

Abstract
The known mode of action of isoniazid (INH) is to inhibit bacterial cell wall synthesis following activation by the bacterial catalase–peroxidase enzyme KatG in Mycobacterium tuberculosis (Mtb). This simplistic model fails to explain (a) how isoniazid penetrates waxy granulomas with its very low lipophilicity, (b) how isoniazid kills latent Mtb lacking a typical cell wall, and (c) why isoniazid treatment time is remarkably long in contrast to most other antibiotics. To address these questions, a novel comprehensive mode of action of isoniazid has been proposed here. Briefly, isoniazid eradicates latent tuberculosis (TB) by prompting slow differentiation of pro‐inflammatory monocytes and providing protection against reactive species‐induced “self‐necrosis” of phagocytes. In the case of active TB, different immune cells form INH‐NAD+ adducts to inhibit Mtb's cell wall biosynthesis. This additionally suggests that the antibacterial properties of INH do not rely on KatG of Mtb. As such, isoniazid‐resistant TB needs to be re‐evaluated.
Abbreviations
- INH
isoniazid
- INH*
isoniazid free radical/isonicotinoyl radical
- INH‐NAD+
isoniazid‐nicotinamide adenine dinucleotide adduct
- iNOS
inducible NOS
- KatG
Mtb catalase–peroxidase
- MPO
myeloperoxidase
- Mtb
Mycobacterium tuberculosis
- NET
neutrophil extracellular traps
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
1. INTRODUCTION
Tuberculosis (TB) is caused by either Mycobacterium tuberculosis (Mtb) or a subspecies of M. tuberculosis complex (i.e., Mycobacterium bovis, Mycobacterium africanum, Mycobacterium pinnipedii, Mycobacterium microti, Mycobacterium caprae, and Mycobacterium canettii). These bacteria were originally believed to be a prehistoric scourge for humans in light of archaeological findings (Gomez i Prat & de Souza, 2003). However, a recent comparative genomic analysis of bacterial DNA collected from thousand‐year‐old mummies instead suggests that TB originated from Africa only within the last 6,000 years (Bos et al., 2014). Although TB is an ancient disease with a plethora of first‐ and second‐line drugs available for various anti‐TB regimens, it remains one of the top 10 major causes of death worldwide. Approximately 10 million new TB cases were reported worldwide in 2017 as per the WHO Report of September 2018 (Organization, 2018). Of these 10 million, 1.3 million died due to TB alone, while another 300,000 died from co‐infection of TB and HIV. In 2015, the World Health Organization (WHO) launched the “End TB Strategy” to address the global burden imposed by TB, prioritizing novel research initiatives alongside other prevention strategies in order to effectively reduce TB deaths by 95% by the year 2035 (http://www.who.int/tb/post2015_strategy/en/). These novel research initiatives include discovery, development, and rapid uptake of new tools, interventions, and strategies. While it does promote the development of new drugs, the End TB Strategy is noteworthy for its commitment to furthering our understanding of existing anti‐TB drugs. Improved understanding of existing drugs would be expected to aid in the adoption of cheap and rapid intervention strategies suited to developing nations most impacted by TB. This is especially pertinent to the cheapest first‐line drug, isoniazid (INH), whose exact mode of action remains unclear. This lack of clarity restricts the application of INH in many situations and makes curbing the TB epidemic expensive as well as challenging. In this review, recent studies on INH in immune modulation will be discussed to propose a novel mode of action on the basis of the interaction between TB and host immunity.
2. TB PATHOPHYSIOLOGY AND THE ROLE OF THE IMMUNE SYSTEM
2.1. First‐line immune defence determines the fate of TB
As with other infections, the human immune system plays a primary role in defending against the progression of TB, whose infectious dose is reported to be between 1 and 200 bacilli. The invasion of Mtb in the lungs is primarily countered by alveolar macrophages, which act to engulf the pathogens. At this early stage of TB infection, neutrophils are then promptly recruited around each alveolar macrophage in response to inflammatory signals (Eruslanov et al., 2005; Fialkow, Wang, & Downey, 2007; Lowe, Redford, Wilkinson, O'Garra, & Martineau, 2012). In healthy patients, macrophages usually kill Mtb with nitric oxide (https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2509) through its https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1250 (iNOS) activity; however, neutrophils cannot kill Mtb as bacterial KatG can neutralize both acidic compounds and ROS through its catalase–peroxidase activity (Forman & Torres, 2001; Voskuil, Bartek, Visconti, & Schoolnik, 2011). If macrophages successfully kill Mtb, they undergo apoptotic cell death (Behar et al., 2011). Neutrophils then engulf these apoptotic cells and carry their contents to lymph nodes to activate T cells (Abadie et al., 2005). These T cells then subsequently defend the host against further TB infection (Sud, Bigbee, Flynn, & Kirschner, 2006). However, another scenario plays out if macrophages fail to kill Mtb. In this case, macrophages form granulomas by recruiting additional macrophages, macrophage‐like immune cells, and a small population of lymphocytes. The formation of a granuloma can effectively stop the dissemination of Mtb throughout the body, a stage known as latent TB (Guirado & Schlesinger, 2013). In the latent stage (within the granuloma), Mtb loses its typical cell wall, where it does not replicate but generates high amounts of energy (Wolfe, Mahaffey, Kruh, & Dobos, 2010). This condition induces host immune cells to generate continuous ROS, which creates a high burden of oxidative stress within the granuloma. Mtb exhibits unusually high tolerance to ROS and thus remains viable (Voskuil et al., 2011). However, such a high concentration of ROS leads to mitochondrial damage followed by necrotic cell death of infected macrophages (Behar et al., 2011; Dallenga et al., 2017). After necrosis, Mtb then escapes the granuloma and emerges as active TB. The success of macrophages in preventing this outcome depends on several factors, such as the number of Mtb and their virulence, the number of macrophages, and the survival capacity of host functional immune cells (Guirado & Schlesinger, 2013; Lowe et al., 2012). Based on individual host variations, the possible outcomes of Mtb infection have been illustrated in Figure 1. Healthy individuals generally possess sufficient immunity to either eradicate or halt the progression of Mtb infection, which results in either complete remedy or latent TB, respectively. If the host's immune system is compromised (due to either HIV infection or malnutrition), the immune system cannot provide sufficient protection against Mtb infection due to both rapid death and slow replenishment of these immune cells. Roughly 5% (excluding HIV co‐infection) of infected individuals meet these criteria and ultimately accede to active TB (Balasubramanian, Wiegeshaus, Taylor, & Smith, 1994; Sakamoto, 2012; Smith, 2003). Thus, the host immune system plays a critical role in defending against TB.
Figure 1.
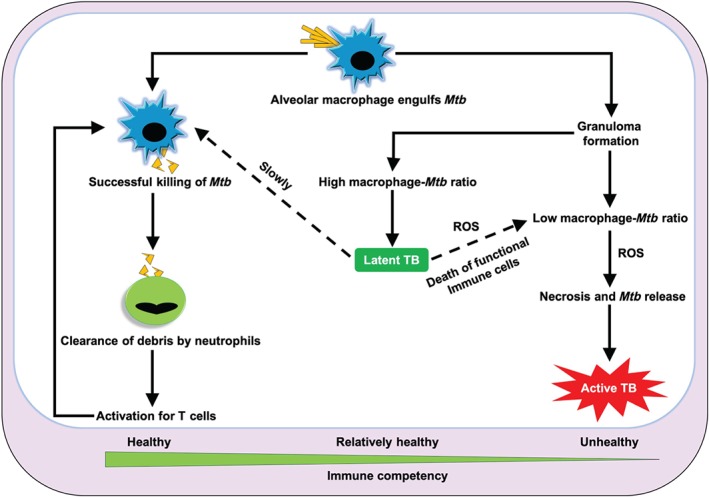
The possible scenarios for tuberculosis (TB) infections based on individual host immunity
2.2. Granuloma formation—Host defence or bacterial survival strategy?
The typical TB granuloma is a caseous structure, composed of inner epithelioid macrophages with other monocyte‐derived cells and neutrophils surrounded by a cellular necrotic region containing https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2995 and CD8+ T cells, as well as a smaller number of B cells (Gonzalez‐Juarrero et al., 2001). If the first line of host defence fails to kill Mtb, the immune system adopts this strategy to prevent the bacteria from spreading out across the body. From this point of view, granuloma formation is a host defence strategy that should provide a suitable niche for the immune system to inhibit Mtb growth and slowly kill the infection. The balance between pro‐ and anti‐inflammatory responses of granuloma‐residing immune cells is considered a crucial factor to control this bacterial growth (Flynn, Chan, & Lin, 2011). In the ideal scenario, granuloma‐residing immune cells execute slow apoptotic killing of bacteria to promote antigen presentation by surrounding T cells. This granuloma appears as non‐caseous. In a caseous granuloma—a hallmark for activation of Mtb from latency—granuloma‐residing immune cells undergo necrotic death and release bacteria. At this stage, Mtb can be spread by coughing (Silva Miranda, Breiman, Allain, Deknuydt, & Altare, 2012). Within the granuloma, the balance between pro‐ and anti‐inflammatory responses is very complex. Initial pro‐inflammatory responses are to kill bacilli. If these fail to kill or halt the growth of bacilli, the pro‐inflammatory responses continue. This recruits additional pro‐inflammatory immune cells that generate excessive ROS within the granuloma. It may in turn cause more necrosis and release of bacilli (given that Mtb's ROS tolerance is high, as discussed later). On the other hand, the shift of immune responses towards anti‐inflammation at the later stage may indicate successful slow killing of bacilli; however, this anti‐inflammatory stage could be beneficial for the survival of the remaining bacilli (Flynn et al., 2011; Lawn, Myer, Edwards, Bekker, & Wood, 2009; Marakalala et al., 2016; Subbian et al., 2015; van Crevel, Ottenhoff, & van der Meer, 2002). For this reason, it remains unclear whether granuloma formation provides host defence or is a bacterial survival strategy. However, it is understood that the successful killing of bacilli inside the granuloma requires pro‐inflammatory responses of immune cells within their tolerance level of ROS in order to ensure their own survival. An in vitro study on INH showed that it increased the ROS tolerability of the human promyelocytic leukaemia (HL‐60) cell line (Khan et al., 2016b; Khan et al., 2016c), which has the potential to differentiate into various immune cells. This increased ROS tolerability of immune cells in response to INH treatment can thus be beneficial to the cells in combatting Mtb.
2.3. Role of monocyte‐derived immune cells in TB
Monocytes are one of the major innate immune cells in humans. Within human blood, there exists three distinct subpopulations of monocytes based on their cell surface markers (mainly the presence of CD14 and CD16) and functionality. CD14+/CD16− monocytes, also known as classical monocytes, are functionally antimicrobial and highly pro‐inflammatory whereas CD14+/CD16++ monocytes, known as nonclassical monocytes, are functionally anti‐inflammatory (Sampath, Moideen, Ranganathan, & Bethunaickan, 2018). Studies on monocytes in TB found that nonclassical monocytes (CD14+/CD16++) expanded with TB progression, whereas anti‐TB treatment reversed this expansion (Castaño, García, & Rojas, 2011; Sánchez et al., 2006). This suggests that the inflammatory function of classical monocytes might play an important role in TB recovery. Interestingly, the aforementioned study on INH's role in inducing monocyte‐like differentiation in the HL‐60 cell line found that differentiation was especially inclined towards classical monocytes (CD14+/CD16−; Babu et al., 2019a). This would certainly be expected to boost immune defence against TB infection. In addition, classical monocytes exhibit up‐regulated https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2789 (MPO) expression (Sampath et al., 2018), which is usually abundant in neutrophils. A recent study revealed that MPO can metabolize INH to form an INH‐NAD+ adduct; the latter is considered an active metabolite of INH, which inhibits the cell wall synthesis of Mtb (Khan et al., 2016d). Therefore, INH may not only increase the classical monocyte population but also use its up‐regulated MPO as a catalyst to kill Mtb. However, this classical area of study has yet to be explored using TB patient's blood samples or animal TB models.
2.4. Neutrophils—Really a Trojan horse?
Fifty to eighty percent of circulating white blood cells are neutrophils. This dominant member of the leukocyte family is well‐equipped with effective phagocytosis mechanisms, and secretes cytokines (such as TNF‐α and IL‐1); they express MPO, https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=2358, and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1632, and produce ROS and neutrophil extracellular traps (NETs) to break up complex structural proteins. Braian and colleagues showed that neutrophils can trap Mtb by NET formation and release heat shock protein 72 to recruit resident alveolar macrophages to the site of infection (Braian, Hogea, & Stendahl, 2013). As such, they are one of the prime innate immune cells. However, their role in TB is controversial for a number of reasons. In humans, the neutrophilic response is found to be positively correlated with the stage of TB and its severity. Sutherland and colleagues found an increased blood neutrophil count with a high neutrophil/lymphocyte ratio in TB patients compared with tuberculin skin test‐positive healthy subjects (i.e., latent TB; Sutherland et al., 2009). Berry and colleagues discovered MPO from blood neutrophil‐specific IFN‐inducible genes as a marker of active TB (Berry et al., 2010). In addition, the overactivity of neutrophils in the pulmonary cavities in active TB is associated with pulmonary destruction (Barry, Breen, Lipman, Johnson, & Janossy, 2009). At this stage, neutrophil‐derived collagenase (also known as MMP‐8) is up‐regulated to breakdown the pulmonary extracellular matrix in TB patients (Ong et al., 2015). Although neutrophils have an effective antibacterial arsenal and also reside in granulomas, evidence suggests that their actions help bacteria transition from the latency stage to the active stage. For this reason, neutrophils are sometimes called “Trojan horses” in TB infection (Warren, Teskey, & Venketaraman, 2017). Mechanistically, neutrophils use MPO to generate ROS as their main antibacterial weapon. Unfortunately, Mtb exhibits high levels of ROS resistance (Voskuil et al., 2011). Neutrophils typically die via self‐necrosis before reaching a high enough concentration to be effective (Dallenga et al., 2017). This ultimately weakens the immune defence of the granuloma, releasing Mtb to kickstart the active TB phase. Therefore, there are two important points to consider regarding neutrophilic immune defence against Mtb: (a) Neutrophils need to implement an alternative to ROS for killing Mtb, and (2) neutrophils must defend against self‐necrosis. Since neutrophils can use MPO to catalyse formation of INH‐NAD+ adducts (Khan et al., 2016d), this may prove a more effective method by which to combat Mtb. However, this is only applicable in active TB (where Mtb has a cell wall) in the absence of self‐destructive actions. A recent study showed that INH could provide cellular defence against ROS‐induced necrosis (Khan et al., 2016b; Khan et al., 2016c). Therefore, under INH treatment, neutrophils would likely be very effective immune cells to eradicate TB infection.
2.5. Eosinophils—Do they fight against TB?
The role of eosinophils in TB pathogenesis has yet to be established. One case study observed eosinophilia (i.e. increased eosinophil count at >1,500 eosinophils/μL) in a TB patient (Garg, Gogia, Kakar, & Miglani, 2017). Another recent clinical study where 44 active TB patients were compared to 44 latent TB patients found that eosinophil‐derived neurotoxin was one of the markers for active TB (Moideen et al., 2018). As such, it is worthwhile to determine whether eosinophils truly play a role in TB pathogenesis. Although eosinophilia is typical in parasitic infections, it remains understudied in TB patients. Todd and colleagues showed that rapid eosinophil infiltration of pulmonary lesions occurs in response to Mtb infection in pigs (Lasco et al., 2004). This suggests that eosinophils actively cooperate with neutrophils and monocytes to defend against Mtb; as eosinophils possess eosinophil peroxidase (which shares similar reaction mechanisms with KatG and MPO), it is possible that they may generate INH‐NAD+ adducts under INH treatment. A study by Babu and colleagues supports this, in which they showed that eosinophil peroxidase possesses the capacity to metabolize INH to form INH‐NAD+ adducts (Babu et al., 2019b). Therefore, eosinophil recruitment to a site of TB infection may present yet another mechanism of host defence against Mtb with INH treatment.
3. IMMUNITY EVASION STRATEGIES OF MTB
Macrophages generally neutralize pathogens via a process involving four distinct phases: (a) surface binding, (b) uptake and vacuole/phagosome formation, (c) maturation by fusion with a lysosome to trigger digestion, and (d) initiation of necrosis. However, Mtb manages to circumvent these defences via direct interference in the phagosome maturation pathway, which begins upon contact of Mtb with the macrophage. The bacterium possesses a number of ligands on its surface, which are used to directly target and bind to the macrophage surface. However, it remains unclear which types of receptors are employed for infiltration, of which macrophages have many. These include complement receptors, mannose receptors, pulmonary surfactant proteins, scavenger receptors, Fcy receptors, and CD14, among others (Ernst, 1998). Receptor expression patterns may vary depending on the stage of differentiation and activation state of the macrophages, making it difficult to ascertain which are important to Mtb infiltration. After surface binding, Mtb enters into the macrophage by forming a fused membrane‐bound vacuole known as a phagosome. Usually, fusion of phagosomes with lysosomes is needed to acquire antimicrobial activity in the form of microorganism digestion. However, Mtb escapes this by adapting/modulating several molecular signals in active TB (Figure 2) to interfere with phagosome maturation and prevent the emergence of its digestive capacity, followed by parasitic hijacking of cell machinery for its own nutrition and propagation. This manipulation takes place in four key phases: (a) inhibition of lysosome formation by the production of eukaryotic‐like signalling molecules (such as kinases or phosphatases) that interfere with host signal trafficking, (b) inhibition of https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=787 to block https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2351 translocation into lysosomes, (c) inhibition of lysosomal maturation and acidification, and (d) Mtb propagation inside macrophages and release through necrosis, which leads to active TB (Hestvik, Hmama, & Av‐Gay, 2005; Sun et al., 2013; Wong, Bach, Sun, Hmama, & Av‐Gay, 2011b).
Figure 2.
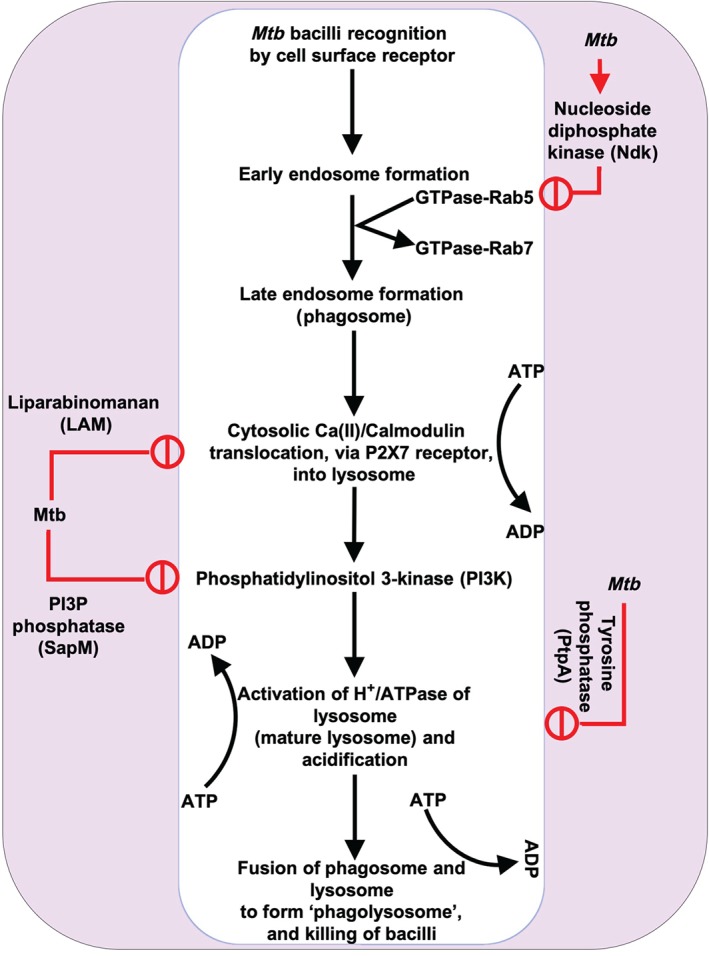
A schematic diagram of phagocytosis and its modulation by Mtb. Mtb produces a number of signalling modulators such as nucleoside diphosphate kinase (Ndk), lipoarabinomannan (LAM), PI3P phosphatase (SapM), and tyrosine phosphate (PtpA), which interfere with phagocytosis and allow them to survive inside phagocytic cells
3.1. Role of ATP in host defence against TB
Following engulfment of pathogens, macrophages acidify the phagosomal lumen by recruiting vacuolar H+‐ATPase (V‐ATPase), a multisubunit protein‐pump complex that actively transports protons across membranes using energy from https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=1713 hydrolysis (and which is structurally similar to mitochondrial ATP synthase). This acidification not only inhibits bacterial growth but also regulates lysosomal fusion through macrophage class C vacuolar protein sorting (C VPS) complex, a key regulator of endosomal membrane fusion. However, Mtb inhibits phagosome acidification and maturation by secreting https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=980 (PtpA), which binds to subunit H of V‐ATPase; this subsequently stops lysosome fusion by inhibiting C VPS complex activity through dephosphorylation of VPS33B, a member of the class C VPS complex and a cognate substrate of PtpA (Wong, Bach, Sun, Hmama, & Av‐Gay, 2011a). A study showed that addition of 3mM ATP induced rapid autophagy (at 30 min post‐treatment) of Mtb‐infected macrophages, which was accompanied by rapid phagolysosomal fusion and loss of mycobacterial viability within infected cells (Fairbairn, Stober, Kumararatne, & Lammas, 2001; Lammas et al., 1997). Therefore, it can be surmised that inhibition of V‐ATPase is the rate‐limiting step of Mtb‐induced phagocytic modulation. However, it is not yet known if INH has any effect on V‐ATPase activity or in ATP biogenesis. A quantitative proteomics study on HL‐60 cells treated with INH revealed that the cytoprotective effect of INH against ROS was partially due to increased ATP biogenesis (Khan, Aljuhani, et al., 2016b). This suggests that INH may have a positive role in the ATP biogenesis process of immune cells, which is essential for phagosome maturation and acidification to kill bacteria.
4. INH, AN ANCIENT MYSTERY
4.1. The general consensus on the mode of action of isoniazid (INH)
INH remains the foremost treatment for both active and latent TB (Gafter, 1999; Sia & Wieland, 2011 ). It was first synthesized by two PhD candidates, Hans Meyer and Josef Mally, at German Charles University in Prague as a part of their doctoral requirements in 1912; however, its enormous potential against TB was unknown at the time. In 1951, the drug's high degree of efficacy in combatting TB was demonstrated by researchers from companies Hoffmann‐La Roche and E.R. Squibb & Sons in the United States, as well as by another group at Bayer in West Germany. INH was later approved as an anti‐TB drug against both active and latent TB by the FDA in 1952 and 1967, respectively (http://www.fda.gov/). Extensive research on the mode of action of INH was launched alongside FDA approval in 1952, taking roughly half a century to establish a general consensus. In the late 20th to early 21st century, the molecular mechanism of INH antibacterial activity was proposed to involve enzymatic oxidation of INH by bacterial catalase–peroxidase KatG to form isonicotinoyl radicals, which in turn forms a key chemical adduct with nicotinamide adenine dinucleotide (INH‐NAD+). This adduct is a potent inhibitor of enoyl acyl‐carrier‐protein reductase, an essential enzyme for mycolic acid biosynthesis of the Mtb cell wall, resulting in bacterial death (Timmins & Deretic, 2006). In drug‐resistant TB, mutations in KatG were confirmed to lower INH‐NAD+ adduct formation and overall antibacterial activity of INH (Ando et al., 2010); this development resulted in the removal of INH from the clinical uses in drug‐resistant TB.
4.2. The limitations of this general consensus
A number of limitations have already been identified that challenge the validity of the consensus model for the mode of action of INH. These limitations have emerged from both long‐standing clinical queries and more recent studies on TB pathophyisology. One such study showed that INH penetration into the granuloma (where Mtb resides in TB) was very low in a rabbit model of TB infection (Kjellsson et al., 2012); this is possibly due to both its low lipophilicity (logP = − 0.70) and very low protein binding activity (0–10%) [DrugBank: Isoniazid (DB00951)]. If INH cannot penetrate into the site of infection, its antibacterial activity would not be expected to effectively kill bacteria. Additionally, while several studies have demonstrated that INH induces rapid killing of Mtb in vitro, it remains puzzling that it takes 6–9 months of treatment time for clinical efficacy (Sia & Wieland, 2011). Another question has arisen in light of recent evidence on the latent stage of Mtb, which has been described as nonreplicating but energy‐generating within granulomas (Ehlers & Schaible, 2012). One study investigated cell wall composition in populations of latent Mtb and found that all populations remained in a cell wall free stage (Velayati et al., 2016). However, INH functions through inhibition of cell wall biosynthesis. Thus, if latent Mtb does not possess a cell wall, how might INH prove effective against latent TB? In light of these observations, it is clear that the general consensus of the mode of action of INH is not sufficient. Additionally, one can only surmise that INH must possess one or more additional mechanisms of action that remain to be identified.
4.3. An alternative mode of action of isoniazid (INH) is NO generation?
Before delving into novel evidence that addresses these limitations, it is worth considering alternative models that have been proposed to capture INH function; two studies can be mentioned here. Both showed that supra‐pharmacological concentration (35 mM) of INH can generate NO through the peroxidation cycle of KatG (Timmins, Master, Rusnak, & Deretic, 2004a; Timmins, Master, Rusnak, & Deretic, 2004b). To produce NO from INH, the one‐electron oxidation of the hydrazide nitrogen atom was proposed, which added oxygen and decomposed to NO and isonicotinyl free radical (INH•). However, a more recent study on the temperature‐dependent rate constants for the hydroxyl radical oxidation and solvated electron reduction of INH disputed this claim. The researchers found the distal nitrogen of the hydrazyl moiety to be the initial oxidation site and revealed the decomposition products to be https://pubchem.ncbi.nlm.nih.gov/compound/123195 (HN═NH) and INH• (Rickman, Swancutt, Mezyk, & Kiddle, 2013). While this NO generation theory offers a useful clarification of INH metabolism, it still fails to address the aforementioned limitations (i.e., granuloma penetration, efficacy against latent TB, and longer treatment time) within INH's proposed mode of action.
5. RECENT KNOWLEDGE ON ISONIAZID (INH)
5.1. KatG‐free INH‐NAD+ adduct formation
As INH is a prodrug, the activation of INH (i.e. formation of isonicotinyl free radical, INH•) requires a one‐electron enzymatic oxidation step. The bacterial catalase–peroxidase enzyme, KatG, possesses the capacity to facilitate this redox reaction. However, there are a number of functionally similar enzymes in the host system such as myeloperoxidase (MPO). MPO, a member of the haem peroxidase‐cyclooxygenase superfamily, is the major component of the azurophilic granules within neutrophils; it is also found to a lesser extent in the lysosomes of monocytes and certain types of macrophages (Tavora, Ripple, Li, & Burke, 2009). When comparing MPO and KatG, MPO possesses approximately 104 times stronger peroxidase activity than KatG (Khan et al., 2016d). However, it was previously unknown whether MPO played a role in INH‐NAD+ adduct formation. This was addressed by a recent study that showed that human neutrophil‐derived MPO can generate INH‐NAD+ adducts (Khan et al., 2016d). This proved to be an important revelation, as neutrophils are recruited to the site of TB infection (granulomas) via cell death signals secreted by infected macrophages, in order to engulf Mtb (Molloy, 2012). Recent studies on eosinophil peroxidase demonstrated that the latter, which is present in eosinophils, is also able to produce INH‐NAD+ adducts (Babu et al., 2019b).
5.2. Isoniazid prevents Mtb‐induced oxidative stress mediated necrosis
Extensive studies on TB pathophysiology have shown that virulent Mtb strains induce necrosis in infected phagocytic cells via mitochondrial inner membrane disruption (Chen, Gan, & Remold, 2006). ROS (e.g. superoxide radical anion, hydroxyl radical, and hydrogen peroxide) are one of the main causes of such mitochondrial damage (Belhadj Slimen et al., 2014). In the case of Mtb infection, host phagocytic cells induce the formation of reactive species to kill bacteria; however, Mtb has extraordinary strategies to defend itself against these species, particularly ROS (Ehrt & Schnappinger, 2009). Therefore, the induction of ROS by phagocytic cells cannot kill Mtb; however, ROS promotes phagocytic self‐destruction through necrosis. To effectively combat Mtb, phagocytic cells must first avoid self‐destruction through oxidative stress‐induced necrosis (oxidative necrosis). A recent study showed that INH prevented oxidative stress‐induced necrosis of the human promyelocytic leukaemia (HL‐60) cell line, a model for human neutrophils (Khan et al., 2016b). This is important given that inhibition of necrotic death in infected neutrophils can effectively stop the onset of active TB.
5.3. Isoniazid induces monocytic differentiation and immunomodulation
For pathogen killing during phagocytosis, macrophages mainly rely on reactive nitrogen species (RNS) production, whereas neutrophils rely on ROS production. Each type of phagocytic cell generally produces nanomolar (nM) to micromolar (μM) amounts of their respective reactive species (RNS/ROS). However, Mtb possesses a very high tolerance for ROS (up to 50‐mM H2O2; Voskuil et al., 2011), whereas tolerance against RNS is relatively low (up to 5 mM of RNS is bacteriostatic; above 5 mM is bactericidal; Voskuil et al., 2011). Additionally, few macrophages merge together to form giant cells within the granuloma, which typically occurs in general circulation (Lay et al., 2007). The formation of the giant cell ceases its microbial uptake function but strengthens its antigen presentation property (similar to dendritic cells). It is necessary to activate T cells that can reactivate the inactivated macrophages within the granuloma (Lay et al., 2007). For these reasons, macrophages provide the main line of defence against TB. In order to amplify the macrophagic response needed to successfully combat Mtb, more monocytes must be recruited from circulation, which necessitates greater monocytic differentiation from granulocyte–monocyte (GM) progeny stem cells. A recent study showed that INH can induce monocytic differentiation in HL‐60 cells, with monocytes comprising approximately one fourth of the population of total cells at the pharmacological concentration of INH (Babu et al., 2019a).
In addition to inducing monocytic differentiation, INH has also been shown to possess several other immunomodulatory roles. Kim et al. have shown that INH induces host immune cell autophagy, which was required to kill Mtb (Kim et al., 2012). Another study by Yamashiro et al. showed that INH blocks IL‐1 receptor and TNF signalling to indirectly inhibit bacterial growth during active TB, which complements KatG‐dependent activation of INH (Yamashiro et al., 2016).
6. PROPOSAL OF A NOVEL MODE OF ACTION OF ISONIAZID (INH)
To address the limitations of the consensus view on INH's mode of action, a series of recent studies investigated INH's conceivable roles in modulating the host immune system. These ultimately yielded three novel pharmacological findings regarding INH's mode of action (Figure 3). In the first study, neutrophil myeloperoxidase (MPO) was shown to metabolize INH (similarly to KatG), which forms the aforementioned antibacterial INH‐NAD+ adduct (an inhibitor of enoyl‐acyl carrier protein reductase, a key enzyme for mycolic acid synthesis). This finding may explain how INH manages to kill Mtb without penetrating the lipophilic granuloma. Since neutrophils engulf granuloma‐escaping Mtb in active TB, their antibacterial INH‐NAD+ adduct formation capacity during INH treatment could play a vital role in killing Mtb directly. Therefore, the previous understanding about the role of neutrophils in TB, termed as “Trojan Horse,” is disputable during INH treatment. In addition, this finding refutes the claim that KatG is the only site of INH activation. Thus, mutations in the KatG gene may not be a critical consideration for INH resistance; however, this claim warrants further investigation.
Figure 3.
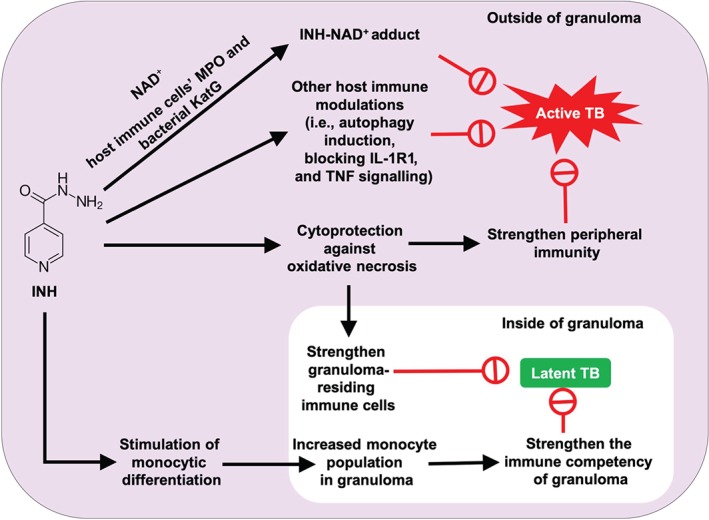
A comprehensive proposed mode of action of isoniazid (INH): In the case of latent tuberculosis (TB) (inside the granuloma) where Mtb does not possess a cell wall, an increased proportion of pro‐inflammatory monocytes in the granuloma is key to slowly killing Mtb. INH permits this via induction of pro‐inflammatory (CD14+/CD16−) monocytic differentiation from progenitor stem cells (Khan et al., 2016a; Babu et al., 2019a). This increased circulatory monocyte population permits the build‐up of appropriate levels of pro‐inflammatory monocytes in the granuloma. Through this mechanism, INH is able to slowly eradicate latent Mtb. The cytoprotective effect of INH further boosts the strength of granuloma‐residing immune cells, especially neutrophils that are prone to self‐necrosis via ROS generation (Khan et al., 2016b). In the case of active TB where Mtb possesses a cell wall and remains outside of the granuloma, the INH‐NAD+ adduct is sufficient to kill the bacteria. Various peroxidase enzymes (i.e., MPO, eosinophil peroxidase, and KatG) produced by different immune cells and Mtb can generate INH‐NAD+ adducts shortly after administration of INH treatment (Babu et al., 2019b; Khan et al., 2016d). INH additionally induces autophagy in infected immune cells (Kim et al., 2012) and inhibits IL‐1R1 and TNF signalling to further combat Mtb (Yamashiro et al., 2016)
The second study revealed the cytoprotective role of INH against oxidative stress‐induced necrosis. This type of oxidative stress‐induced necrosis is the underlying cause of phagocytic cell death and is followed by granuloma degradation in TB. The cytoprotective role of INH may strengthen granuloma‐mediated host immune defence and induce latency of Mtb. The proposed possible mechanisms for this cytoprotection were (a) an increase in cell replication, (b) an increase in ATP synthesis, and (c) an increase in the expression of structural maintenance proteins, as well as inhibition of pro‐death signals (Khan et al., 2016b).
The third study revealed the capacity of INH to induce monocytic differentiation in a model cell line for GM progenitor stem cells (HL‐60 cells). This finding is important since monocyte‐derived macrophages are known to play a major role in killing both latent and active Mtb due to their capacity to generate NO, for which Mtb does not possess any defence mechanism. Since this INH‐induced monocytic differentiation is very weak (i.e., one fourth the strength of known inducer https://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2779), it takes considerably longer to increase the population of circulating monocytes to high enough levels for granuloma recruitment, which is needed to effectively kill latent Mtb; this view thus offers an possible explanation for the prolonged treatment time of INH.
7. CONCLUDING REMARKS
In light of the limitations of the consensus view on INH's mode of action, novel pharmacological findings on INH have been combined to propose a revised comprehensive mode of action, as seen in Figure 3. This proposed mode of action can explain how INH may (a) exert antibacterial action without penetrating into the granuloma, (b) prove effective against latent TB, and (c) why INH treatment time is relatively long. Furthermore, this novel proposed mode of action of INH also disputes the justification for halting INH treatment in cases of TB deemed to be INH‐resistant, which is considered to be the first step preceding all other types of drug resistances. Drug resistance is usually diagnosed in vitro through either a phenotypic drug susceptibility test or a PCR‐based molecular drug susceptibility test (http://www.cdc.gov/tb/), both of which completely overlook the drug's systemic effects (e.g. host immune modulation). Moreover, mutation of KatG has been identified in around 45% of total clinical INH‐resistant TB cases (Guo, Seet, Denkin, Parsons, & Zhang, 2006), which is suggested to reduce INH efficiency by interfering with KatG binding (Yu, Girotto, Lee, & Magliozzo, 2003). However, as both neutrophilic MPO and eosinophil peroxidase were found to be an alternative to KatG for INH activation, this provides additional reason to employ INH in cases of INH‐resistant TB, particularly when KatG mutations are present. It is worth noting that INH was found to be ineffective against murine macrophages infected with KatG‐mutant Mtb in vitro in the absence of NAD+ and MPO activators (Skinner, Furney, Kleinert, & Orme, 1995). This aspect of INH warrants further investigation since MPO is not activated normally in immune cells and NAD+ is critical for INH‐NAD+ adduct formation. The previous consensus on the mode of action of INH was based on in vitro biochemical behaviour of INH. This proposal builds upon that model with more recent in vitro biochemical studies of INH with different enzyme systems (i.e., MPO and eosinophil peroxidase) and cells (neutrophils, eosinophils, and HL‐60 cells). This ultimately provides a theoretical background for further studies using immune cells from TB patients.
7.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
ACKNOWLEDGEMENT
S.R.K. is currently a Diabetes Canada post‐doctoral fellow at the University of Toronto.
Khan SR, Manialawy Y, Siraki AG. Isoniazid and host immune system interactions: A proposal for a novel comprehensive mode of action. Br J Pharmacol. 2019;176:4599–4608. 10.1111/bph.14867
Saifur R. Khan and Arno G. Siraki are joint senior authors.
REFERENCES
- Abadie, V. , Badell, E. , Douillard, P. , Ensergueix, D. , Leenen, P. J. , Tanguy, M. , … Winter, N. (2005). Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood, 106(5), 1843–1850. [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … Davies, J. A. (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174(S1), S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando, H. , Kondo, Y. , Suetake, T. , Toyota, E. , Kato, S. , Mori, T. , & Kirikae, T. (2010). Identification of katG mutations associated with high‐level isoniazid resistance in Mycobacterium tuberculosis . Antimicrobial Agents and Chemotherapy, 54(5), 1793–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu, D. , Khan, S. R. , Srivastava, N. , Kyoung Suh, L. Y. , Morgan, A. G. , Aljuhani, N. , … Siraki, A. G. (2019a). Isoniazid induces a monocytic‐like phenotype in HL‐60 cells. Archives of Biochemistry and Biophysics, 664, 15–23. [DOI] [PubMed] [Google Scholar]
- Babu, D. , Morgan, A. G. , Reiz, B. , Whittal, R. M. , Almas, S. , Lacy, P. , & Siraki, A. G. (2019b). Eosinophil peroxidase oxidizes isoniazid to form the active metabolite against M. tuberculosis, isoniazid‐NAD+ . Chemico‐Biological Interactions, 305, 48–53. [DOI] [PubMed] [Google Scholar]
- Balasubramanian, V. , Wiegeshaus, E. H. , Taylor, B. T. , & Smith, D. W. (1994). Pathogenesis of tuberculosis: Pathway to apical localization. Tubercle and Lung Disease, 75(3), 168–178. [DOI] [PubMed] [Google Scholar]
- Barry, S. , Breen, R. , Lipman, M. , Johnson, M. , & Janossy, G. (2009). Impaired antigen‐specific CD4+ T lymphocyte responses in cavitary tuberculosis. Tuberculosis, 89(1), 48–53. [DOI] [PubMed] [Google Scholar]
- Behar, S. M. , Martin, C. J. , Booty, M. G. , Nishimura, T. , Zhao, X. , Gan, H. X. , … Remold, H. G. (2011). Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis . Mucosal Immunology, 4(3), 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belhadj Slimen, I. , Najar, T. , Ghram, A. , Dabbebi, H. , Ben Mrad, M. , & Abdrabbah, M. (2014). Reactive oxygen species, heat stress and oxidative‐induced mitochondrial damage. A review. International Journal of Hyperthermia, 30(7), 513–523. [DOI] [PubMed] [Google Scholar]
- Berry, M. P. R. , Graham, C. M. , McNab, F. W. , Xu, Z. , Bloch, S. A. A. , Oni, T. , … Quinn, C. (2010). An interferon‐inducible neutrophil‐driven blood transcriptional signature in human tuberculosis. Nature, 466, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos, K. I. , Harkins, K. M. , Herbig, A. , Coscolla, M. , Weber, N. , Comas, I. , … Campbell, T. J. (2014). Pre‐Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature, 514(7523), 494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braian, C. , Hogea, V. , & Stendahl, O. (2013). Mycobacterium tuberculosis‐induced neutrophil extracellular traps activate human macrophages. Journal of Innate Immunity, 5(6), 591–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño, D. , García, L. F. , & Rojas, M. (2011). Increased frequency and cell death of CD16+ monocytes with Mycobacterium tuberculosis infection. Tuberculosis, 91(5), 348–360. [DOI] [PubMed] [Google Scholar]
- Chen, M. , Gan, H. , & Remold, H. G. (2006). A mechanism of virulence: Virulent Mycobacterium tuberculosis strain H37Rv, but not attenuated H37Ra, causes significant mitochondrial inner membrane disruption in macrophages leading to necrosis. The Journal of Immunology, 176(6), 3707–3716. [DOI] [PubMed] [Google Scholar]
- Dallenga, T. , Repnik, U. , Corleis, B. , Eich, J. , Reimer, R. , Griffiths, G. W. , & Schaible, U. E. (2017). M. tuberculosis‐induced necrosis of infected neutrophils promotes bacterial growth following phagocytosis by macrophages. Cell Host & Microbe, 22(4), 519, e513–530. [DOI] [PubMed] [Google Scholar]
- Ehlers, S. , & Schaible, U. E. (2012). The granuloma in tuberculosis: Dynamics of a host–pathogen collusion. Frontiers in Immunology, 3, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrt, S. , & Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: Defense against host stresses. Cellular Microbiology, 11(8), 1170–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst, J. D. (1998). Macrophage receptors for Mycobacterium tuberculosis . Infection and Immunity, 66(4), 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eruslanov, E. B. , Lyadova, I. V. , Kondratieva, T. K. , Majorov, K. B. , Scheglov, I. V. , Orlova, M. O. , & Apt, A. S. (2005). Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infection and Immunity, 73(3), 1744–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbairn, I. P. , Stober, C. B. , Kumararatne, D. S. , & Lammas, D. A. (2001). ATP‐mediated killing of intracellular mycobacteria by macrophages is a P2X7‐dependent process inducing bacterial death by phagosome‐lysosome fusion. The Journal of Immunology, 167(6), 3300–3307. [DOI] [PubMed] [Google Scholar]
- Fialkow, L. , Wang, Y. , & Downey, G. P. (2007). Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radical Biology & Medicine, 42(2), 153–164. [DOI] [PubMed] [Google Scholar]
- Flynn, J. L. , Chan, J. , & Lin, P. L. (2011). Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunology, 4, 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman, H. J. , & Torres, M. (2001). Redox signaling in macrophages. Molecular Aspects of Medicine, 22(4‐5), 189–216. [DOI] [PubMed] [Google Scholar]
- Gafter, A. K. U. (1999). Tuberculosis prophylaxis for the chronically dialysed patient—Yes or no? Nephrology, Dialysis, Transplantation, 14(12), 2857–2859. [DOI] [PubMed] [Google Scholar]
- Garg, G. , Gogia, A. , Kakar, A. , & Miglani, P. (2017). Persistent marked peripheral eosinophilia due to tuberculosis: A case report. Iranian Journal of Medical Sciences, 42(1), 102–105. [PMC free article] [PubMed] [Google Scholar]
- Gomez i Prat, J. , & de Souza, S. M. (2003). Prehistoric tuberculosis in America: Adding comments to a literature review. Memórias do Instituto Oswaldo Cruz, 98(Suppl 1), 151–159. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Juarrero, M. , Turner, O. C. , Turner, J. , Marietta, P. , Brooks, J. V. , & Orme, I. M. (2001). Temporal and spatial arrangement of lymphocytes within lung granulomas induced by aerosol infection with Mycobacterium tuberculosis . Infection and Immunity, 69(3), 1722–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirado, E. , & Schlesinger, L. S. (2013). Modeling the Mycobacterium tuberculosis granuloma—The critical battlefield in host immunity and disease. Frontiers in Immunology, 4, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, H. , Seet, Q. , Denkin, S. , Parsons, L. , & Zhang, Y. (2006). Molecular characterization of isoniazid‐resistant clinical isolates of Mycobacterium tuberculosis from the USA. Journal of Medical Microbiology, 55(Pt 11), 1527–1531. [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … Bryant, C. (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to immunopharmacology. Nucleic Acids Research, 46(D1), D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestvik, A. L. , Hmama, Z. , & Av‐Gay, Y. (2005). Mycobacterial manipulation of the host cell. FEMS Microbiology Reviews, 29(5), 1041–1050. [DOI] [PubMed] [Google Scholar]
- Khan, S. R. (2016a). The novel pharmacological actions of isoniazid: A proposal for its mode of action. PhD, University of Alberta, era.library.ualberta.ca.
- Khan, S. R. , Aljuhani, N. , Morgan, A. G. M. , Baghdasarian, A. , Fahlman, R. P. , & Siraki, A. G. (2016b). Cytoprotective effect of isoniazid against H2O2 derived injury in HL‐60 cells. Chemico‐Biological Interactions, 244, 37–48. [DOI] [PubMed] [Google Scholar]
- Khan, S. R. , Baghdasarian, A. , Fahlman, R. P. , & Siraki, A. G. (2016c). Global protein expression dataset acquired during isoniazid‐induced cytoprotection against H2O2 challenge in HL‐60 cells. Data in Brief, 6, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, S. R. , Morgan, A. G. M. , Michail, K. , Srivastava, N. , Whital, R. M. , Aljuhani, N. , & Siraki, A. G. (2016d). Metabolism of isoniazid by neutrophil myeloperoxidase leads to INH‐NAD+ adduct formation: A comparison of the reactivity of isoniazid with its known human metabolites. Biochemical Pharmacology, 106, 46–55. [DOI] [PubMed] [Google Scholar]
- Kim, J.‐J. , Lee, H.‐M. , Shin, D.‐M. , Kim, W. , Yuk, J.‐M. , Jin, H. S. , … Shin, S. J. (2012). Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host & Microbe, 11(5), 457–468. [DOI] [PubMed] [Google Scholar]
- Kjellsson, M. C. , Via, L. E. , Goh, A. , Weiner, D. , Low, K. M. , Kern, S. , … Dartois, V. (2012). Pharmacokinetic evaluation of the penetration of antituberculosis agents in rabbit pulmonary lesions. Antimicrobial Agents and Chemotherapy, 56(1), 446–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammas, D. A. , Stober, C. , Harvey, C. J. , Kendrick, N. , Panchalingam, S. , & Kumararatne, D. S. (1997). ATP‐induced killing of mycobacteria by human macrophages is mediated by purinergic P2Z(P2X7) receptors. Immunity, 7(3), 433–444. [DOI] [PubMed] [Google Scholar]
- Lasco, T. M. , Turner, O. C. , Cassone, L. , Sugawara, I. , Yamada, H. , McMurray, D. N. , & Orme, I. M. (2004). Rapid accumulation of eosinophils in lung lesions in guinea pigs infected with Mycobacterium tuberculosis . Infection and Immunity, 72(2), 1147–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawn, S. D. , Myer, L. , Edwards, D. , Bekker, L.‐G. , & Wood, R. (2009). Short‐term and long‐term risk of tuberculosis associated with CD4 cell recovery during antiretroviral therapy in South Africa. AIDS (London, England), 23(13), 1717–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lay, G. , Poquet, Y. , Salek‐Peyron, P. , Puissegur, M. P. , Botanch, C. , Bon, H. , … Altare, F. (2007). Langhans giant cells from M. tuberculosis‐induced human granulomas cannot mediate mycobacterial uptake. The Journal of Pathology, 211(1), 76–85. [DOI] [PubMed] [Google Scholar]
- Lowe, D. M. , Redford, P. S. , Wilkinson, R. J. , O'Garra, A. , & Martineau, A. R. (2012). Neutrophils in tuberculosis: Friend or foe? Trends in Immunology, 33(1), 14–25. [DOI] [PubMed] [Google Scholar]
- Marakalala, M. J. , Raju, R. M. , Sharma, K. , Zhang, Y. J. , Eugenin, E. A. , Prideaux, B. , … Eum, S. Y. (2016). Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nature Medicine, 22, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moideen, K. , Kumar, N. P. , Nair, D. , Banurekha, V. V. , Bethunaickan, R. , & Babu, S. (2018). Heightened systemic levels of neutrophil and eosinophil granular proteins in pulmonary tuberculosis and reversal following treatment. Infection and Immunity, 86(6), e00008–e00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molloy, S. (2012). Host response: Double trouble for TB. Nature Reviews. Microbiology, 10(11), 730–731. [DOI] [PubMed] [Google Scholar]
- Ong, C. W. M. , Elkington, P. T. , Brilha, S. , Ugarte‐Gil, C. , Tome‐Esteban, M. T. , Tezera, L. B. , … Gilman, R. H. (2015). Neutrophil‐derived MMP‐8 drives AMPK‐dependent matrix destruction in human pulmonary tuberculosis. PLoS Pathogens, 11(5), e1004917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organization, W. H. (2018). Global tuberculosis report 2018. Geneva. [Google Scholar]
- Rickman, K. A. , Swancutt, K. L. , Mezyk, S. P. , & Kiddle, J. J. (2013). Isoniazid: Radical‐induced oxidation and reduction chemistry. Bioorganic & Medicinal Chemistry Letters, 23(10), 3096–3100. [DOI] [PubMed] [Google Scholar]
- Sakamoto, K. (2012). The pathology of Mycobacterium tuberculosis infection. Veterinary Pathology, 49(3), 423–439. [DOI] [PubMed] [Google Scholar]
- Sampath, P. , Moideen, K. , Ranganathan, U. D. , & Bethunaickan, R. (2018). Monocyte subsets: Phenotypes and function in tuberculosis infection. Frontiers in Immunology, 9, 1726–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez, M. D. , García, Y. , Montes, C. , París, S. C. , Rojas, M. , Barrera, L. F. , … García, L. F. (2006). Functional and phenotypic changes in monocytes from patients with tuberculosis are reversed with treatment. Microbes and Infection, 8(9), 2492–2500. [DOI] [PubMed] [Google Scholar]
- Sia, I. G. , & Wieland, M. L. (2011). Current concepts in the management of tuberculosis. Mayo Clinic Proceedings, 86(4), 348–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Miranda, M. , Breiman, A. , Allain, S. , Deknuydt, F. , & Altare, F. (2012). The tuberculous granuloma: An unsuccessful host defence mechanism providing a safety shelter for the bacteria? Clinical and Developmental Immunology, 2012, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner, P. S. , Furney, S. K. , Kleinert, D. A. , & Orme, I. M. (1995). Comparison of activities of fluoroquinolones in murine macrophages infected with Mycobacterium tuberculosis . Antimicrobial Agents and Chemotherapy, 39(3), 750–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, I. (2003). Mycobacterium tuberculosis pathogenesis and molecular determinants of virulence. Clinical Microbiology Reviews, 16(3), 463–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbian, S. , Tsenova, L. , Kim, M.‐J. , Wainwright, H. C. , Visser, A. , Bandyopadhyay, N. , … Russell, D. G. (2015). Lesion‐specific immune response in granulomas of patients with pulmonary tuberculosis: A pilot study. PLoS ONE, 10(7). e0132249‐e0132249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sud, D. , Bigbee, C. , Flynn, J. L. , & Kirschner, D. E. (2006). Contribution of CD8+ T cells to control of Mycobacterium tuberculosis infection. The Journal of Immunology, 176(7), 4296–4314. [DOI] [PubMed] [Google Scholar]
- Sun, J. , Singh, V. , Lau, A. , Stokes, R. W. , Obregon‐Henao, A. , Orme, I. M. , … Hmama, Z. (2013). Mycobacterium tuberculosis nucleoside diphosphate kinase inactivates small GTPases leading to evasion of innate immunity. PLoS Pathogens, 9(7), e1003499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland, J. S. , Jeffries, D. J. , Donkor, S. , Walther, B. , Hill, P. C. , Adetifa, I. M. O. , … Ota, M. O. (2009). High granulocyte/lymphocyte ratio and paucity of NKT cells defines TB disease in a TB‐endemic setting. Tuberculosis, 89(6), 398–404. [DOI] [PubMed] [Google Scholar]
- Tavora, F. R. , Ripple, M. , Li, L. , & Burke, A. P. (2009). Monocytes and neutrophils expressing myeloperoxidase occur in fibrous caps and thrombi in unstable coronary plaques. BMC Cardiovascular Disorders, 9, 27–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins, G. S. , & Deretic, V. (2006). Mechanisms of action of isoniazid. Molecular Microbiology, 62(5), 1220–1227. [DOI] [PubMed] [Google Scholar]
- Timmins, G. S. , Master, S. , Rusnak, F. , & Deretic, V. (2004a). Nitric oxide generated from isoniazid activation by KatG: Source of nitric oxide and activity against Mycobacterium tuberculosis . Antimicrobial Agents and Chemotherapy, 48(8), 3006–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmins, G. S. , Master, S. , Rusnak, F. , & Deretic, V. (2004b). Requirements for nitric oxide generation from isoniazid activation in vitro and inhibition of mycobacterial respiration in vivo. Journal of Bacteriology, 186(16), 5427–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Crevel, R. , Ottenhoff, T. H. M. , & van der Meer, J. W. M. (2002). Innate immunity to Mycobacterium tuberculosis . Clinical Microbiology Reviews, 15(2), 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayati, A. A. , Abeel, T. , Shea, T. , Konstantinovich Zhavnerko, G. , Birren, B. , Cassell, G. H. , … Farnia, P. (2016). Populations of latent Mycobacterium tuberculosis lack a cell wall: Isolation, visualization, and whole‐genome characterization. International Journal of Mycobacteriology., 5, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voskuil, M. I. , Bartek, I. , Visconti, K. , & Schoolnik, G. K. (2011). The response of Mycobacterium tuberculosis to reactive oxygen and nitrogen species. Frontiers in Microbiology, 2, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, E. , Teskey, G. , & Venketaraman, V. (2017). Effector mechanisms of neutrophils within the innate immune system in response to Mycobacterium tuberculosis infection. Journal of Clinical Medicine, 6(2), 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe, L. M. , Mahaffey, S. B. , Kruh, N. A. , & Dobos, K. M. (2010). Proteomic definition of the cell wall of Mycobacterium tuberculosis . Journal of Proteome Research, 9(11), 5816–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, D. , Bach, H. , Sun, J. , Hmama, Z. , & Av‐Gay, Y. (2011a). Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar‐H+‐ATPase to inhibit phagosome acidification. Proceedings of the National Academy of Sciences of the United States of America, 108(48), 19371–19376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, D. , Bach, H. , Sun, J. , Hmama, Z. , & Av‐Gay, Y. (2011b). Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar‐H+–ATPase to inhibit phagosome acidification. Proceedings of the National Academy of Sciences, 108(48), 19371–19376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro, L. H. , Eto, C. , Soncini, M. , Horewicz, V. , Garcia, M. , Schlindwein, A. D. , … Báfica, A. (2016). Isoniazid‐induced control of Mycobacterium tuberculosis by primary human cells requires interleukin‐1 receptor and tumor necrosis factor. European Journal of Immunology, 46(8), 1936–1947. [DOI] [PubMed] [Google Scholar]
- Yu, S. , Girotto, S. , Lee, C. , & Magliozzo, R. S. (2003). Reduced affinity for isoniazid in the S315T mutant of Mycobacterium tuberculosis KatG is a key factor in antibiotic resistance. Journal of Biological Chemistry, 278(17), 14769–14775. [DOI] [PubMed] [Google Scholar]