAD4 is the central mediator of TGF-β/SMAD signaling and shuttling between the cytoplasm and the nucleus.16,17 The SMAD4 protein is modified in the post-translational level by SUMO proteins in the TGF-β signaling pathway, whereby SMAD4 is SUMOylated, and SUMO-1 forms complexes with SMAD4. The accumulation of SUMO-1 is directly related to SMAD4 SUMOylation; however, these findings indicate an opposite effect of SUMOylation on SMAD4 activity, which can play a positive or negative role in regulating TGF-β/SMAD signaling.18 Thus, an in-depth analysis of SMAD4 targets is needed to better understand the molecular mechanism by which the canonical TGF-β signaling pathway controls cell function.16
Ginkgolic acid (GA) exists in leaves, nuts, and external seed coatings of ginkgo. GA has been shown to exhibit various pharmacological activities, such as antitumor, anti-depressant, anti-fungal, and anti-microbial effects.19, 20, 21, 22, 23 By directly binding to E1, GA impaired SUMOylation by blocking the formation of an E1-SUMO thioester complex.11,15 Prior studies have also indicated that GA could inhibit the proliferative effect of human cancer cells through the induction of apoptosis.23 However, the role of GA as a SUMO scavenger for impacting cancer cell migration has yet to be explored, which is appealing given the possible regulation of the TGF-β/SMAD pathway by SUMOylation. Therefore, in this study, we screened for inhibitors of protein SUMOylation from a botanical extract library by using an in situ SUMOylation screening system. We found the inhibitory activity of protein SUMOylation in the extract of ginkgo biloba leaves and identified GA as an inhibitor. GA and its structural analog inhibited SUMOylation both in vitro and in vivo. The discovery of a low-molecular inhibitor of protein SUMOylation is particularly useful for drug development.11
Results
GA Suppresses the Viability of OSCC Cell Lines
Prior studies have indicated that GA could inhibit the growth of various tumorigenic cell lines.24 To analyze the effect of GA against Tca8113 and Cal-27 cells, we treated the cells with various concentrations (2, 5, 10, 20, and 40 μM) for 24 h and then assessed cell viability using the cell counting kit-8 (CCK-8) assay. Treatment of cells with GA led to growth inhibition in a dose-dependent manner in both cell lines. Tca8113 cell viability decreased by approximately 31.23% and 41.79% in cells incubated with GA (5 μM and 10 μM, respectively) for 24 h. Meanwhile, GA (5 μM and 10 μM) inhibited Cal-27 cell viability by approximately 31.13% and 53.53%, respectively (Figures 1A and 1B). The results demonstrate that the historicity of the GA IC50 value is less than or equal to a 10 μM concentration (Figures 1A and 1B). Therefore, all in vitro migration studies were performed below the 10 μM dose level. GA can significantly reduce cell proliferation in both Tca8113 and Cal-27 cells in a dose- and time-dependent manner.
Figure 1.

GA Inhibits Cell Viability and Induces Cyto-apoptosis of OSCC
(A and B) Tca8113 cells (A) and Cal-27 cells (B) were incubated with increasing concentrations of GA for 24 h. Relative or percent cell viability was determined by CCK-8 assay and based on the OD (optical density) values as indicated in the Materials and Methods. Data are expressed as the mean ± SEM of three independent experiments. Statistically significant differences are marked with *p <0.05, **p <0.01, and ***p <0.001 by one-way ANOVA Tukey’s test. (C) Representative morphological changes of apoptotic cells were determined by electron microscope analysis. Magnification: ×5,000. Scale bar, 2 μm. (D) Apoptotic cells were determined by TUNEL fluorescence staining on Tca8113 cells and Cal-27 cells. The nuclei were stained blue with DAPI. Blue, DAPI; red, TUNEL. Magnification: ×200. Scale bar, 10 μm. (E-F) The rate of apoptotic positive cells in GA(10 **p <0.01 versus control group.
GA Induces Cyto-apoptosis in OSCC Cell Lines
Previous reports have demonstrated that GA might have an anti-proliferative effect against human cancer cells through the induction of apoptosis.23 GA treatment might cause apoptosis that inhibits Tca8113 and Cal-27 cell survival. As shown in Figures 1C and 1D, a significant accumulation of apoptosis cells is induced by GA at 10 μM. We also examined the micro-morphological changes induced by GA using an electron microscope at a magnification of 10,000 to provide an alternative indication of apoptosis. As demonstrated in Figure 1C, GA resulted in micro-structure alterations of the cell, such as cell shrinkage, nuclear condensation fragmentation, and formation of separated apoptotic bodies, at 24 h. Subsequently, we used the TUNEL assay to measure apoptosis after treating the OSCC cells with GA. As depicted in Figure 1D, the treatment of OSCC cells with 10 μM GA for 24 h induced significant cell apoptosis. GA at a concentration of 10 μM led to 35.51% and 51.15% promotion of apoptosis in Tca8113 and Cal-27 cells, respectively (Figures 1C–1F). The extent of TUNEL-positive cells also increased as compared to the control.
GA Represses Migration of OSCC Cell Lines
The potential effects of GA on cancer cell invasion and migration are not fully understood, nor are the possible molecular mechanism(s) regulating the PI3K/Akt/mTOR pathway. GA could inhibit invasion and migration, and EMT in lung cancer cells, suggesting that GA exerts both antimetastatic and/or anti-EMT effects.24 Therefore, a Transwell migration assay was performed to determine the anti-migration effect of GA in Tca8113 and Cal-27 cells. As depicted in Figure 2A, GA significantly inhibited the migration activity of Tca8113 and Cal-27 cells. Additionally, we investigated whether GA reduces the migration of Tca8113 and Cal-27 cells using a wound-healing assay. Figure 2B demonstrates that Tca8113 and Cal-27 cells migrated faster than the cells treated with GA (5 μM, 10 μM). In particular, GA at a concentration of 5 μM and 10 μM led to an inhibition of migration in TGF-induced Tca8113 cells of 66.3% and 96.1%, respectively. Meanwhile, GA at a concentration of 5 μM and 10 μM led to a 29.55% and 55.89% inhibition of migration in TGF-induced Cal27 cells, respectively, in a concentration-dependent manner (Figure 2A).
Figure 2.

GA Represses Migration of OSCC Cell Lines
(A) Migration capacity of oral squamous cancer cells (Tca8113 and Cal-27) cultured with TGF-β1 and treated with GA was determined by the in vitro transwell migration system. Representative photographs of migratory cells on the membrane are shown. Scale bar, 10 μm. (B) GA significantly suppressed the migration of Tca8113 cells and Cal-27 cells as reported by the wound-healing assay. Scale bar, 100 μm. (C and D) Averaged data (mean ± SEM, n = 3) from transwell migration assay showing the concentration-dependent suppression of migration. Statistically significant differences are marked with #p < 0.05, ##p < 0.01, and ###p <0.001 by one-way ANOVA Tukey’s test, compared with TGF-β1. (E and F) Statistical quantification of the wound healing assays. The results shown are the mean ± SEM of three independent experiments. *p < 0.05, compared to control; ###p <0.001, compared with TGF-β1.
GA Represses TGF-β1-Induced EMT in OSCC Cell Lines
It has been well established that EMT is a critical process for the aggressive metastatic dissemination of carcinomas. EMT undergoes multiple and dynamic transitional states from epithelial to mesenchymal phenotypes.25 Decreases in the mesenchymal markers N-cadherin and Vimentin and increases in epithelial markers E-cadherin and α-catenin indicate dampening of EMT and vice versa.26 Following TGF-β1 and GA treatment, both Tca8113 and Cal-27 cells showed morphological changes. The GA-only treatment induced more cobblestone-shaped cells than the control. Treatment of TGF-β1 resulted in many more fusiform-shaped cells than the control. In addition, GA decreased the ratio of fusiform-shaped cells that was caused by the treatment of TGF-β1, as more cobblestone-shaped cells were present (Figure 3A). We then explored whether EMT plays a role in mediating the anti-metastasis property of GA by measuring the expression changes of these biomarkers using western blot analysis. As illustrated in Figures 3B and 3C, the GA only treatment showed inhibition of EMT. Notably, GA treatment reversed TGF-β1 (10 ng mL-1)-induced EMT at 5 μM, as indicated by the downregulation of N-cadherin and Vimentin expression, and induced a concomitant upregulation of E-cadherin and α-catenin expression in Tca8113 and Cal-27 cells (Figures 3D–3K). Immunofluorescent staining also demonstrated that when GA was applied to OSCC cells 1 h before TGF-β1 (10 ng mL-1) treatment, which lasted for 48 h, there was a significant decrease in Vimentin expression and an increase in E-cadherin expression relative to the control (Figure 3B). As shown in Figures 3B–3K, the GA only treatment versus GA+TGF-β1 group comparison confirmed that GA depended on TGF-β1 in the regulation of EMT-related proteins.
Figure 3.

GA Abolished TGF-β1-Induced EMT in OSCC Cell Lines
(A) Effects of GA inhibition on TGF-β1-induced mesenchymal. Phenotype in OSCC cells by morphology analysis. TGF-β1 induced a marked mesenchymal spindle-like morphology. The inhibition of GA markedly attenuated the TGF-β1-induced morphological changes. Scale bar, 10 μm. (B) GA significantly upregulated E-cadherin and downregulated Vimentin in OSCC cells by immunofluorescence assay. Blue, DAPI; red, E-cadherin; green, Vimentin. Scale bar, 10 μm. (C) The protein levels of EMT markers were compared and quantified in OSCC cells incubated with GA (5 μM) by western blot assay. (D–K) The histogram shows data from five independent western blot analyses of epithelial and mesenchymal markers proteins, indicated as mean ± SEM, normalized to GAPDH as a loading control. Data are presented as mean ± SEM of 3 independent experiments. *p < 0.01, **p < 0.01, ***p < 0.001.
GA Mediates TGF-β1-Induced SMAD4 SUMOylation in OSCC Cells
SUMO represents a class of ubiquitin-like proteins that are conjugated by a set of enzymes to cellular regulatory proteins, much like ubiquitin.27 Exogenous TGF-β1 induced the upregulation of SUMOs in a time-dependent manner15. To quantify the TGF-β1-mediated SUMO upregulation and determine the optimal time of TGF-β1 required to induce the maximal SUMOs expression in OSCC, we conducted western blot analysis. The results indicated that the addition of TGF-β1 induced SUMO protein expressions in a dose- (10 ng mL-1) and time- (0, 4, 6, 12, 24, 48 h) dependent manner (Figures 4A and 4B). The optimal dose of TGF-β1 was 10 ng mL-1 with a peaked protein expression at 12 h. Moreover, TGF-β1 (10 ng mL-1, 12 h) also induced the maximum protein expression of SUMO-1 and SUMO-2/3 (Figures 4A and 4B). Therefore, 12 h and 10 ng mL-1 TGF-β1 were adopted as the optimal time point and dosage in the following experiments. Since GA is involved in repressing migration and TGF-β1-induced EMT of lung cancer cells through PI3K/AKT/mTOR inactivation,24 we investigated whether GA could regulate EMT through TGF-β/SMAD/SUMOs in OSCC cells. If so, GA would provide evidence of direct inhibition of the TGF-β-induced SUMOs upregulation in the TGF-β/SMAD signaling pathways. As expected, GA eliminated the TGF-β-induced SUMOs (Figure 4C). Meanwhile, the TGF-β1-mediated phosphorylation of SMAD2/3 protein is also reduced following the application of GA (Figure S1). On the contrary, SMAD4 protein levels increased following the application of GA (Figures 4C and S1). SMAD4, which is a substrate for ubiquitin modification, is often modified by SUMOylation, raising the possibility that TGF-β/SMADs could be targeted for SUMOylation.28 To illustrate further this phenomenon, we used immunoprecipitation (IP) to detect the interaction of SUMO-1 with SMAD4. As anticipated, coIP experiments confirmed that anti-SMAD4 antibodies were able to pull down associated proteins, and the GA treatment reversed TGF-β1-induced SUMO1 conjunction to SMAD4 (Figure 4D). Of note, a clear interaction between the specific SMAD4 isoform and SUMO-1 in a molecular mass of 80 kDa to 100 kDa occurred, which is the most well-defined and extensively studied isoform.29 Consistent with the immunoblotting data from SMAD4 SUMOylation, immunofluorescence analysis revealed that after the addition of TGF-β1, the levels of SMAD4 SUMOylation in the nucleus increased significantly (Figure 4E). 12 h after TGF-β1 treatment (Figure 4A), SMAD4 SUMOylation accumulated at the highest density during the 12 h of TGF-β1 application. Subsequently, GA treatment reversed TGF-β1-induced SUMO-1 conjunction to SMAD4 (Figures 4D and 4E), indicating that GA can counteract TGF-β1-triggered SMAD4 SUMOylation in vitro.
Figure 4.

GA Regulates TGF-β1-Induced SMAD4 SUMOylation in OSCC Cells
(A and B) Western blot was performed to detect the expression of SUMOylation proteins in OSCC cell lines. Representative western blots and their densitometries from three independent experiments. (C) The western blotting analysis demonstrates that GA increased the SMAD4 protein level but reduced the TGF-β1-mediated phosphorylated SMAD2/3 protein expressions. Averaged data (mean ± SEM) from 3 independent experiments. (D) The amount of the SUMO and SMAD4 binding was markedly decreased in Tca8113 cells by coIP analysis after GA administration. (E) GA treatment reversed TGF-β1 induced SUMO-1 conjunction of SMAD4 by immunofluorescence assay. Blue, DAPI; red, SUMO-1; green, SMAD4. Scale bar, 10 μm.
GA Suppresses Tumor Growth of Tca8113 Cells in a Xenograft Model
Based on the findings in vitro, we conducted a follow-up in vivo experiment to confirm the effect of GA. The average tumor volume, tumor weight, and body weight were measured twice a week. Following a single dosage of 20 or 50 mg kg−1 (body weight) by oral gavage, both doses of GA effectively suppressed the growth of tumors, showing greater antitumor activity than the control, which showed no effect (Figures 5A–5D). GA effectively suppressed the growth of tumors, GA with 50 mg kg-1 showing greater antitumor activity (tumor weight IR% = 71.38%, tumor volume IR% = 68.51%) than 20 mg kg-1 (tumor weight IR% = 17.25%, tumor volume IR% = 30.42%; Figures 5A and 5B). The antitumor activities of GA are summarized in Table 1. In a follow-up western blot study, the epithelial marker E-cadherin was upregulated, while mesenchymal markers, namely Vimentin and N-cadherin, were downregulated by GA (20 or 50 mg kg-1, Figures 5E and S3). Mesenchymal and epithelial markers have been demonstrated to promote tumor progression and are implicated in EMT.5 In this study, as shown by western blot, the degradation of phosphorylated SMAD2/3/SUMO-1/SUMO-2/3 proteins was inhibited by GA in the tumors of the GA group. On the contrary, the SMAD4 protein level increased after GA application (Figures 5F and S2). As expected, and consistent with the coIP data in vitro, GA treatment significantly attenuated SUMO-1 conjunction of SMAD4 compared with the control (Figure 5G).
Figure 5.

GA Retards Tumor Growth of Tca8113 Cells In Vivo
(A) Photographs of Tca8113 residual tumor xenografts treated with saline or GA (20 mg kg-1 and 50 mg kg-1) by oral gavage. Scale bar, 1 cm. (B–D) Tumor growth curve is showing the suppression of growth of Tca8113 residual tumor xenograft tumors by GA. The average tumor volumes in the GA groups were reduced, and the tumor weight was decreased by GA. **p < 0.01 and ***p < 0.001 by one-way ANOVA; Dunnett, compared with control. (E) The protein levels of EMT markers were compared and quantified in Tca8113 residual tumor xenograft tumors by western blot assay. (F) GA increased the SMAD4 protein level but reduced the phosphorylated SMAD2/3 protein expressions in the Tca8113 residual tumor xenograft tumors. (G) coIP analysis indicated GA treatment reversed SUMO1 conjunction of SMAD4.
Table 1.
Antitumor Activity of GA on Postsurgical Residual Tumor Xenografts of Human Oral Squamous Cancer Cell in Nude Mice
| Drug Administration |
Toxicity |
Anticancer Activity |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Group | Dose (mg·kg−1) | Schedule | Route |
Average Body Weight (g, ± s) |
Death | Tumor Weight (g) | IR (%) | Tumor Volume (mm3) | IR (%) | |
| start | stop | |||||||||
| Control | − | QD × 21 | i.g. | 22.6 ± 0.9 | 24 ± 0.7 | 0/8 | 0.48 ± 0.27 | − | 522.77 ± 228.39 | − |
| GA | 20 | QD × 21 | i.g. | 22.6 ± 1.2 | 22.8 ± 1.0 | 0/8 | 0.4 ± 0.16 | 17.25 | 363.74 ± 118.99 | 30.42 |
| GA | 50 | QD × 21 | i.g. | 22.4 ± 1.5 | 20.1 ± 2.9 | 0/8 | 0.14 ± 0.09∗∗ | 71.38 | 164.63 ± 93.29*** | 68.51 |
p < 0.01, ***p < 0.001. The significance of differences (versus control) was determined by one-way ANOVA and Student’s t test. QD, every day; IR, inhibitory rate; i.g. represents gastric gavage daily. n = 8, ± s.
GA Moderately Affects the Proliferation and Migration of Tca8113 Cell Line Induced by the Knockdown of SMAD4
Based on the data mentioned above, we postulated that SUMOylated SMAD4 mediates migration by EMT. To test this hypothesis, we investigated the possible regulation of migration by SMAD4 using small interfering RNA (siRNA) oligonucleotides to knock down SMAD4 expression in Tca8113. Successful knockdown of SMAD4 into the cells was confirmed by western blot (Figures 6A and 6B). The CCK-8 assay demonstrated that GA treatment particularly inhibited the accelerated Tca8113 cell proliferation promoted by the knockdown of SMAD4. The GA only treatment could reduce cell viability by 38.59% compared to the control. However, knockdown of SMAD4 attenuated the effect of GA on cell viability. The viability of the cells in the siRNA group increased by 13.33% compared to the GA group (Figure 6G). After TGF-β1 and GA treatment, transwell migration and wound-healing assays demonstrated that SMAD4 silencing increased the migration of Tca8113 cells. GA treatment could reduce cell migration by 62.30% compared to TGF-β1. However, knockdown of SMAD4 attenuated the effect of GA on cell migration. The migration capacity of the cells in the siRNA group increased by 52.66% compared to the GA group (Figures 6C–6F). Meanwhile, si-SMAD4 attenuated the GA-induced E-cadherin upregulation and Vimentin downregulation in Tca8113 cells (Figures 6H and S4). Knockdown of SMAD4 abolished the reducing viability of GA in Tca8113 (Figure 6G). These data suggest that TGF-β1-induced SMAD4 SUMOylation is involved in OSCC cell proliferation and migration (Figure 7). Moreover, GA reduces TGF-β1-induced SMAD4 SUMOylation. Consequently, proliferation and migration were inhibited in the Tca8113 cell line.
Figure 6.

Knockdown of SMAD4 Attenuates the Inhibition of Migration and Viability Caused by GA in Tca8113
(A) Si-SMAD4 and negative-control expression vectors were transfected into Tca8113 cells. (B) Western blot assay showing a successful SMAD4 knockdown of Tca8113 cells compared with control. **p < 0.01 and ***p < 0.001 by one-way ANOVA. (C) After TGF-β1 and GA treatment, wound-healing assay demonstrated that SMAD4 silencing increased the number of Tca8113 cells migration compared with control. Scale bar, 100 μm. (D) Transwell migration assay showed that the SMAD4 knockdown promoted the migration ability of Tca8113 cells compared with control. Scale bar, 10 μm. (E) Averaged data (mean ± SEM, n = 3) from wound-healing assay showing the acceleration of migration by si-SMAD4. *p < 0.05, compared with control. ###p <0.001, compared with TGF-β1. (F) Averaged data (mean ± SEM, n = 3) from transwell migration assay showing the acceleration of migration by si-SMAD4. ***p <0.001, compared with control. ###p <0.001, compared with TGF-β1. (G) Knockdown SMAD4 of Tca8113 cells were incubated with GA (10 μM) for 24 h. Relative or percent cell viability was determined by CCK-8 assay. Data are expressed as the mean ± SEM of 3 independent experiments. Statistically significant differences are marked with ***p <0.001 compared with control, ###p <0.001 compared with GA (10 μM). (H) The protein levels of EMT markers by knockdown SMAD4 were compared and quantified in OSCC cells incubated with GA (5 μM) by western blot assay. TGF-β1 increased N-cadherin and Vimentin protein levels and decreased E-cadherin protein levels compared with the control. Data are presented as mean ± SEM of 3 independent experiments.
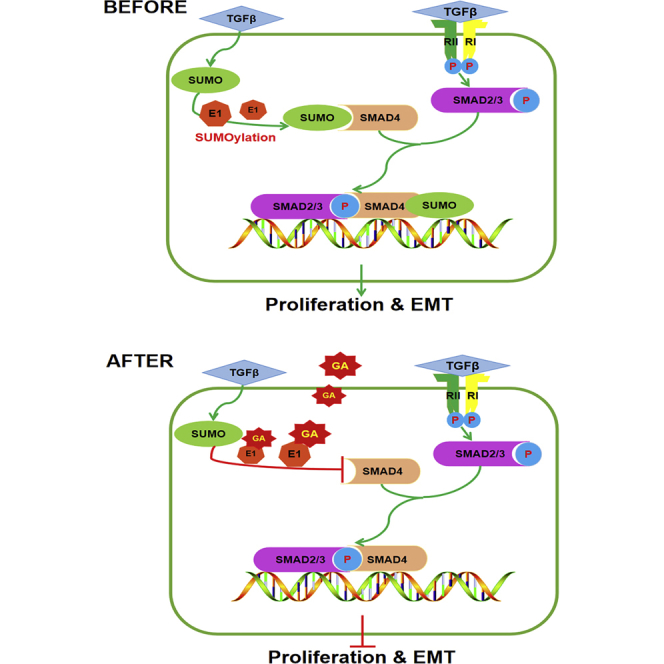
Figure 7.

Mechanism of GA Inhibiting SUMOylation of SMAD4 and Inhibiting Proliferation and EMT of OSCC
TGF-β1 is probably a novel enhancer or inducer of SMAD4 SUMOylation and forms a passive feedback loop in the TGF-β1/SMAD signaling pathway and subsequently accelerates the proliferation and migration of OSCC cells in vivo and in vitro. However, GA treatment significantly attenuated TGF-β1-induced SMAD4 SUMOylation. Meanwhile, GA released SMAD4, which associates with SMAD2/3 to form a heterooligomeric complex that is then translocated to the nucleus where it activates the transcription of various target genes to reduce proliferation and migration of OSCC cells.
Discussion
The purpose of this investigation was to demonstrate, for the first time, that TGF-β1 is a novel enhancer or inducer of SMAD4 SUMOylation. Furthermore, TGF-β1 forms a passive feedback loop in the TGF-β/SMAD signaling pathway and subsequently accelerates OSCC cell proliferation and migration in vivo and in vitro. However, GA treatment significantly attenuated TGF-β1-induced SMAD4 SUMOylation. The dissociative SMAD4 associates with SMAD2/3 and forms a heterooligomeric complex, which translocates to the nucleus where it activates the transcription of various target genes to reduce the proliferation and migration of OSCC cells. GA has antitumor activities, including the inhibition of the proliferation and migration of OSCC cells.
Recently, the antitumor efficacy of GA has gained considerable attention.19,30 Our detailed mechanistic study revealed that GA had significant antitumor effects in both OSCC cell lines and a xenograft nude mouse model. The inhibition of the expression of critical genes involved in proliferation and migration could be the basis of these effects induced by GA. Recent reports have demonstrated that GA suppresses the proliferation, migration, and invasion, as well as the de novo lipogenesis of cancer cells that are likely induced by the activation of AMPK.31 Additionally, GA may negatively regulate the TGF-β-induced PI3K/AKT/mTOR signaling pathway, which results in inhibition of EMT-related protein synthesis and therefore has antimetastatic and anti-EMT effects.24 Furthermore, Zhou et al.23 showed that GA reduced the viability of various types of cancer cells by inhibiting division, reducing the progress of the cell cycle, and inducing apoptosis without affecting the viability of non-tumorigenic cells. The application of GA was explored for antitumor effects on human colon cancer both in vitro and in vivo. These findings suggest that GA reduced cell viability, migration, and invasion in various cell lines of colon cancer. GA also induced apoptosis and autophagy in colon cancer cells regulated by reactive oxidative stress.32
The cytotoxicity of GA (20 μM) in primary rat hepatocytes was lower than in HepG2 cells. GA demonstrated less cytotoxicity in 4-day-cultured primary rat hepatocytes than in 20 h cultured cells. HepG2 cells were more sensitive to the cytotoxicity of GA than primary rat hepatocytes, and GA had time- and dose-dependent cytotoxic and growth inhibitory effects on a human tongue squamous carcinoma cell line.33 Cancer cell lines underwent growth arrest after treatment with GA in doses equivalent to the IC50 values of 20 μM.23 Kim et al.34 showed that GA (5 μM, 10 μM, and 30 μM) inhibited SUMO-1 conjugations in a dose-dependent manner. Thus, GA prevented the migration of breast cancer cell lines without reducing cell viability after a 72 h treatment, and GA at a dose of 10 μM significantly inhibited SUMO-1 modifications.35
In the present investigation, the cytotoxicity of GA was assessed using CCK-8 assays in a time- and dose-dependent manner, and we found that this compound has the ability to reduce the viability of OSCC cells. The historicity of the GA IC50 value is less than or equal to a 10 μM concentration. GA treatment at 5 μM reversed the EMT induced by TGF-β1 (10 ng mL-1), as indicated by the downregulation of N-cadherin and Vimentin expression, and the concomitant upregulation of E-cadherin and α-cadherin expression in OSCC cell lines. We conclude that GA revealed considerable anti-migratory activity at 5 μM concentrations, indicating antimetastatic activity with low toxicity. Additionally, a 10 μM GA treatment led to a significant accumulation of apoptotic cells.
Earlier studies demonstrated that protein modification by SUMOs controls a diverse array of cellular functions. Dysregulation of SUMOylation or deSUMOylation processes have been implicated in the development of cancer. The inhibition of protein SUMOylation both in vitro and in vivo by GA is explained by its direct binding to E1, which prevents the formation of the E1-SUMO intermediate.11 Hamdoun et al.35 revealed that GA inhibited NEMO SUMOylation, leading to the inhibition of IκBα degradation and consequently a reduction in nuclear factor κB (NF-κB) activity. This led to the downregulation of metastasis-related genes, including uPA, PAI-1, CXCR4, and MMP-9. Interestingly, Qiu et al.14 proposed an anti-fibrotic effect of GA and an underlying mechanism that may involve the downregulation of the TGF-β1 pathway, which may at least partially inhibit PML SUMOylation. Our data provide direct evidence that GA eliminated TGF-β1-induced SUMOs elevation. Our results indicate that GA reverses TGF-β1-triggered SMAD4 SUMOylation, and, consequently, reduces the proliferation and migration of OSCC cells in vitro and in vivo.
It has recently been demonstrated that the TGF-β/SMAD signaling pathway is altered by a genetic mutation that contributes to the carcinogenesis of HNSCC.36 Numerous studies have revealed that TGF-β1 stimulates the EMT process in certain epithelial cells.37 TGF-β1 drives cancer progression by inducing EMT; epithelial cells acquire a mesenchymal phenotype, leading to their enhanced motility and invasiveness. TGF-β1 directly activates the expression of EMT transcription factors, which are induced by TGF-β/SMAD signaling and play critical roles in TGF-β1-induced EMT.38,39
SMAD4, a downstream mediator of the TGF-β/SMAD signaling pathway, was first described in 1996 by Hahn and colleagues.40 SMAD4 was originally thought to regulate pancreatic cell proliferation and apoptosis via the TGF-β pathway.41,42 Furthermore, a loss of SMAD4 expression is observed in about half of all patients diagnosed with pancreatic cancer, and SMAD4 inactivation is associated with a worse prognosis and with the pattern of disease recurrence/metastatic progression in localized pancreatic cells.43 More specifically, SMAD4 is a critical transcriptional factor of TGF-β signaling and acts as a tumor suppressor.44
TGF-β and TGF-β-related factors regulate cell growth, differentiation, and apoptosis, and play critical roles in normal development and tumorigenesis. TGF-β family-induced changes in gene expression are mediated by SMADs as intracellular effectors. Receptor-activated SMADs are combined with a common SMAD4 to translocate into the nucleus where they cooperate with other transcription factors to activate or repress transcription. The activities of the receptor-activated SMADs are controlled by post-translational modifications such as SUMOylation.28 SIRT1 downregulation by hypoxia in a SUMOylation-dependent manner promotes EMT and eventually leads to tumor metastasis.45 Here we show that SMAD4 is modified by SUMOylation. SUMO represents a class of ubiquitin-like proteins that can regulate the function of a protein by changing its subcellular localization, protein-protein interactions, oncogenes, and tumor suppressor genes, all of which play key roles in the control of cell growth, differentiation, and apoptosis. Lin et al.29 revealed that the SUMOs dysfunction of SMAD4 plays a critical role in carcinogenesis, whereas the involvement of SUMOylation of SMAD4 in the tumor migration and proliferation of OSCC remains largely unknown. The present investigation provides the first evidence that exogenous TGF-β1 induces the upregulation of SUMOs in a time-dependent manner. To quantify the TGF-β1-mediated SUMO upregulation and determine the optimal time required for TGF-β1 to induce the maximum SUMOs expression in OSCC, we adopted an optimal dose of TGF-β1 at 10 ng mL-1 with the peaked time for protein expression at 12 h. TGF-β1 (10 ng mL-1, 12 h) also induced the maximum protein expression of SUMO-1 and SUMO-2/3 (Figures 4A and 4B). GA treatment reversed TGF-β1-induced SUMOs/p-SMAD2/3 increasing and SUMO-1 conjunction to SMAD4 (Figure 4D, bottom panel), indicating that GA presumably reverses the TGF-β1-triggered SMAD4 SUMOylation and releases SMAD4. Finally, the transwell migration and wound-healing assay demonstrated that siSMAD4 improved cell migration and viability, which was inhibited by GA in Tca8113 cells. At the same time, siSMAD4 attenuated GA-induced E-cadherin upregulation and Vimentin downregulation in Tca8113 cells.
OSCC is a highly recurrent and aggressive metastatic tumor that requires a better understanding of the complex molecular pathways and its various regulatory molecules.46 It is worth noting that a large number of studies in the last decade have shown the role of the SUMOylation system in several types of tumors other than OSCC, such as the function of Piasy (one of the SUMOylation factors) in human skin squamous cell carcinoma (SCC).47 Katayama et al.48 reported that SUMO-1 was expressed at much higher levels in OSCC tissue and OSCC cell lines than in normal oral epithelium. Transfection of the anti-SUMO-1 antisense oligonucleotide to OSCC cells significantly reduced the proliferation of the cells. Ding et al.49 clarified the relationship between overexpression of SUMO-1 and SENP5 in OSCC. The relationship between overexpression of SUMO-1 and SENP5 in the maintenance of mitochondrial morphology in OSCC has also been described previously. As mentioned above, our reports indicate that SUMOs play a crucial role in modifying tumorigenesis and tumor progression in OSCC.
Collectively, these results suggest that the effects of GA in vitro are reproducible in xenograft studies in both preventive and treatment regimens, showing an inhibition of OSCC tumor xenograft growth in nude mice, which was associated with high levels of damage and a marked induction in migration and proliferation of tumor cells. Overall, our results demonstrate that GA has a strong antagonistic effect on the migration and proliferation of OSCC in vitro and in vivo. More specifically, GA produces these inhibitory effects on OSCC via its selective ability to induce migration-inhibition and apoptosis. Importantly, the anticancer effects of GA and all associated mechanisms are a consequence of the SUMOylation status of SMAD4, which is one of the primary causes of cellular migration and proliferation in OSCC. However, how SMAD4 promotes angiogenesis and lymph node metastasis in OSCC remains unknown and needs further study. Considering these findings, more studies to evaluate both anticancer and chemo-preventive efficacy of GA in relevant preclinical models are warranted to establish its potential usefulness against human OSCC.
Materials and Methods
Cell Lines, Culture Conditions, and Reagents
OSCC cell lines of human origin (Tca8113, Cal-27) were procured from the Chinese Academy of Science Shanghai Cell Bank (Shanghai, China). All cells were cultured in RPMI 1640 medium or DMEM (HyClone Laboratories, Logan, UT, USA), and both were supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA), 100 U/mL penicillin, and 100 μg/mL streptomycin in a humidified chamber of 5% CO2 at 37°C. After adjusting cell density to 80%, OSCC cells were then treated with TGF-β1 (#AF-100-21C; PeproTech, Rock Hill, NJ, USA) for 12 h after starvation in the serum-free medium. GA (MedChem Express, Monmouth Junction, NJ, USA) was dissolved in DMSO and diluted with normal saline (final concentration of DMSO: 0.1%), and a specific SUMOs inhibitor was applied to OSCC cells 30 min before the TGF-β1 treatment.
Cell Viability by CCK-8 Assay
Tca8113 and Cal-27 cells were plated in 96-well plates at a density of 4–8 × 103 per well. After the cells reached about 80% confluency, they were starved overnight. The cells were then exposed to different concentrations of GA (2, 5, 10, 20, and 40 μM) for 24 h, and RPMI 1640 medium was used as a negative control. After incubation, CCK-8 (10 μL/well) was added and the plates were incubated for 1.5 h. The absorbance was then measured at 490 nm using a PowerWave HT microplate spectrophotometer (BioTek, USA). The data were normalized to the negative control in each group.
Electron Microscopy
Tca8113 and Cal-27 cells were cultured in the presence of GA (10 μM) for 24 h and were then harvested and fixed in 2.5% glutaraldehyde (pH 7.4) overnight. Then, cells were immersed in 0.1 M cacodylate buffer with 1% osmium tetroxide for 1 h. Samples were dehydrated with a concentration gradient of ethanol and then embedded in Epon medium and dissected into 60–70 nm sections. After being stained with uranyl acetate and lead citrate, sections were observed with a JEOL 1200 electron microscope (JEOL, Tokyo, Japan).
TUNEL Assay
Tca8113 and Cal-27 cells cultured in the presence of GA (10 μM) for 24 h were analyzed for apoptosis with TUNEL assay by using the In Situ Cell Death Detection Kit (Cat. No. 11684817910, Roche, Mannheim, Germany) according to the manufacturer’s instructions. Cells were fixed with 4% paraformaldehyde, penetrated with 0.1% Triton X-100, and incubated with TUNEL Reaction Mixture for 1 h at 37°C in the dark. After a 10 min DAPI (4’,6-diamidino-2-phenylindole) counterstain at room temperature, cells were photographed with a fluorescence microscope. The assay was then repeated five times (Zeiss, Jena, Germany).
Wound-Healing Assay
Tca8113 and Cal-27 cells were seeded onto a 6-well plate at a density of 5 × 105/mL until they reached full confluency. OSCC cells were then treated with TGF-β1 (#AF-100-21C; PeproTech, Rock Hill, NJ, USA) at 12 h after starvation in the serum-free medium. GA (MedChem Express, Monmouth Junction, NJ, USA) was dissolved in DMSO and diluted with RPMI 1640 medium (final concentration of DMSO: 0.1%) that was applied to OSCC cells 30 min before TGF-β1 treatment. The cells were cultured for 24 h in the presence of varying concentrations of GA. RPMI 1640 medium was used as a negative control. In another experiment, Tca8113 cells transfected with a siSMAD4 expression vector or a control vector were seeded. Photographs were captured by a digital single-lens reflex camera (Canon, Japan) at 0 h and 24 h. The migration gap area of the cells was measured by ImageJ software (https://imagej.nih.gov/ij/; Center for Information Technology, National Institute of Health, Bethesda, MA, USA). Each measurement was repeated five times.
Transwell Migration Assay
Tca8113 and Cal-27 cells (4–5 × 104/well) were seeded on upper chambers (6.5 mm Transwell with 8.0 μm Pore Polycarbonate Membrane Insert, Corning, NY, USA) in serum-free medium with 0.1% BSA. Other transwell assays using a siSMAD4 expression vector or the control vector-transfected Tca8113 cells were carried out using the same procedure. A total of 600 μL medium containing 10% FBS was added to the lower chambers. After incubation for 24 h, non-migrating cells were gently removed by a cotton swab. The cells were then fixed with methanol for 30 min and stained with 0.1% crystal violet for another 20 min. The area of dyed pores was calculated under a microscope at a magnification of 400×. Five views were selected randomly for analysis. Each measurement was repeated five times.
Western Blot Analysis
Whole-cell lysates were extracted by Radio Immunoprecipitation Assay (RIPA) lysis buffer (Beyotime, China) with protease (Roche, USA). Phosphatase inhibitors (Roche, USA) were added for primary antibodies against p-SMAD2/3. Protein samples of 70 μg per lane were loaded onto 10% SDS-PAGE gels and transferred onto nitrocellulose membranes (PALL, Germany) for 90 min. For immunodetection, the membranes were incubated with primary antibodies at 4°C overnight: E-cadherin (ab76055, Abcam, Cambridge, UK) at 1:1,000 dilution, α-catenin (ab52227, Abcam, Cambridge, UK) at 1:10,000 dilution, Vimentin (HPA001762, Sigma-Aldrich, USA) at 1:250 dilution, N-cadherin (C3865, Sigma-Aldrich, USA) at 1:500 dilution, SMAD4 (sc-7966, Santa Cruz, USA) at 1:200 dilution, p-SMAD2/3 (sc-11769, Santa Cruz, CA, USA) at 1:200 dilution, or SMAD2/3 (sc-133098, Santa Cruz, CA, USA) at 1:200 dilution, SUMO-1 (#sc-9060; Santa Cruz Biotechnology, CA, USA) at a 1:200 dilution, or SUMO-2/3 (#ab3742; Abcam, Cambridge, MA, USA), at a 1:500 dilution. After incubation with a secondary antibody and gentle shaking at room temperature for 30–60 min, the membranes were scanned using an Odyssey Infrared Imaging System (Li-COR, USA). All experiments were done at least five times. The values of protein band densities were normalized to those of the solution or blank control group.
Immunofluorescent Staining for E-cadherin, Vimentin, SMAD4, and SUMO-1
Tca8113 and Cal-27 cells were plated onto a 24-well plate, and GA was dissolved in DMSO and diluted with RPMI 1640 medium, which was applied to OSCC cells 30 min before treatment with TGF-β1. The cells were cultured for 24 h in the presence of varying concentrations of GA (5 μM). The cells were washed with cold sterilized PBS for five times, fixed in 4% paraformaldehyde for 1 h, penetrated by 0.5% Triton X-100 (Sigma-Aldrich, MO, USA) for 15 min, and blocked with 0.1% bovine serum albumin (BSA) for 1 h. The cells were incubated at 4°C overnight with E-cadherin (ab76055, Abcam, Cambridge, UK) at 1:200 dilution, Vimentin (HPA001762, Sigma-Aldrich, MO, USA) at 1:200 dilution, SMAD4 (sc-7966, Santa Cruz, CA, USA) at 1:50 dilution, and SUMO-1 (1:50; #sc-9060; Santa Cruz Biotechnology, CA, USA). The samples were incubated with goat anti-mouse IgG (H+L) Highly cross-adsorbed secondary antibody, Alexa Fluor 488 (Invitrogen, Thermo Fisher, USA), or goat anti-rabbit IgG (H+L) highly cross-adsorbed secondary antibody, Alexa Fluor 594 (Invitrogen, Thermo Fisher, USA) at 1:200 dilution for 1 h. Nuclei were stained with DAPI (Beyotime, China) at room temperature for 5 min. The samples were visualized under a fluorescence microscope (Nikon 80i, Japan).
CoIP
Lysates were incubated with specific antibodies and then conjugated with protein A/G agarose beads. After centrifugation, the beads were collected and rinsed gently. The samples were denatured and then analyzed using SDS-PAGE to detect the interaction between two protein partners.
Transfection of siRNA
SMAD4 inhibitor (siSMAD4) and the SMAD4 negative control (NC) were synthesized by Shanghai GenePharma (China). siSMAD4: sense, 5′-GCU CCU AGA CGA AGU ACU UTT-3′; anti-sense, 5′-AAG UAC UUC GUC UAG GAG CTT-3′. Tca8113 cells were plated onto a 6-well plate at 5 × 104 in 2 mL culture medium overnight until they achieved the desired density of 50%–70% confluency. Then, they were transfected with siRNA and NC the using X-tremeGENE siRNA Transfection Reagent (Roche, USA) following the manufacturer’s instructions. Transfection efficiency was assessed by western blot.
In Vivo Animal Experiment
Our protocol for animal use was approved by the Institution Animal Care and Use Committee of Harbin Medical University, and all animal experiments were carried out according to the Guide for the Care and Use of Laboratory Animals, and in strict accordance with the People’s Republic of China Legislation Regarding the Use and Care of Laboratory Animals. All studies involving animals follow the ARRIVE guidelines for reporting experiments involving animals.
In Vivo Tumor Growth Inhibition by GA in Xenografts
Adult female athymic BALB/c nude mice (18–20 g of 4-week-old mice were purchased from Beijing Vital River Laboratory Animal Technology (Certificate No. SCXK [Jing] 2016-0011; No. 11400700161156). The animals were housed in a controlled environment at 23°C ± 2°C and 40%–70% humidity under a 12 h dark/light cycle with free access to irradiated food and sterile water. They were housed in individually ventilated cages: five per cage, with 4–6 mm of corncob bedding after 60Co radiation disinfection. The Tca8113 tumor cells (over 5 × 107/mL) were harvested and washed with sodium chloride. A cell suspension of 1 × 106 in 0.2 mL was injected subcutaneously into the right axillary of all mice to establish the traditional tumor xenograft nude mouse model. After 1 week, animals were randomly divided into three groups, the first group received vehicle (100 μL saline) by oral gavage (control group, n = 8); another two groups were administered with GA (suspended in saline, 20 mg kg-1, 50 mg kg-1) via gastric gavage daily for 4 weeks (GA group, n = 8). The tumors’ dimensions were monitored with Vernier calipers every 2 days throughout the experiment, and tumor volume was calculated using the following formula: V (tumor volume) = d (shorter diameter) 2 × D (longer diameter) × 0.5. At the end of the 4-week experimental period, the mice were euthanized and individual tumor weights were measured. The tumor samples were fixed with 4% paraformaldehyde and embedded in paraffin.
Statistical Analysis
Statistical comparisons between two groups were made with a Student’s t test and multiple groups were determined by one-way analysis of variance (ANOVA) followed by the Tukey post hoc analysis for comparisons of mean values (p < 0.05 was considered statistically significant). Results are expressed as mean ± the standard error of the mean (SEM).
Survival curves were plotted with the Kaplan-Meier technique and compared with the log-rank test. Results are expressed as the mean of at least five times or more determinations. Statistical analysis was performed by using GraphPad Software 5.0 (GraphPad Software, San Diego, CA, USA).
Author Contributions
X.-F.W. designed the experiments; K.L. composed the draft and analyzed the data; W.-F.C. and D.Z. revised the manuscript; K.L., X.W. and D.L. conducted most experiments; and D.X. and D.L. performed animal models. Z.L. was responsible for cells culture.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (81672827, 81770281, and 31871175).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2019.12.005.
Supplemental Information
References
- 1.Jaime-Ramirez A.C., Yu J.G., Caserta E., Yoo J.Y., Zhang J., Lee T.J., Hofmeister C., Lee J.H., Kumar B., Pan Q. Reolysin and Histone Deacetylase Inhibition in the Treatment of Head and Neck Squamous Cell Carcinoma. Mol. Ther. Oncolytics. 2017;5:87–96. doi: 10.1016/j.omto.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tyagi A., Gu M., Takahata T., Frederick B., Agarwal C., Siriwardana S., Agarwal R., Sclafani R.A. Resveratrol selectively induces DNA Damage, independent of Smad4 expression, in its efficacy against human head and neck squamous cell carcinoma. Clin. Cancer Res. 2011;17:5402–5411. doi: 10.1158/1078-0432.CCR-11-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eckert A.W., Kappler M., Schubert J., Taubert H. Correlation of expression of hypoxia-related proteins with prognosis in oral squamous cell carcinoma patients. Oral Maxillofac. Surg. 2012;16:189–196. doi: 10.1007/s10006-012-0335-8. [DOI] [PubMed] [Google Scholar]
- 4.Heo K., Kim Y.H., Sung H.J., Li H.Y., Yoo C.W., Kim J.Y., Park J.Y., Lee U.L., Nam B.H., Kim E.O. Hypoxia-induced up-regulation of apelin is associated with a poor prognosis in oral squamous cell carcinoma patients. Oral Oncol. 2012;48:500–506. doi: 10.1016/j.oraloncology.2011.12.015. [DOI] [PubMed] [Google Scholar]
- 5.Siegel P.M., Massagué J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat. Rev. Cancer. 2003;3:807–821. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 6.Zavadil J., Böttinger E.P. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 7.Chiao P.J., Hunt K.K., Grau A.M., Abramian A., Fleming J., Zhang W., Breslin T., Abbruzzese J.L., Evans D.B. Tumor suppressor gene Smad4/DPC4, its downstream target genes, and regulation of cell cycle. Ann. N Y Acad. Sci. 1999;880:31–37. doi: 10.1111/j.1749-6632.1999.tb09507.x. [DOI] [PubMed] [Google Scholar]
- 8.Peng B., Fleming J.B., Breslin T., Grau A.M., Fojioka S., Abbruzzese J.L., Evans D.B., Ayers D., Wathen K., Wu T. Suppression of tumorigenesis and induction of p15(ink4b) by Smad4/DPC4 in human pancreatic cancer cells. Clin. Cancer Res. 2002;8:3628–3638. [PubMed] [Google Scholar]
- 9.Zhao X., Feng T., Chen H., Shan H., Zhang Y., Lu Y., Yang B. Arsenic trioxide-induced apoptosis in H9c2 cardiomyocytes: implications in cardiotoxicity. Basic Clin. Pharmacol. Toxicol. 2008;102:419–425. doi: 10.1111/j.1742-7843.2007.00150.x. [DOI] [PubMed] [Google Scholar]
- 10.Bornstein S., White R., Malkoski S., Oka M., Han G., Cleaver T., Reh D., Andersen P., Gross N., Olson S. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J. Clin. Invest. 2009;119:3408–3419. doi: 10.1172/JCI38854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fukuda I., Ito A., Hirai G., Nishimura S., Kawasaki H., Saitoh H., Kimura K., Sodeoka M., Yoshida M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009;16:133–140. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 12.Johnson E.S. Protein modification by SUMO. Annu. Rev. Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y., Zhao D., Qiu F., Zhang L.L., Liu S.K., Li Y.Y., Liu M.T., Wu D., Wang J.X., Ding X.Q. Manipulating PML SUMOylation via Silencing UBC9 and RNF4 Regulates Cardiac Fibrosis. Mol. Ther. 2017;25:666–678. doi: 10.1016/j.ymthe.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu F., Dong C., Liu Y., Shao X., Huang D., Han Y., Wang B., Liu Y., Huo R., Paulo P. Pharmacological inhibition of SUMO-1 with ginkgolic acid alleviates cardiac fibrosis induced by myocardial infarction in mice. Toxicol. Appl. Pharmacol. 2018;345:1–9. doi: 10.1016/j.taap.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 15.Wu D., Huang D., Li L.L., Ni P., Li X.X., Wang B., Han Y.N., Shao X.Q., Zhao D., Chu W.F., Li B.Y. TGF-β1-PML SUMOylation-peptidyl-prolyl cis-trans isomerase NIMA-interacting 1 (Pin1) form a positive feedback loop to regulate cardiac fibrosis. J. Cell. Physiol. 2019;234:6263–6273. doi: 10.1002/jcp.27357. [DOI] [PubMed] [Google Scholar]
- 16.Du X., Pan Z., Li Q., Liu H., Li Q. SMAD4 feedback regulates the canonical TGF-β signaling pathway to control granulosa cell apoptosis. Cell Death Dis. 2018;9:151. doi: 10.1038/s41419-017-0205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pierreux C.E., Nicolás F.J., Hill C.S. Transforming growth factor beta-independent shuttling of Smad4 between the cytoplasm and nucleus. Mol. Cell. Biol. 2000;20:9041–9054. doi: 10.1128/mcb.20.23.9041-9054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abramczyk M.J., Forrester T. Utilization review: a psychiatric perspective. Nurs. Manage. 1989;20:46–48. [PubMed] [Google Scholar]
- 19.Itokawa H., Totsuka N., Nakahara K., Takeya K., Lepoittevin J.P., Asakawa Y. Antitumor principles from Ginkgo biloba L. Chem. Pharm. Bull. (Tokyo) 1987;35:3016–3020. doi: 10.1248/cpb.35.3016. [DOI] [PubMed] [Google Scholar]
- 20.Kalkunte S.S., Singh A.P., Chaves F.C., Gianfagna T.J., Pundir V.S., Jaiswal A.K., Vorsa N., Sharma S. Antidepressant and antistress activity of GC-MS characterized lipophilic extracts of Ginkgo biloba leaves. Phytother. Res. 2007;21:1061–1065. doi: 10.1002/ptr.2212. [DOI] [PubMed] [Google Scholar]
- 21.Lee J.H., Kim Y.G., Ryu S.Y., Cho M.H., Lee J. Ginkgolic acids and Ginkgo biloba extract inhibit Escherichia coli O157:H7 and Staphylococcus aureus biofilm formation. Int. J. Food Microbiol. 2014;174:47–55. doi: 10.1016/j.ijfoodmicro.2013.12.030. [DOI] [PubMed] [Google Scholar]
- 22.Oh J., Hwang I.H., Hong C.E., Lyu S.Y., Na M. Inhibition of fatty acid synthase by ginkgolic acids from the leaves of Ginkgo biloba and their cytotoxic activity. J. Enzyme Inhib. Med. Chem. 2013;28:565–568. doi: 10.3109/14756366.2012.658786. [DOI] [PubMed] [Google Scholar]
- 23.Zhou C., Li X., Du W., Feng Y., Kong X., Li Y., Xiao L., Zhang P. Antitumor effects of ginkgolic acid in human cancer cell occur via cell cycle arrest and decrease the Bcl-2/Bax ratio to induce apoptosis. Chemotherapy. 2010;56:393–402. doi: 10.1159/000317750. [DOI] [PubMed] [Google Scholar]
- 24.Baek S.H., Ko J.H., Lee J.H., Kim C., Lee H., Nam D., Lee J., Lee S.G., Yang W.M., Um J.Y. Ginkgolic Acid Inhibits Invasion and Migration and TGF-β-Induced EMT of Lung Cancer Cells Through PI3K/Akt/mTOR Inactivation. J. Cell. Physiol. 2017;232:346–354. doi: 10.1002/jcp.25426. [DOI] [PubMed] [Google Scholar]
- 25.Polyak K., Weinberg R.A. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat. Rev. Cancer. 2009;9:265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 26.Nieto M.A., Huang R.Y., Jackson R.A., Thiery J.P. Emt: 2016. Cell. 2016;166:21–45. doi: 10.1016/j.cell.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 27.Alarcon-Vargas D., Ronai Z. SUMO in cancer--wrestlers wanted. Cancer Biol. Ther. 2002;1:237–242. doi: 10.4161/cbt.74. [DOI] [PubMed] [Google Scholar]
- 28.Lee P.S., Chang C., Liu D., Derynck R. Sumoylation of Smad4, the common Smad mediator of transforming growth factor-beta family signaling. J. Biol. Chem. 2003;278:27853–27863. doi: 10.1074/jbc.M301755200. [DOI] [PubMed] [Google Scholar]
- 29.Lin X., Liang M., Liang Y.Y., Brunicardi F.C., Melchior F., Feng X.H. Activation of transforming growth factor-beta signaling by SUMO-1 modification of tumor suppressor Smad4/DPC4. J. Biol. Chem. 2003;278:18714–18719. doi: 10.1074/jbc.M302243200. [DOI] [PubMed] [Google Scholar]
- 30.DeFeudis F.V., Drieu K. Ginkgo biloba extract (EGb 761) and CNS functions: basic studies and clinical applications. Curr. Drug Targets. 2000;1:25–58. doi: 10.2174/1389450003349380. [DOI] [PubMed] [Google Scholar]
- 31.Ma J., Duan W., Han S., Lei J., Xu Q., Chen X., Jiang Z., Nan L., Li J., Chen K. Ginkgolic acid suppresses the development of pancreatic cancer by inhibiting pathways driving lipogenesis. Oncotarget. 2015;6:20993–21003. doi: 10.18632/oncotarget.3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y., Yang B., Zhang L., Cong X., Liu Z., Hu Y., Zhang J., Hu H. Ginkgolic acid induces interplay between apoptosis and autophagy regulated by ROS generation in colon cancer. Biochem. Biophys. Res. Commun. 2018;498:246–253. doi: 10.1016/j.bbrc.2018.01.091. [DOI] [PubMed] [Google Scholar]
- 33.Liu Z.H., Zeng S. Cytotoxicity of ginkgolic acid in HepG2 cells and primary rat hepatocytes. Toxicol. Lett. 2009;187:131–136. doi: 10.1016/j.toxlet.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 34.Kim Y.S., Nagy K., Keyser S., Schneekloth J.S., Jr. An electrophoretic mobility shift assay identifies a mechanistically unique inhibitor of protein sumoylation. Chem. Biol. 2013;20:604–613. doi: 10.1016/j.chembiol.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hamdoun S., Efferth T. Ginkgolic acids inhibit migration in breast cancer cells by inhibition of NEMO sumoylation and NF-κB activity. Oncotarget. 2017;8:35103–35115. doi: 10.18632/oncotarget.16626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Báez A., Cantor A., Fonseca S., Marcos-Martinez M., Mathews L.A., Muro-Cacho C.A., Muñoz-Antonia T. Differences in Smad4 expression in human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck squamous cell carcinoma. Clin. Cancer Res. 2005;11:3191–3197. doi: 10.1158/1078-0432.CCR-04-1299. [DOI] [PubMed] [Google Scholar]
- 37.Moustakas A., Heldin C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512–1520. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moustakas A., Heldin C.H. Induction of epithelial-mesenchymal transition by transforming growth factor β. Semin. Cancer Biol. 2012;22:446–454. doi: 10.1016/j.semcancer.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 39.Xu J., Lamouille S., Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Voorneveld P.W., Jacobs R.J., Kodach L.L., Hardwick J.C. A Meta-Analysis of SMAD4 Immunohistochemistry as a Prognostic Marker in Colorectal Cancer. Transl. Oncol. 2015;8:18–24. doi: 10.1016/j.tranon.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hahn S.A., Schutte M., Hoque A.T., Moskaluk C.A., da Costa L.T., Rozenblum E., Weinstein C.L., Fischer A., Yeo C.J., Hruban R.H., Kern S.E. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 42.Xia X., Wu W., Huang C., Cen G., Jiang T., Cao J., Huang K., Qiu Z. SMAD4 and its role in pancreatic cancer. Tumour Biol. 2015;36:111–119. doi: 10.1007/s13277-014-2883-z. [DOI] [PubMed] [Google Scholar]
- 43.Iacobuzio-Donahue C.A., Fu B., Yachida S., Luo M., Abe H., Henderson C.M., Vilardell F., Wang Z., Keller J.W., Banerjee P. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009;27:1806–1813. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogawa R., Yamamoto T., Hirai H., Hanada K., Kiyasu Y., Nishikawa G., Mizuno R., Inamoto S., Itatani Y., Sakai Y., Kawada K. Loss of SMAD4 promotes colorectal cancer progression by recruiting tumor-associated neutrophils via CXCL1/8-CXCR2 axis. Clin. Cancer Res. 2019;25:2887–2899. doi: 10.1158/1078-0432.CCR-18-3684. [DOI] [PubMed] [Google Scholar]
- 45.Sun L., Li H., Chen J., Dehennaut V., Zhao Y., Yang Y., Iwasaki Y., Kahn-Perles B., Leprince D., Chen Q. A SUMOylation-dependent pathway regulates SIRT1 transcription and lung cancer metastasis. J. Natl. Cancer Inst. 2013;105:887–898. doi: 10.1093/jnci/djt118. [DOI] [PubMed] [Google Scholar]
- 46.Joseph J.P., Harishankar M.K., Pillai A.A., Devi A. Hypoxia induced EMT: A review on the mechanism of tumor progression and metastasis in OSCC. Oral Oncol. 2018;80:23–32. doi: 10.1016/j.oraloncology.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 47.Albor A., Kulesz-Martin M. Novel initiation genes in squamous cell carcinomagenesis: a role for substrate-specific ubiquitylation in the control of cell survival. Mol. Carcinog. 2007;46:585–590. doi: 10.1002/mc.20344. [DOI] [PubMed] [Google Scholar]
- 48.Katayama A., Ogino T., Bandoh N., Takahara M., Kishibe K., Nonaka S., Harabuchi Y. Overexpression of small ubiquitin-related modifier-1 and sumoylated Mdm2 in oral squamous cell carcinoma: possible involvement in tumor proliferation and prognosis. Int. J. Oncol. 2007;31:517–524. [PubMed] [Google Scholar]
- 49.Ding X., Sun J., Wang L., Li G., Shen Y., Zhou X., Chen W. Overexpression of SENP5 in oral squamous cell carcinoma and its association with differentiation. Oncol. Rep. 2008;20:1041–1045. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.