

Abstract
Background and Purpose
The morbidity and mortality associated with recreational use of synthetic cannabinoid receptor agonists (SCRAs) may reflect strong activation of CB1 receptors and is a major health concern. The properties of SCRA at CB1 receptors are not well defined. Here we have developed an assay to determine acute CB1 receptor efficacy using receptor depletion with the irreversible CB1 receptor antagonist AM6544, with application of the Black and Leff operational model to calculate efficacy.
Experimental Approach
Receptor depletion in mouse AtT‐20 pituitary adenoma cells stably expressing human CB1 receptors was achieved by pretreatment of cells with AM6544 (10 μM, 60 min). The CB1 receptor‐mediated hyperpolarisation of AtT‐20 cells was measured using fluorescence‐based membrane potential dye. From data fit to the operational model, the efficacy (τ) and affinity (K A) parameters were obtained for each drug.
Key Results
AM6544 did not affect the potency or maximal effect of native somatostatin receptor‐induced hyperpolarization. The τ value of ∆9‐THC was 80‐fold less than the reference CB receptor agonist CP55940 and 260‐fold less than the highest efficacy SCRA, 5F‐MDMB‐PICA. The operational efficacy of SCRAs ranged from 233 (5F‐MDMB‐PICA) to 28 (AB‐PINACA), with CP55940 in the middle of the efficacy rank order. There was no correlation between the τ and K A values.
Conclusions and Implications
All SCRAs tested showed substantially higher efficacy at CB1 receptors than ∆9‐THC, which may contribute to the adverse effects seen with these drugs but not ∆9‐THC.
Abbreviations
- AtT‐20‐CB1
mouse pituitary tumour cells stably transfected with HA‐tagged human CB1 receptors
- GIRK
G protein‐coupled inwardly rectifying potassium channel
- NPS
new psychoactive substances
- PTX
Pertussis toxin
- RAi
relative agonist activity
- SCRAs
synthetic cannabinoid receptor agonists
- SRIF
somatotropin release‐inhibiting factor
1. INTRODUCTION
Synthetic cannabinoid receptor agonists (SCRAs) are a large class of new psychoactive substances (NPS), notionally designed to mimic the effects of http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2424 (∆9‐THC), the main psychoactive ingredient in cannabis (Wiley, Marusich, & Huffman, 2014). SCRAs have been marketed as herbal incense blends (often known as Spice or K2) and legal cannabis substitutes which are undetectable using conventional drug tests (Auwärter et al., 2009). Since the first generation of generally available SCRAs (including http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9706, JWH‐073, JWH‐200, and CP47497) were detected in herbal blends in 2008, more than 250 SCRAs have been reported in over 100 countries (Banister & Connor, 2018; United Nations Office on Drugs and Crime, 2018). SCRA use has been associated with adverse health effects including hundreds of hospitalisations and dozens of fatalities (Adams et al., 2017; Trecki, Gerona, & Schwartz, 2015). The most commonly reported adverse effects are psychosis, anxiety, agitation, seizures, tachycardia, hypothermia, and kidney injury (Tait, Caldicott, Mountain, Hill, & Lenton, 2016). In addition to these life‐threatening effects, daily SCRA use has been linked to dependence and withdrawal (Cooper, 2016).
SCRAs activate http://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=13, with their psychoactive effects caused by activation of CB1 receptors (Pacher, Bátkai, & Kunos, 2006). While cannabinoids, including SCRA, have been reported to have activity at a variety of ion channels and GPCRs other than CB1 and CB2 receptors (De Petrocellis & Di Marzo, 2010), the relevance of these interactions to the effects of cannabinoids in humans remains to be established, and their potential role in SCRA toxicity is unknown. In rodents, both JWH‐018‐ and http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=9705‐induced seizures are mediated by CB1 receptors (Funada & Takebayashi‐Ohsawa, 2018; Malyshevskaya et al., 2017; Vigolo et al., 2015). SCRA‐induced hypothermia and bradycardia are also CB1 receptor‐dependent (Banister et al., 2013; Banister et al., 2015; Banister et al., 2016). Intriguingly, a recent report suggests that the hypertensive effects of some SCRAs in rats may be independent of CB1 receptors (Schindler, Gramling, Justinova, Thorndike, & Baumann, 2017).
Most SCRAs studied to date activate CB1 receptors with greater potency and efficacy than ∆9‐THC in [35S]GTPγS binding assay (Gamage et al., 2018; Wiley et al., 2015), https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=257 assay (Costain et al., 2018; Hess, Schoeder, Pillaiyar, Madea, & Müller, 2016), β‐arrestin 2 recrutiment assay using NanoLuc binary technology (Cannaert, Storme, Franz, Auwärter, & Stove, 2016), and fluorescence‐based membrane potential assay (Banister et al., 2013; Banister et al., 2015; Banister et al., 2016; Banister, Stuart, et al., 2015). However, there is little quantitative information about the efficacy of SCRA at CB1 receptors. A substantial component of SCRA toxicity may be mediated through activation of CB1 receptors (Krishna Kumar et al., 2019), and defining the efficacy of SCRAs is an important step towards understanding possible mechanisms of CB1 receptor‐mediated toxicity. The similar maximal effects of several SCRAs reported in these assays may reflect receptor reserve, with only submaximal receptor occupancy by agonists needed to achieve their maximal response. Depleting receptor reserve can allow for quantitative determination of efficacy, by fitting concentration–response data before and after receptor depletion to the operational model of Black and Leff (Black & Leff, 1983). We have used the irreversible CB1 receptor antagonist AM6544 (Finlay et al., 2017) to facilitate quantitative measure of SCRAs efficacy to produce CB1 receptor‐dependent hyperpolarisation of intact AtT‐20 cells expressing human CB1 receptors. We determined the efficacy of a library of the most prevalent SCRAs identified in the NPS market since 2008 and found that the SCRAs we tested had up to 300 times the efficacy of ∆9‐THC, which may contribute to the apparently greater toxicity of these drugs. In this study, we have established an assay that can be used to quantitate CB1 receptor efficacy efficiently, and which is readily adaptable to the study of other CB1 receptor signalling pathways.
2. METHODS
2.1. Cell culture
Experiments used mouse AtT‐20 pituitary tumour cells (RRID:CVCL_4109) engineered to express FLP recombination site were transfected with human CB1 receptors as previously described (Banister et al., 2016). Cells were cultured in DMEM (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 10% FBS (Sigma‐Aldrich, St. Louis, MO, USA), 100 units·ml−1 of penicillin, 100 μg·ml−1 of streptomycin (Thermo Fischer Scientific, Waltham, MA, USA), and 80 μg·ml−1 of hygromycin (InvivoGen, San Diego, CA, USA). The cells were grown and maintained in 75 cm2 flask and passaged at 80% confluency or grown to 90% confluency for assay. Cells were incubated at 37°C in a humidified 5% CO2 atmosphere.
2.2. Achieving receptor depletion in a membrane potential assay
Cannabinoid receptors couple to https://www.guidetopharmacology.org/GRAC/FamilyIntroductionForward?familyId=74 (GIRK) channels in several types of neurons (Bacci, Huguenard, & Prince, 2004; Marinelli, Pacioni, Cannich, Marsicano, & Bacci, 2009). This coupling reflects a close association between CB receptors, G proteins, and channels (Guo & Ikeda, 2004), and all the components, other than the receptor, are naturally expressed in the AtT‐20 cells. Endogenous expression of GIRK channels (https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=434 and https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=435) in AtT‐20 cells is crucial for the main signalling assay performed during this work, and the direct activation of these channels in AtT‐20 cells by CB1 receptors has been studied repeatedly (Garcia, Brown, Hille, & Mackie, 1998; Mackie, Lai, Westenbroek, & Mitchell, 1995). Changes in the membrane potential of cells in response to GIRK activation were measured using the fluorometric imaging plate reader (FLIPR) membrane potential (blue) assay kit (Molecular Devices, Sunnyvale, CA) as previously described (Knapman et al., 2013). Cells were detached from the flask using trypsin/EDTA (Sigma‐Aldrich), and the pellet was resuspended in 10 ml Leibovitz's (L‐15) media supplemented with 1% FBS, 100 units·ml−1 of penicillin, 100 μg·ml−1 of streptomycin, and 15 mM glucose. The cells were seeded in a volume of 90 μl in poly‐d‐lysine (Sigma‐Aldrich) coated, black wall, clear bottom 96 well microplates. Cells were incubated overnight at 37°C in ambient CO2.
We used a receptor depletion assay to quantitatively determine the efficacy of a range of SCRAs. This approach involves irreversible binding of an antagonist to the orthosteric binding site of the receptors, thus permanently occluding a fraction of functional receptors available to an orthosteric ligand (Besse & Furchgott, 1976). We used the new CB1 receptor irreversible antagonist AM6544, synthesised at the Center for Drug Discovery, Northeastern University (Patent US8084451, 2011) to systematically reduce active receptor number in the AtT‐20‐CB1 cell expression system. The day after plating, AM6544 (10 μM) was prepared in HBSS composed of (mM) NaCl 145, HEPES 22, Na2HPO4 0.338, NaHCO3 4.17, KH2PO4 0.441, MgSO4 0.407, MgCl2 0.493, CaCl2 1.26, glucose 5.56 (pH 7.4, osmolarity 315 ± 15), and supplemented with 0.1% BSA. Receptor depletion was achieved following pretreatment of cells with AM6544 (10 μM) in parallel to the vehicle (control) for 60 min after removal of the L‐15, at 37°C in ambient CO2. The concentration of DMSO (0.1%) was kept constant for AM6544‐treated and control cells. Cells were then washed twice with warm HBSS and loaded with 90 μl per well of L‐15 media and 90 μl per well of reconstituted FLIPR dye. The cell plate was incubated at 37°C in ambient CO2 for 1 hr prior to measuring the fluorescence using a FlexStation 3 microplate reader (Molecular Devices). The AM6544‐treated and control cells were compared side by side. The cells were excited at a wavelength of 530 nm and emission measured at 565 nm, with cut‐off at 550 nm, and the readings were made every 2 s. Baseline readings were taken for 2 min after which 20 μl of drug (10×) was added to each well to give the desired concentration. The drugs of various concentrations were prepared in HBSS containing 0.1% BSA and 1% DMSO. The final concentration of DMSO in each well was always 0.1%. A concentration–response curve (CRC) for http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=730 was performed each day for quality control. On rare occasions where AM6544 pretreatment failed to produce a substantial shift in responses to CP55940, results were discarded, as they probably indicated experimenter error.
SCRAs were synthesised as previously described by Banister, Moir, et al. (2015), Banister, Stuart, et al. (2015), and Banister et al. (2016). Chemical structure of SCRAs can be found in Figure S1. The functional activity (EC50) of SCRAs at CB1 receptors were compared to ∆9‐THC and CP55940 (see Table S1). We have previously shown that the effects of SCRAs in AtT‐20‐CB1 cells were blocked by http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=743, a CB1 receptor antagonist, and that none of the SCRAs produced a significant change in the membrane potential of AtT‐20 wild‐type cells (Banister et al., 2016; Banister, Moir, et al., 2015; Banister, Stuart, et al., 2015). SCRA‐mediated hyperpolarisation of AtT‐20‐CB1 cells is also Pertussis toxin (PTX) sensitive, confirming that the response is Gi/o‐dependent (Banister et al., 2016, Banister, Moir, et al., 2015, Banister, Stuart, et al., 2015).
2.3. Data analysis
2.3.1. Operational model analysis
Drug responses are reported as percentage change of baseline fluorescence, following correction for the vehicle responses (0.1% DMSO). The hyperpolarisation of the cells produces a decrease in fluorescence. For convenience, values are expressed such that a change of 30% means a reduction in fluorescence of 30%. Data for individual experiments were analysed and the CRC before and after receptor depletion was fitted with the Black and Leff operational model in PRISM (Graph Pad Software Inc., San Diego, CA; RRID:SCR_002798), using five‐parameter non‐linear regression (Basal, K A, Effect max, τ, and transducer slope) to fit the operational model‐receptor depletion equation (Motulsky & Christopoulos, 2004).
The equation for operational model‐depletion presented in the same style as Prism:
where the maximal response of the system is given by Effect max. The parameter, τ, equals the total concentration of receptor in the system divided by the concentration of agonists occupied receptors that are required to produce half‐maximal tissue response. The parameter, K A, is define as the equilibrium K D for agonist binding to the receptors, while n is the slope factor of the transducer function.
From the operational model, efficacy (τ) and affinity (K A) parameters were obtained for each drug. The basal parameter was constrained to zero as the basal activity (without drug) was routinely subtracted from the measurements. The transducer slope n of all the agonist CRCs was constrained to 1 (after initial fits showed this to be a good approximation). The parameter Effect max is tissue specific and thus shared by all agonists acting on CB1 receptors through a given pathway for that day. The parameter, K A, is ligand–receptor specific, whereas τ has ligand‐specific elements (efficacy of ligand) and system‐specific elements (coupling efficiency of receptors to signalling pathway). Thus, for individual drugs, K A was shared between the AM6544‐treated and control state, but the separate best fit values of τ were determined for each data set. The τ value in the control state was used to measure the CB1 receptor agonist efficacy. This procedure serves to measure the efficacy of a group of agonists on a per‐day basis obtained from fitting data simultaneously to the operational model, with mean and SEM calculated using individual values for each experiment.
2.4. Estimate of relative agonist activity (RA i)
After determining the efficacy and affinity of SCRAs from the operational model under control and AM6544‐treated conditions, the data were used for the calculation of the initial estimate of RA i value. The relative affinities of agonists for the active state of a receptor (RA i) expressed relative to that of a reference agonist as described previously by Ehlert (2008) was calculated.
In this equation, τ A and τ B denote the intrinsic efficacies, and K A and K B denote the K D of reference and test agonist, obtained earlier from the operational model. CP55940 was used as the reference agonist to define the RA i of the SCRAs.
We also estimated RA i values of eight SCRAs from four published studies to compare these with the initial estimate of RA i values determined from the data generated in our laboratory. RA i values were estimated from studies on the [35S]GTPγS binding assay in HEK cells (RRID:CVCL_0045) by Ford et al. (2017), Thomas et al. (2017), Gamage et al. (2018), and Wiley et al. (2015). We used a simple calculation for the estimation of RA i as only the EC50 and the E max values of SCRAs were available from the literature (Ehlert, Griffin, Sawyer, & Bailon, 1999; Griffin, Figueroa, Liller, & Ehlert, 2007).
in which the subscript refers to the parameters of reference (A) and tests (B) agonists. In all instances, it was impossible to extract SEM for these data sets, as we do not have access to their raw data. The rank order of agonist activity based upon RA i values calculated from the literature for [35S]GTPγS binding assay was compared to our results for membrane potential assay. Finally, for each agonist, the RA i value for membrane potential assay was divided by the RA i value of GTPγS binding assay to estimate the bias factor.
Unless otherwise stated, the data represent mean ± SEM of at least six independent experiments, each conducted in duplicate. The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Statistical significance is defined as P < .05.
2.5. Materials
CP55940, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=729, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=2364, and CUMYL‐4CN‐BINACA were purchased from Cayman Chemical Company (Ann Arbour, MI, USA); ∆9‐THC was obtained from The Lambert Initiative (Sydney, NSW, Australia). AM6544 was a gift from laboratory of Professor Alexandros Makriyannis (Northeastern University, Massachusetts, USA). All the SCRAs, unless otherwise stated, were synthesised by Samuel D. Banister and Mitchell Longworth in the lab of Professor Michael Kassiou at Sydney University (Sydney, NSW, Australia). All the drugs were stored in aliquots of 30 mM at −80°C until needed.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org/, the common portal of data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/2018 (Alexander, Christopoulos, et al., 2017; Alexander, Fabbro et al., 2017).
3. RESULTS
3.1. Specificity of AM6544: a new irreversible antagonist of CB1 receptors
In vitro, AM6544 behaves as an irreversible antagonist of CB1 receptors , as established by Finlay et al. (2017) using radioligand binding assays in whole cells (pEC50 5.45 ± 0.11). To confirm that AM6544 does not non‐specifically interfere with receptor signalling mechanisms in AtT‐20‐CB1 cells, we examined the effect of AM6544 on the activation of native http://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=356. Pretreatment with AM6544 (10 μM, 60 min) had no effect on the potency or maximal effect of SRIF‐induced hyperpolarisation when compared to vehicle‐treated cells (Control, pEC50 9.13 ± 0.05, E max 38 ± 1%; AM6544‐treated pEC50 9.18 ± 0.04, E max 39 ± 0.7%, Figure 1), indicating that AM6544 did not interfere with either SRIF receptors or their signalling pathways likely to be shared with CB1 receptors in AtT‐20 cells. We also examined the possibility that AM6544 could affect the membrane potential of the cells prior to agonist addition. Application of AM6544 for 60 min at concentration up to 10 μM did not significantly affect the membrane potential of the AtT20‐CB1 cells by itself, nor did it modify the GIRK‐mediated hyperpolarisation produced by SRIF (Figure S2, P > .05).
Figure 1.
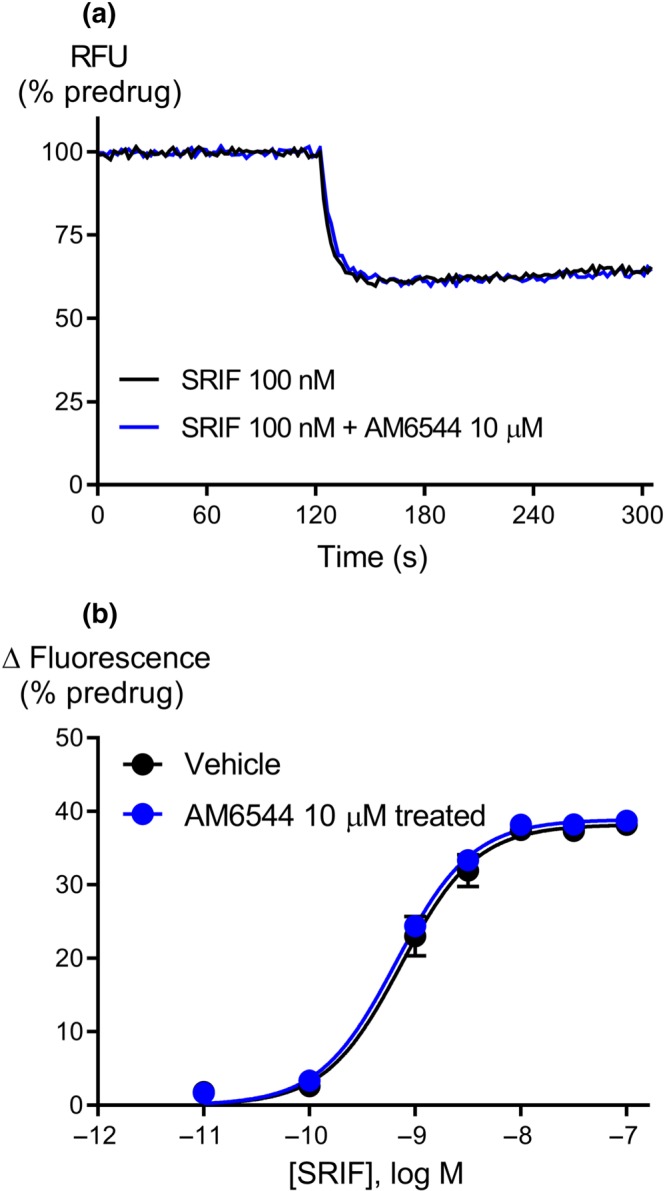
AM6544 is a specific irreversible antagonist of CB1 receptors. (a) Raw trace showing the change in fluorescence normalised to the predrug baseline for SRIF on AtT‐20‐CB1 cells pretreated for 60 min with vehicle or AM6544 (10 μM) and then washed twice before incubation with MPA dye. The traces are representative of at least six independent experiments. (b) Concentration–response curve for SRIF mediated hyperpolarisation of AtT‐20‐CB1 cells following pretreatment with AM6544 (10 μM) or vehicle. Data represent the mean ± SEM of six independent determinations performed in duplicate. There was no difference in the potency or maximal effect of SRIF between vehicle or following pretreatment with AM6544.
3.2. Functional activity of cannabinoids after receptor depletion with AM6544
The efficacy of the classical CB1 receptor agonist, CP55940, was measured after the pharmacological knockdown of CB1 receptors with AM6544 (10 μM, 60 min). The maximal response of CP55940 (10 μM) was reduced after AM6544 pretreatment compared to vehicle‐treated cells (Control, E max 33 ± 2; AM6544‐treated E max 26 ± 2, P < .05, Figure 2a). The τ value for CP55940 was reduced 10‐fold in AM6544 pretreated cells compared to vehicle cells (Table 1), suggesting that AM6544 can effectively deplete the receptors available to high efficacy SCRAs. From the operational model, the pK A of CP55940 was estimated to be 5.8 ± 0.1 (Table 1, n = 20).
Figure 2.
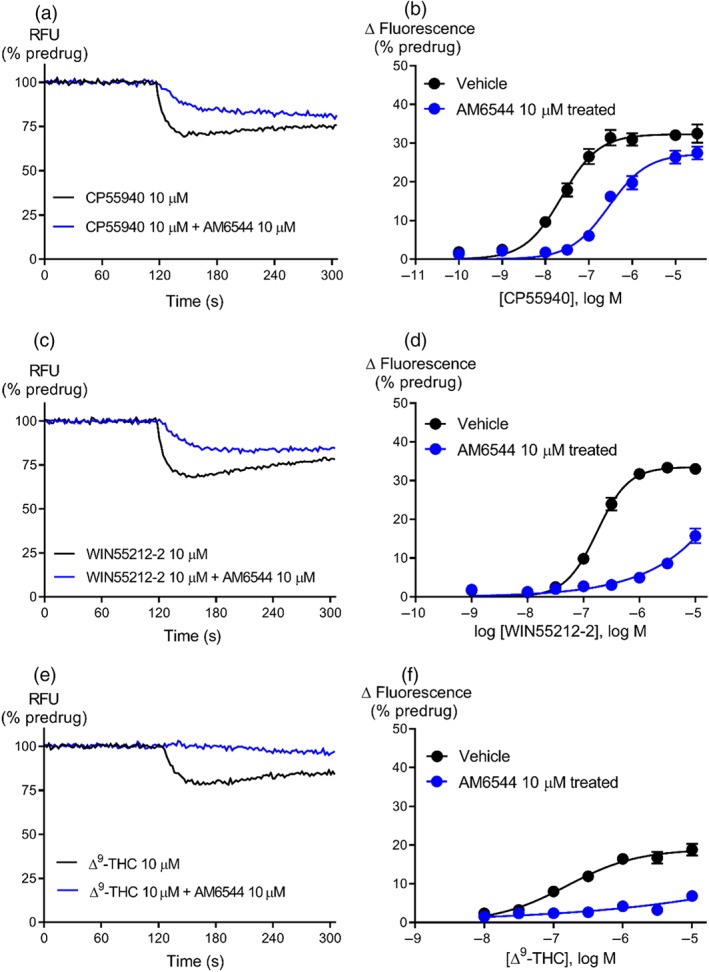
Representative traces for research cannabinoids CP55940 (a), WIN55212‐2 (c), and ∆9‐THC (e) following pretreatment with vehicle or AM6544 (10 μM) on AtT‐20‐CB1 cells. Raw trace showing reduction in hyperpolarisation induced by maximally effective concentration (10 μM) of CP55940, WIN55212‐2, and ∆9‐THC after AM6544 pretreatment compared to vehicle. Concentration–response curves for (b) CP55940 (n = 20), (d) WIN55212‐2 (n = 7), and (f) ∆9‐THC (n = 6) were plotted using five‐parameter non‐linear regression to fit the operational model‐receptor depletion equation with basal constrained to 0. Data represent the mean ± SEM of technical replicates. For some points, the error bars are smaller than the height of the symbol.
Table 1.
Efficacy and functional affinity of CP55940, ∆9‐THC, and other SCRAs
| Compound | Operational efficacy τ | Functional affinity pK A (±SEM) | |
|---|---|---|---|
| Control (±SEM) | AM6544‐treated (±SEM) | ||
| CP55940 | 72 (30) | 7 (3) | 5.78 (0.09) |
| WIN55212‐2 | 57 (41) | 3 (1) | 4.84 (0.24) |
| ∆9‐THC | 0.9 (0.1) | 0.3 (0.1) | 6.54 (0.07) |
| 2‐AG | 60 (27) | 3 (1) | 5.16 (0.08) |
| AEA | 5 (2) | 2 (2) | 5.17 (0.10) |
| JWH‐018 | 43 (21) | 2.5 (1) | 5.72 (0.21) |
| AM‐2201 | 32 (6) | 2 (0.4) | 6.53 (0.08) |
| UR‐144 | 36 (7) | 3 (0.5) | 4.51 (0.15) |
| XLR‐11 | 152 (73) | 13 (9) | 4.68 (0.26) |
| PB‐22 | 44 (10) | 3.2 (1) | 6.73 (0.10) |
| 5F‐PB‐22 | 102 (48) | 7 (5) | 7.05 (0.09) |
| AB‐CHMINACA | 92 (42) | 4 (1) | 6.98 (0.12) |
| AB‐PINACA | 28 (7) | 2 (0.4) | 6.90 (0.21) |
| MDMB‐CHMICA | 79 (32) | 8 (4) | 6.35 (0.17) |
| MDMB‐FUBINACA | 103 (62) | 5 (1.4) | 7.12 (0.10) |
| 5F‐MDMB‐PICA | 233 (65) | 14 (3) | 6.71 (0.13) |
| CUMYL‐4CN‐BINACA | 70 (28) | 4 (1) | 7.48 (0.07) |
Note. Values were calculated using the operational model of pharmacological agonism following CB1 receptor depletion with AM6544, as outlined in Section 2.
We also determined the efficacy of some frequently used research cannabinoids ‐ http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=733, the main psychoactive phytocannabinoid ∆9‐THC, and the endogenous cannabinoids 2‐arachidonolylglycerol and anandamide ‐ on CB1 receptors after receptor depletion with AM6544. The hyperpolarisation produced by WIN55212‐2 was strongly inhibited by AM6544 pretreatment (10 μM, 60 min) compared to vehicle‐treated cells (Figure 2). The τ for WIN55212‐2 was reduced 1.5‐fold compared to CP55940 but was 63‐fold greater than ∆9‐THC (Table 1). The efficacy of endogenous cannabinoids, 2‐arachidonolylglycerol and anandamide, was respectively 1.2‐ and 14‐fold less than that of CP55940 (Figure 3; Table 1).
Figure 3.
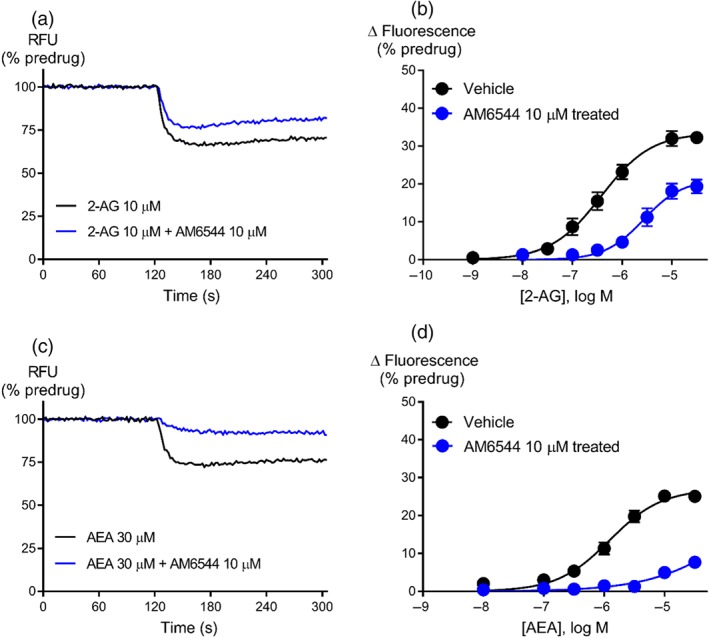
Representative traces for endogenous cannabinoids (a) 2‐arachidonolylglycerol (2‐AG) and (c) anandamide (AEA) after pretreatment with vehicle or AM6544 (10 μM) on AtT‐20‐CB1 cells. Raw trace showing reduction in hyperpolarisation induced by maximally effective concentration of 2‐AG (10 μM) and AEA (30 μM) after AM6544 pretreatment compared to vehicle. Concentration–response curves for (b) 2‐AG (n = 7) and (d) AEA (n = 7) were plotted using four parameter non‐linear regression to fit the operational model‐receptor depletion equation with basal constrained to 0. Illustrates the increase in efficacy of 2‐AG as compared to AEA. Data represent the mean ± SEM of technical replicates. For some points, the error bars are smaller than the height of the symbol.
We assessed the relative efficacy of SCRAs to provide insight into potential mechanisms of toxicity and the functional consequences of the evolution of SCRA structures over time. The efficacy of SCRAs was determined following receptor depletion with AM6544. Example traces and CRC are shown for JWH‐018, http://www.guidetopharmacology.org/GRAC/LigandDisplayForward?ligandId=10178, and XLR‐11 (Figure 4). The efficacy for all the drugs we examined are found in Table 1. The τ of SCRAs tested ranged from 28 to 233, with two of the 13 CRAs having τ values greater than 150 (5F‐MDMB‐PICA and XLR‐11). The first SCRA to be identified in Spice, JWH‐018, exhibited 2‐fold less τ than CP55940 but 43‐fold higher τ than ∆9‐THC. Only two of the SCRAs (MDMB‐CHMICA and CUMYL‐4CN‐BINACA) exhibited similar efficacy to CP55940 (Table 1), whereas four of the SCRAs (PB‐22, UR‐144, AM‐2201, and AB‐PINACA) had approximately 50% of the efficacy of CP55940. The least efficacious SCRA, AB‐PINACA, showed 3‐fold less τ than CP55940 while the most efficacious SCRA, 5F‐MDMB‐PICA, showed 3‐fold higher τ than CP55940 (Table 1).
Figure 4.
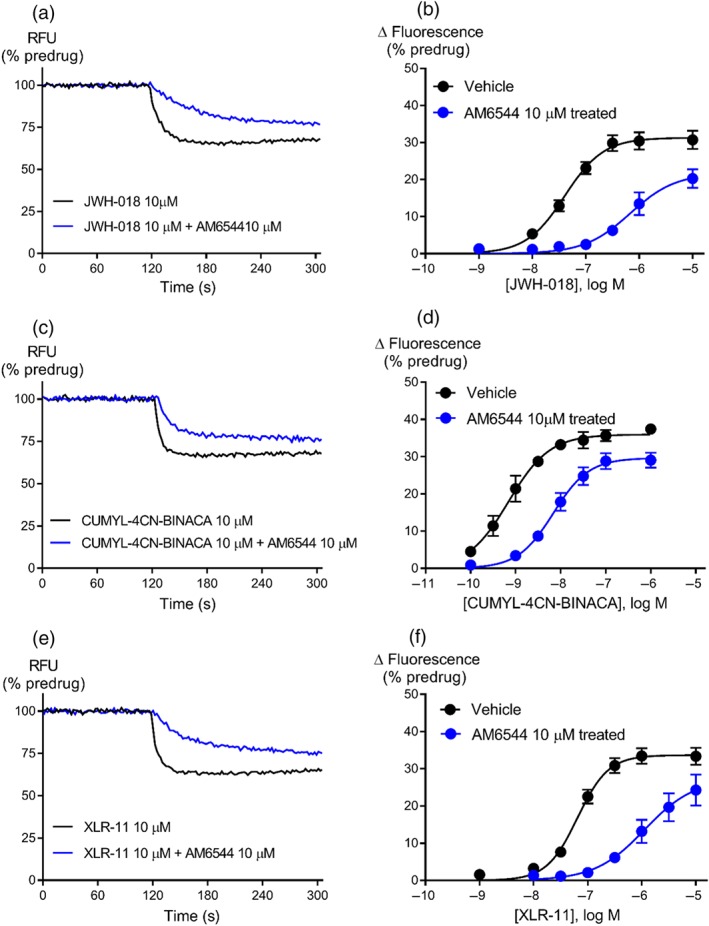
Representative traces for JWH‐018 (a), CUMYL‐4CN‐BINACA (c), and XLR‐11 (e) following pretreatment with vehicle or AM6544 (10 μM) on AtT‐20‐CB1 cells. Raw trace showing reduction in hyperpolarisation induced by maximally effective concentration of JWH‐018 (10 μM), CUMYL‐4CN‐BINACA (10 μM), and XLR‐11 (10 μM) after AM6544 pretreatment compared to vehicle. Concentration–response curves for (b) JWH‐018 (n = 8), (d) CUMYL‐4CN‐BINACA (n = 7), and (f) XLR‐11 (n = 7) were plotted using four parameter non‐linear regression to fit the operational model‐receptor depletion equation with basal constrained to 0. Data represent the mean ± SEM of technical replicates. For some points, the error bars are smaller than the height of the symbol.
We calculated the functional affinity of SCRAs at CB1 receptors using the operational model (Table 1). In the present study, the K A of SCRAs ranged from 33 nM to 31 μM, where three of the 13 SCRAs had K A values less than 100 nM (CUMYL‐4CN‐BINACA, MDMB‐FUBINACA, and 5F‐PB‐22) and three demonstrated micromolar affinities (JWH‐018, XLR‐11, and UR‐144). Most of the SCRAs had a higher affinity for CB1 receptors compared to CP55940, with the exception of UR‐144 and XLR‐11, which had 19‐ and 13‐fold lower affinity respectively (Table 1). No correlation was found between the operational efficacy and affinity obtained for SCRAs (Figure 5, r 2 = .0004, P > .05).
Figure 5.
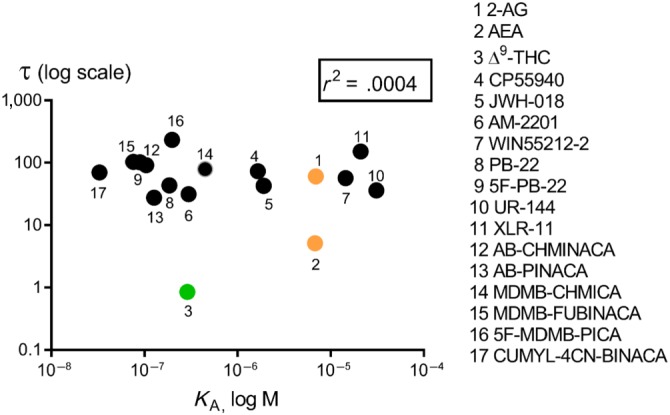
Correlation of operational efficacy (τ) and functional affinity (K A) for CB1 receptor agonists on Gi‐dependent activation of GIRK channel in AtT‐20 cells. Representative data are presented, demonstrating a non‐significant value of r 2 of .0004, where τ and K A values shown are the fitted values from the operational analysis.
To determine the percentage of CB1 receptors available after AM6544 pretreatment, the ratio of τ post‐ and pre‐receptor depletion were measured for each SCRA. The ratio of τ (depleted) to τ (control) for each drug reflects the reduction in the total functional receptor concentration [R0] due to AM6544‐treatment. The average value of τ post‐ and pre‐receptor depletion curves for SCRAs tested was found to be 0.07 ± 0.005, indicating that AM6544 caused an overall 93% reduction in receptors available to CB1 receptor agonists.
3.3. Quantification of relative agonist activity (RA i)
RA i values of the SCRAs for stimulating [35S]GTPγS binding assay and membrane potential assay were calculated in an attempt to compare the relative efficacy of these compounds in two very different assays of receptor activation. Activity was calculated with reference to that of the CP55940 and summarised in Table 2. A rank order of agonist activity, based on selectivity for the [35S]GTPγS binding assay is AB‐CHMINACA > PB‐22 > MDMB‐FUBINACA > AB‐PINACA > JWH‐018 > XLR‐11 > UR‐144 > ∆9‐THC. By contrast, relative activity of SCRAs in the membrane potential assay is MDMB‐FUBINACA > AB‐CHMINACA > PB‐22 > AB‐PINACA > JWH‐018 > XLR‐11 > ∆9‐THC > UR‐144. The pattern of selectivity that we observed in our studies is consistent with the data from the literature for [35S]GTPγS binding assay, with the striking exception of MDMB‐FUBINACA, which exhibited 37‐fold greater RA i value at membrane potential assay than those calculated for [35S]GTPγS binding assay. Most of the SCRAs had a higher RA i value at membrane potential assay compared to [35S]GTPγS binding assay, with the exception of UR144, which had a higher activity at [35S]GTPγS binding assay with a bias factor of 0.21 (Table 2). UR144 had a RA i value lower than ∆9‐THC in the membrane potential assay, despite the much higher efficacy of UR144 calculated using the operational model; this presumably relates to the very low functional affinity of UR144 in membrane potential assay.
Table 2.
Comparison of human CB1 receptor functional efficacy for selected SCRAs at CB1 receptors, measured using [35S]GTPγS binding assay and membrane potential assay
| Compound | GTPγS binding assay, RA i | Membrane potential assay, RA i | Bias factor |
|---|---|---|---|
| ∆9‐THC | 32 (27–36)a | 1 (0.1) | 5.0 |
| 0.02 | 0.1 | ||
| 3 | 6 | ||
| JWH‐018 | 1.02 (±0.10)b | 43 (21) | 1.1 |
| 0.45 | 0.5 | ||
| 4 | 8 | ||
| UR‐144 | 193 (164–221)c | 36 (7) | 0.21 |
| 0.14 | 0.03 | ||
| 5 | 6 | ||
| XLR‐11 | 205 (177–233)c | 152 (73) | 1.3 |
| 0.16 | 0.2 | ||
| 5 | 7 | ||
| PB‐22 | 415 (373–458)c | 44 (10) | 5.9 |
| 0.91 | 5.4 | ||
| 2 | 6 | ||
| AB‐CHMINACA | 205 (±14)d | 92 (42) | 3.8 |
| 5.21 | 20 | ||
| 6 | 7 | ||
| AB‐PINACA | 192 (±25)d | 28 (7) | 9.8 |
| 0.51 | 5.0 | ||
| 6 | 7 | ||
| MDMB‐FUBINACA | 75 (68–82)a | 103 (62) | 37 |
| 0.83 | 31 | ||
| 3 | 9 |
Note. Agonist activity (RA i) of SCRAs for eliciting different responses in assays for CB1 receptors are expressed relative to CP55940 is shown in bold below the E max (±SEM) for GTPγS binding assay or τ (±SEM) for membrane potential assay. The bias factor is expressed as the ratio of RA i‐membrane potential assay to RA i‐GTPγS binding assay. For each measure, the number of replicates n is shown below the relative efficacy.
Values from Gamage et al (2018) represent E max (95% confidence interval) for percentage increase over basal stimulation.
Values from Ford et al. (2017) represent E max (±SEM) are presented as the fraction of the effect produced by reference agonist CP55940.
Values from Thomas et al (2017) represent E max (95% confidence interval) for percentage [35S]GTPγS with basal globally shared at 100%.
Values from Wiley et al (2015) represent E max (±SEM) for percentage increase over basal stimulation.
4. DISCUSSION
We have measured the efficacy of a wide range of SCRA‐induced activation of native GIRK channels in an intact AtT‐20‐CB1 cells. To achieve this, we have employed high throughput assay technology to construct full concentration–response data following receptor depletion with the irreversible CB1 receptor antagonist AM6544 fitted to the operational model of pharmacological agonism to calculate the efficacy (τ) and affinity (K A) of these SCRAs. The principal finding of this study is that all the SCRAs tested showed substantially higher agonist activity at CB1 receptors than ∆9‐THC (τ, 0.9 ± 0.1), with τ that ranged between 28 and 233. 5F‐MDMB‐PICA and XLR‐11 exhibited the highest efficacies from the SCRAs tested. However, there was no correlation between the τ and K A of SCRAs, and no obvious trend for decreasing/increasing τ over time.
We have used the new CB1 receptor irreversible antagonist, AM6544, to specifically deplete the CB1 receptor reserve from the pool available for orthosteric agonist binding. The specificity of AM6544 was confirmed by showing the lack of effect of AM6544 pretreatment on the activation of native SRIF receptors in the same cells. AM6544 treatment effectively blocked CB1 receptors in AtT20 cells, although as it does not have a high affinity at these receptors on intact cells (pEC50 5.45, Finlay et al., 2017), a relatively high concentration had to be used. Under these conditions, the receptors are sufficiently depleted to ensure that the high efficacy agonist can no longer yield a system maximum at saturating concentrations. Other irreversible CB1 receptor antagonists have been identified, but they are not well suited for use in the kind of studies described here. Methyl arachidonyl flurophosphate can act at several components of the cannabinoid system including https://www.guidetopharmacology.org/GRAC/ObjectDisplayForward?objectId=1400, an enzyme involved in the degradation of endogenous cannabinoids (Fernando & Pertwee, 1997); falcarinol is very unstable (Leonti et al., 2010), and previously known irreversible analogues of SR141716A displayed lower affinities for CB1 receptors or acted as partial agonists (Howlett et al., 2000). AM6544 was used by Finlay et al. (2017) as an irreversible antagonist to deplete CB1 receptors in order to study the effect of receptor number on G protein preference in coupling to AC and, here, we have shown that it can also be used to study coupling to K channels through PTX‐sensitive G proteins. These kinds of quantitative pharmacological studies were not possible for CB1 receptors before AM6544 became available, and it is likely to be a useful compound in future experiments examining CB1 receptors.
The efficacy of SCRAs has principally been measured using [35S]GTPγS binding assays, which measures the accumulated activation of G proteins in membranes over a period of 30–60 min (De Luca et al., 2016; Gamage et al., 2018; Thomas et al., 2017; Wiley et al., 2015). In these assays, the maximum response is used as the measure of efficacy, with the assumption that this maximum response is not constrained—that there is an excess of G‐proteins relative to CB1 receptors. Given the high levels of receptor expression that can be achieved in recombinant systems, this assumption may not be valid (Gamage et al., 2018). In our study, we have circumvented this limitation by reducing receptor number, and we have been able to measure a very wide range of apparent efficacies to produce acute hyperpolarisation of AtT‐20‐CB1 cells (>250‐fold), compared with a twofold to threefold difference in the maximum response to agonists in CB1 receptor GTPγS assays (Table 2). Estimation of RA i values of SCRAs in GTPγS and membrane potential assay was undertaken in order to further observe an effect of different assays on the relative agonist activity of these compounds and also to quantify the functional selectivity of SCRAs for different receptor active states (Table 2). The rank order of agonist activity that we observed in our studies is generally similar with the data from the GTPγS assay. Only three of the SCRAs (AB‐CHMINACA, PB‐22, and MDMB‐FUBINACA) exhibited higher E max relative to CP55940, whereas all the other SCRAs presented very similar maximal response as CP55940 in GTPγS assays. Our assay seems sensitive to differences in efficacy, probably because GIRK activation is relatively poorly amplified, probably requiring 4 Gβγ subunits to simultaneously bind to each channel complex to open it (Whorton & MacKinnon, 2013), in contrast to the single ligand–receptor G protein complex required for stimulation of effectively irreversible GTPγS binding. In contrast to the 4 Gβγ subunits required to fully activate GIRK, inhibition of AC or voltage‐gated calcium channels by CB receptor activation requires only 1 Gα or Gβγ subunit respectively. Given that each ligand‐bound receptor is likely to activate multiple G protein heterotrimers, there will be significantly greater signal amplification when AC or I Ca are used as readouts. Activation of kinases such as https://www.guidetopharmacology.org/GRAC/FamilyDisplayForward?familyId=514 are usually multistep processes, and ERK activation can also be stimulated by several upstream signalling pathways (Jain, Watson, Vasudevan, & Saini, 2018), meaning that amplification can occur at several points. CB1 receptor‐dependent activation of GIRK has been reported in several types of neuron, and it represents a naturalistic, if understudied, signalling pathway for CB receptors (Azad et al., 2003; Daniel, Rancillac, & Crepel, 2004; Marinelli et al., 2009). Our work represents acute activation of one pathway; GTPγS assays provide a more general measure of Gαi/o‐subunit activation, but uncoupled from signalling pathways (Ibsen, Connor, & Glass, 2017). Neither assay effectively captures CB1 receptor coupling to Gαs or Gαq, or non‐G protein mediated pathways, such as those dependent on arrestin, but together they re‐enforce the quantitative differences in receptor activity between ∆9‐THC and more recently encountered cannabinoid agonists. The activity of SCRA‐induced arrestin recruitment by CB1 receptors has been studied using NanoLuc binary technology, and many SCRAs showed strong activation of CB1 receptors in this assay compared to ∆9‐THC (Cannaert et al., 2016; Noble, Cannaert, Linnet, & Stove, 2018), consistent with its low relative efficacy reported in the present work and GTPγS binding assays.
The first generation of SCRAs, JWH‐018, JWH‐073, JWH‐200, and CP47497 were detected in herbal blends in 2008 (Auwärter et al., 2009; Banister & Connor, 2018), and since then, there has been a rapid increase in structurally diverse sets of compounds with relatively unknown pharmacology and toxicology that continues to this day (European Monitoring Centre for Drugs and Drug Addiction, 2018; United Nations Office on Drugs and Crime, 2018). The toxicity associated with emerging SCRA has been reviewed elsewhere (Hermanns‐Clausen, Kneisel, Szabo, & Auwärter, 2013). There is no information as to whether the toxic effects of SCRAs may be mediated via direct CB1 receptor activation in humans, but studies in animals and cell lines indicate that seizures and effects on the kidney associated with SCRA may depend on CB1 receptor activation (Silva, Carmo, & Carvalho, 2018; Wiley, Barrett, Lowe, Balster, & Martin, 1995). All 13 SCRAs tested in this study had a much higher efficacy than ∆9‐THC, suggesting that adverse effects produced by ∆9‐THC intake may provide a limited guide to the potential consequences of CB1 receptor activation with high efficacy agonists. SCRAs produce CB1 receptor‐mediated seizures in animals, in addition to the well‐characterised CB1 receptor‐mediated “tetrad” of hypolocomotion, catalepsy, anti‐nociception, and hypothermia, and these could conceivably account for some of the adverse effects of SCRAs in humans (Vigolo et al., 2015). We also measured the efficacy of two principal endocannabinoids: 2‐AG and anandamide. Our results are consistent with previous reports, showing that 2‐AG is a higher efficacy agonist of CB1 receptors compared to anandamide (Di Marzo & De Petrocellis, 2012) and that both have a higher efficacy than ∆9‐THC (Pertwee, 1997). Thus, ∆9‐THC, but not most of the SCRAs investigated here, is likely to act as an antagonist of 2‐AG modulation of neuronal activity in vivo (Pertwee, 2008; Straiker & Mackie, 2005).
Cannabinoid interactions with renal and cardiovascular systems have also been described (Pacher, Steffens, Haskó, Schindler, & Kunos, 2018), but the degree to which these interactions are influenced by agonist efficacy is unknown. A specific toxicity attributed to a particular SCRA was the acute kidney injury linked to the use of XLR‐11 (Thornton, Wood, Friesen, & Gerona, 2013). The present study shows that XLR‐11 had a high operational efficacy, which together with its relative non‐selectivity for CB1 over CB2 receptors (Banister, Stuart, et al., 2015) may contribute to its unique toxicological profile. XLR‐11 affects kidney cells via CB receptors on mitochondria, rather than through the plasma‐membrane delimited pathway we have examined, and both CB1 and CB2 receptors were reported to be involved in the toxic effects of XLR‐11 in vitro (Silva et al., 2018). Thus, toxicity for individual SCRAs potentially involves a complex interplay between activity at both CB1 and CB2 receptors as well as efficacy at CB1 receptors, cellular and subcellular distribution, access to receptors to different body and cellular compartments, and the formation of bioactive drug metabolites (Fantegrossi, Moran, Radominska‐pandya, & Prather, 2014).
The emergence of new psychoactive substances provides a continual challenge to the development of targeted interventions and novel therapeutics to help minimise the adverse effects associated with their use (European Monitoring Centre for Drugs and Drug Addiction, 2018). Although some SCRAs were mined from older patents (AM2201, AB‐CHMINACA, AB‐FUBINACA, UR144, etc.), newer drugs have unprecedented structures (Banister & Connor, 2018). We assessed a diversity of SCRAs identified in the NPS market, from the earliest to most recent examples. There was no obvious trend for decreasing/increasing τ over time and τ or functional affinity, suggesting that SCRAs are not designed to be more efficacious over time. Our data also show no obvious relationship between the efficacy of SCRA to activate native GIRK channels and their reported adverse effects. It is not immediately apparent what causes the toxic effects of SCRAs and whether signalling of SCRAs at Gαs, Gαq or arrestins, rather than Gαi/o‐dependent CB1 receptor signalling is important. However, it is clear that these drugs are likely to have very different pharmacological profiles to the commonly consumed cannabinoid, ∆9‐THC. This was highlighted in a recent study where the crystal structure of CB1 receptors bound to MDMB‐FUBINACA demonstrated a “toggle twin switch” interaction that ∆9‐THC did not. This might explain the low efficacy activity of ∆9‐THC compared to the high efficacy of MDMB‐FUBINACA when activating CB1 receptors (Krishna Kumar et al., 2019). Furthermore, CB1 receptors are known to exert pleotropic effects by virtue of its ability to interact with multiple G‐proteins. A recent study reported that AB‐CHMINACA showed specific CB1 receptor‐dependent activation of Gαs signalling (Costain et al., 2018). These observations highlight the complexity of the pharmacology of SCRAs‐mediated activation of different signalling pathways downstream of CB1 receptors . Structural examination of CB1 receptors for ligand efficacy and G‐protein recruitment provides molecular insights into the active state of the receptor (Krishna Kumar et al., 2019) and is a first step in informing us about the diverse physiological consequences resulting from CB1 receptor activation by high efficacy agonists.
AUTHOR CONTRIBUTIONS
S.S. designed and performed experiments, analysed the data, and wrote the manuscript. Data analysis was performed by M.C. and S.S. K.V. and A.M. generated the AM6544 compound. The synthesis of drugs was carried out by S.D.B. and M.L. with direction from M.K. M.S. made and characterised CB1 cells. The manuscript was drafted by S.S. and M.C. with contributions from S.D.B., K.V., and M.K. M.C. supervised the study and revised the manuscript. All the authors have given approval to the final version of manuscript.
CONFLICT OF INTEREST
Two authors (A.M. and K.V.) are co‐inventors for a patent which encompasses AM6544‐heteropyrrole analogues acting on cannabinoid receptors, US Patent US8084451B2, 2011. There are no other conflicts of interest to declare.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for https://bpspubs.onlinelibrary.wiley.com/doi/full/10.1111/bph.14207, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1. Structure of selected SCRAs
Figure S2. Supporting information
Table S1. Functional activity of CP55940, Δ9‐THC, and other SCRAs. Values were calculated using the four‐parameter nonlinear regression to fit the concentration response curves.
ACKNOWLEDGEMENTS
This work was supported by NHMRC Project Grant 1107088 awarded to M.K. and M.C. and National Institutes of Health Grant P01DA009158 to A.M. Work was also supported in part by the European Union's Seventh Framework Programme (FP7/2007‐103) InMind (Grant Agreement HEALTH‐F2‐2011‐278850) and R21DA045882 to K.V. S.S. was supported by a Macquarie University Doctoral Scholarship. We would like to thank Dr Chris Bladen and Alexander Gillis for their insightful discussions regarding work described in the draft manuscript.
Sachdev S, Vemuri K, Banister SD, et al. In vitro determination of the efficacy of illicit synthetic cannabinoids at CB1 receptors. Br J Pharmacol. 2019;176:4653–4665. 10.1111/bph.14829
REFERENCES
- Adams, A. J. , Banister, S. D. , Irizarry, L. , Trecki, J. , Schwartz, M. , & Gerona, R. (2017). “Zombie” outbreak caused by the synthetic cannabinoid AMB‐FUBINACA in New York. New England Journal of Medicine, 376, 235–242. 10.1056/NEJMoa1610300 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Marrion, N. V. , Peters, J. A. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: G protein‐coupled receptors. British Journal of Pharmacology, 174, S17–S129. 10.1111/bph.13878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. British Journal of Pharmacology, 174, S272–S359. 10.1111/bph.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auwärter, V. , Dresen, S. , Weinmann, W. , Müller, M. , Pütz, M. , & Ferreirós, N. (2009). Spice'and other herbal blends: Harmless incense or cannabinoid designer drugs? Journal of Mass Spectrometry, 44, 832–837. 10.1002/jms.1558 [DOI] [PubMed] [Google Scholar]
- Azad, S. C. , Eder, M. , Marsicano, G. , Lutz, B. , Zieglgänsberger, W. , & Rammes, G. (2003). Activation of the cannabinoid receptor type 1 decreases glutamatergic and GABAergic synaptic transmission in the lateral amygdala of the mouse. Learning & Memory, 10, 116–128. 10.1101/lm.53303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci, A. , Huguenard, J. R. , & Prince, D. A. (2004). Long‐lasting self‐inhibition of neocortical interneurons mediated by endocannabinoids. Nature, 431, 312–316. 10.1038/nature02913 [DOI] [PubMed] [Google Scholar]
- Banister, S. D. , & Connor, M. (2018). The chemistry and pharmacology of synthetic cannabinoid receptor agonists as new psychoactive substances: Origins. Handbook of experimental pharmacology, 252, 165–190. 10.1007/164_2018_144 [DOI] [PubMed] [Google Scholar]
- Banister, S. D. , Longworth, M. , Kevin, R. , Sachdev, S. , Santiago, M. , Stuart, J. , … Kassiou, M. (2016). Pharmacology of valinate and tert‐leucinate synthetic cannabinoids 5F‐AMBICA, 5F‐AMB, 5F‐ADB, AMB‐FUBINACA, MDMB‐FUBINACA, MDMB‐CHMICA, and their analogues. ACS Chemical Neuroscience, 7, 1241–1254. 10.1021/acschemneuro.6b00137 [DOI] [PubMed] [Google Scholar]
- Banister, S. D. , Moir, M. , Stuart, J. , Kevin, R. C. , Wood, K. E. , Longworth, M. , … Kassiou, M. (2015). Pharmacology of Indole and Indazole Synthetic Cannabinoid Designer Drugs AB‐FUBINACA, ADB‐FUBINACA, AB‐PINACA, ADB‐PINACA, 5F‐AB‐PINACA, 5F‐ADB‐PINACA, ADBICA, and 5F‐ADBICA. ACS Chemical Neuroscience, 6, 1546–1559. 10.1021/acschemneuro.5b00112 [DOI] [PubMed] [Google Scholar]
- Banister, S. D. , Stuart, J. , Kevin, R. C. , Edington, A. , Longworth, M. , Wilkinson, S. M. , … Kassiou, M. (2015). Effects of bioisosteric fluorine in synthetic cannabinoid designer drugs JWH‐018, AM‐2201, UR‐144, XLR‐11, PB‐22, 5F‐PB‐22, APICA, and STS‐135. ACS Chemical Neuroscience, 6, 1445–1458. 10.1021/acschemneuro.5b00107 [DOI] [PubMed] [Google Scholar]
- Banister, S. D. , Wilkinson, S. M. , Longworth, M. , Stuart, J. , Apetz, N. , English, K. , … Kassiou, M. (2013). The synthesis and pharmacological evaluation of adamantane‐derived indoles: Cannabimimetic drugs of abuse. ACS Chemical Neuroscience, 4, 1081–1092. 10.1021/cn400035r [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besse, J. C. , & Furchgott, R. F. (1976). Dissociation constants and relative efficacies of agonists acting on alpha adrenergic receptors in rabbit aorta. Journal of Pharmacology and Experimental Therapeutics, 197, 66–78. [PubMed] [Google Scholar]
- Black, J. W. , & Leff, P. (1983). Operational models of pharmacological agonism. Proceedings of the Royal Society of London B, 220, 141–162. 10.1098/rspb.1983.0093 [DOI] [PubMed] [Google Scholar]
- Cannaert, A. , Storme, J. , Franz, F. , Auwärter, V. , & Stove, C. P. (2016). Detection and activity profiling of synthetic cannabinoids and their metabolites with a newly developed bioassay. Analytical Chemistry, 88, 11476–11485. 10.1021/acs.analchem.6b02600 [DOI] [PubMed] [Google Scholar]
- Cooper, Z. D. (2016). Adverse effects of synthetic cannabinoids: Management of acute toxicity and withdrawal. Current Psychiatry Reports, 18, 52 10.1007/s11920-016-0694-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costain, W. J. , Rasquinha, I. , Comas, T. , Hewitt, M. , Aylsworth, A. , Rouleau, Y. , … Tauskela, J. S. (2018). Analysis of the pharmacological properties of JWH‐122 isomers and THJ‐2201, RCS‐4 and AB‐CHMINACA in HEK293T cells and hippocampal neurons. European Journal of Pharmacology, 823, 96–104. 10.1016/j.ejphar.2018.01.043 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel, H. , Rancillac, A. , & Crepel, F. (2004). Mechanisms underlying cannabinoid inhibition of presynaptic Ca2+ influx at parallel fibre synapses of the rat cerebellum. The Journal of Physiology, 557, 159–174. 10.1113/jphysiol.2004.063263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Luca, M. A. , Castelli, M. P. , Loi, B. , Porcu, A. , Martorelli, M. , Miliano, C. , … Di Chiara, G. (2016). Native CB1 receptor affinity, intrinsic activity and accumbens shell dopamine stimulant properties of third generation SPICE/K2 cannabinoids: BB‐22, 5F‐PB‐22, 5F‐AKB‐48 and STS‐135. Neuropharmacology, 105, 630–638. 10.1016/j.neuropharm.2015.11.017 [DOI] [PubMed] [Google Scholar]
- De Petrocellis, L. , & Di Marzo, V. (2010). Non‐CB 1, non‐CB 2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: Focus on G‐protein‐coupled receptors and transient receptor potential channels. Journal of Neuroimmune Pharmacology, 5, 103–121. 10.1007/s11481-009-9177-z [DOI] [PubMed] [Google Scholar]
- Di Marzo, V. , & De Petrocellis, L. (2012). Why do cannabinoid receptors have more than one endogenous ligand? Phil. Trans. R. Soc. B, 367, 3216–3228. 10.1098/rstb.2011.0382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlert, F. J. (2008). On the analysis of ligand‐directed signaling at G protein‐coupled receptors. Naunyn‐Schmiedeberg's Archives of Pharmacology, 377, 549–577. 10.1007/s00210-008-0260-4 [DOI] [PubMed] [Google Scholar]
- Ehlert, F. J. , Griffin, M. T. , Sawyer, G. W. , & Bailon, R. (1999). A simple method for estimation of agonist activity at receptor subtypes: Comparison of native and cloned M3 muscarinic receptors in guinea pig ileum and transfected cells. Journal of Pharmacology and Experimental Therapeutics, 289, 981–992. [PubMed] [Google Scholar]
- European Monitoring Centre for Drugs and Drug Addiction (2018). Fentanils and synthetic cannabinoids: Driving greater complexity into the drug situation. An update from the EU early warning system June 2018, Publications Office of the European Union, Luxembourg.
- Fantegrossi, W. E. , Moran, J. H. , Radominska‐pandya, A. , & Prather, P. L. (2014). Distinct pharmacology and metabolism of K2 synthetic cannabinoids compared to Δ9‐THC: Mechanism underlying greater toxicity? Life Sciences, 97, 45–54. 10.1016/j.lfs.2013.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando, S. R. , & Pertwee, R. G. (1997). Evidence that methyl arachidonyl fluorophosphonate is an irreversible cannabinoid receptor antagonist. British Journal of Pharmacology, 121, 1716–1720. 10.1038/sj.bjp.0701303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay, D. B. , Cawston, E. E. , Grimsey, N. L. , Hunter, M. R. , Korde, A. , Vemuri, V. K. , … Glass, M. (2017). Gαs signalling of the CB1 receptor and the influence of receptor number. British Journal of Pharmacology, 174, 2545–2562. 10.1111/bph.13866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, B. M. , Franks, L. N. , Tai, S. , Fantegrossi, W. E. , Stahl, E. L. , Berquist, M. D. , … Prather, P. L. (2017). Characterization of structurally novel G protein biased CB1 agonists: Implications for drug development. Pharmacological Research, 125, 161–177. 10.1016/j.phrs.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funada, M. , & Takebayashi‐Ohsawa, M. (2018). Synthetic cannabinoid AM2201 induces seizures: Involvement of cannabinoid CB1 receptors and glutamatergic transmission. Toxicology and Applied Pharmacology, 338, 1–8. 10.1016/j.taap.2017.10.007 [DOI] [PubMed] [Google Scholar]
- Gamage, T. F. , Farquhar, C. E. , Lefever, T. W. , Marusich, J. A. , Kevin, R. C. , McGregor, I. S. , … Thomas, B. F. (2018). Molecular and behavioral pharmacological characterization of abused synthetic cannabinoids MMB‐and MDMB‐FUBINACA, MN‐18, NNEI, CUMYL‐PICA, and 5‐Fluoro‐CUMYL‐PICA. Journal of Pharmacology and Experimental Therapeutics, 365, 437–446. 10.1124/jpet.117.246983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia, D. , Brown, S. , Hille, B. , & Mackie, K. (1998). Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. Journal of Neuroscience, 18, 2834–2841. 10.1523/JNEUROSCI.18-08-02834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin, M. T. , Figueroa, K. W. , Liller, S. , & Ehlert, F. J. (2007). Estimation of agonist activity at G protein‐coupled receptors: Analysis of M2 muscarinic receptor signaling through Gi/o, Gs, and G15 . Journal of Pharmacology and Experimental Therapeutics, 321, 1193–1207. 10.1124/jpet.107.120857 [DOI] [PubMed] [Google Scholar]
- Guo, J. , & Ikeda, S. R. (2004). Endocannabinoids modulate N‐type calcium channels and G‐protein‐coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Molecular Pharmacology, 65, 665–674. 10.1124/mol.65.3.665 [DOI] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermanns‐Clausen, M. , Kneisel, S. , Szabo, B. , & Auwärter, V. (2013). Acute toxicity due to the confirmed consumption of synthetic cannabinoids: Clinical and laboratory findings. Addiction, 108, 534–544. 10.1111/j.1360-0443.2012.04078.x [DOI] [PubMed] [Google Scholar]
- Hess, C. , Schoeder, C. T. , Pillaiyar, T. , Madea, B. , & Müller, C. E. (2016). Pharmacological evaluation of synthetic cannabinoids identified as constituents of spice. Forensic Toxicology, 34, 329–343. 10.1007/s11419-016-0320-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett, A. C. , Wilken, G. H. , Pigg, J. J. , Houston, D. B. , Lan, R. , Liu, Q. , & Makriyannis, A. (2000). Azido‐ and isothiocyanato‐substituted aryl pyrazoles bind covalently to the CB1 cannabinoid receptor and impair signal transduction. Journal of Neurochemistry, 74, 2174–2181. [DOI] [PubMed] [Google Scholar]
- Ibsen, M. S. , Connor, M. , & Glass, M. (2017). Cannabinoid CB1 and CB2 receptor signaling and bias. Cannabis and Cannabinoid Research, 2, 48–60. 10.1089/can.2016.0037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, R. , Watson, U. , Vasudevan, L. , & Saini, D. K. (2018). ERK activation pathways downstream of GPCRs. International Review of Cell and Molecular Biology, 338, 79–109. 10.1016/bs.ircmb.2018.02.003 [DOI] [PubMed] [Google Scholar]
- Knapman, A. , Santiago, M. , Du, Y. P. , Bennallack, P. R. , Christie, M. J. , & Connor, M. (2013). A continuous, fluorescence‐based assay of μ‐opioid receptor activation in AtT‐20 cells. Journal of Biomolecular Screening, 18, 269–276. 10.1177/1087057112461376 [DOI] [PubMed] [Google Scholar]
- Krishna Kumar, K. , Shalev‐Benami, M. , Robertson, M. J. , Hu, H. , Banister, S. D. , Hollingsworth, S. A. , … Skiniotis, G. (2019). Structure of a signaling cannabinoid receptor 1‐G protein complex. Cell, 176, 448–458.e12. 10.1016/j.cell.2018.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonti, M. , Casu, L. , Raduner, S. , Cottiglia, F. , Floris, C. , Altmann, K.‐H. , & Gertsch, J. (2010). Falcarinol is a covalent cannabinoid CB1 receptor antagonist and induces pro‐allergic effects in skin. Biochemical Pharmacology, 79, 1815–1826. 10.1016/j.bcp.2010.02.015 [DOI] [PubMed] [Google Scholar]
- Mackie, K. , Lai, Y. , Westenbroek, R. , & Mitchell, R. (1995). Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q‐type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. Journal of Neuroscience, 15, 6552–6561. 10.1523/JNEUROSCI.15-10-06552.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malyshevskaya, O. , Aritake, K. , Kaushik, M. K. , Uchiyama, N. , Cherasse, Y. , Kikura‐Hanajiri, R. , & Urade, Y. (2017). Natural (∆9‐THC) and synthetic (JWH‐018) cannabinoids induce seizures by acting through the cannabinoid CB 1 receptor. Scientific Reports, 7, 10516 10.1038/s41598-017-10447-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli, S. , Pacioni, S. , Cannich, A. , Marsicano, G. , & Bacci, A. (2009). Self‐modulation of neocortical pyramidal neurons by endocannabinoids. Nature Neuroscience, 12, 1488–1490. 10.1038/nn.2430 [DOI] [PubMed] [Google Scholar]
- Motulsky, H. , & Christopoulos, A. (2004). Fitting models to biological data using linear and nonlinear regression: A practical guide to curve fitting. San Diego, CA, USA: Oxford University Press. [Google Scholar]
- Noble, C. , Cannaert, A. , Linnet, K. , & Stove, C. P. (2018). Application of an activity‐based receptor bioassay to investigate the in vitro activity of selected indole‐and indazole‐3‐carboxamide‐based synthetic cannabinoids at CB1 and CB2 receptors. Drug Testing and Analysis, 11, 501–511. 10.1002/dta.2517 [DOI] [PubMed] [Google Scholar]
- Pacher, P. , Bátkai, S. , & Kunos, G. (2006). The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacological Reviews, 58, 389–462. 10.1124/pr.58.3.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher, P. , Steffens, S. , Haskó, G. , Schindler, T. H. , & Kunos, G. (2018). Cardiovascular effects of marijuana and synthetic cannabinoids: the good, the bad, and the ugly. Nature Reviews Cardiology, 15, 151–166. 10.1038/nrcardio.2017.130 [DOI] [PubMed] [Google Scholar]
- Pertwee, R. G. (1997). Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacology & Therapeutics, 74, 129–180. 10.1016/S0163-7258(97)82001-3 [DOI] [PubMed] [Google Scholar]
- Pertwee, R. G. (2008). The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9‐tetrahydrocannabinol, cannabidiol and Δ9‐tetrahydrocannabivarin. British Journal of Pharmacology, 153, 199–215. 10.1038/sj.bjp.0707442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler, C. W. , Gramling, B. R. , Justinova, Z. , Thorndike, E. B. , & Baumann, M. H. (2017). Synthetic cannabinoids found in “spice” products alter body temperature and cardiovascular parameters in conscious male rats. Drug & Alcohol Dependence, 179, 387–394. 10.1016/j.drugalcdep.2017.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva, J. P. , Carmo, H. , & Carvalho, F. (2018). The synthetic cannabinoid XLR‐11 induces in vitro nephrotoxicity by impairment of endocannabinoid‐mediated regulation of mitochondrial function homeostasis and triggering of apoptosis. Toxicology Letters, 287, 59–69. 10.1016/j.toxlet.2018.01.023 [DOI] [PubMed] [Google Scholar]
- Straiker, A. , & Mackie, K. (2005). Depolarization‐induced suppression of excitation in murine autaptic hippocampal neurones. The Journal of Physiology, 569, 501–517. 10.1113/jphysiol.2005.091918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait, R. J. , Caldicott, D. , Mountain, D. , Hill, S. L. , & Lenton, S. (2016). A systematic review of adverse events arising from the use of synthetic cannabinoids and their associated treatment. Clinical Toxicology, 54, 1–13. 10.3109/15563650.2015.1110590 [DOI] [PubMed] [Google Scholar]
- Thomas, B. F. , Lefever, T. W. , Cortes, R. A. , Grabenauer, M. , Kovach, A. L. , Cox, A. O. , … Wiley, J. L. (2017). Thermolytic degradation of synthetic cannabinoids: Chemical exposures and pharmacological consequences. Journal of Pharmacology and Experimental Therapeutics, 361, 162–171. 10.1124/jpet.116.238717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton, S. L. , Wood, C. , Friesen, M. W. , & Gerona, R. R. (2013). Synthetic cannabinoid use associated with acute kidney injury. Clinical Toxicology, 51, 189–190. 10.3109/15563650.2013.770870 [DOI] [PubMed] [Google Scholar]
- Trecki, J. , Gerona, R. R. , & Schwartz, M. D. (2015). Synthetic cannabinoid‐related illnesses and deaths. The New England Journal of Medicine, 373, 103–107. 10.1056/NEJMp1505328 [DOI] [PubMed] [Google Scholar]
- United Nations Office on Drugs and Crime (2018). World Drug Report. United Nations publication.978–92–1‐148291‐1.
- Vigolo, A. , Ossato, A. , Trapella, C. , Vincenzi, F. , Rimondo, C. , Seri, C. , … Marti, M. (2015). Novel halogenated derivates of JWH‐018: Behavioral and binding studies in mice. Neuropharmacology, 95, 68–82. 10.1016/j.neuropharm.2015.02.008 [DOI] [PubMed] [Google Scholar]
- Whorton, M. R. , & MacKinnon, R. (2013). X‐ray structure of the mammalian GIRK2–βγ G‐protein complex. Nature, 498, 190–197. 10.1038/nature12241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, J. , Barrett, R. , Lowe, J. , Balster, R. , & Martin, B. (1995). Discriminative stimulus effects of CP 55,940 and structurally dissimilar cannabinoids in rats. Neuropharmacology, 34, 669–676. 10.1016/0028-3908(95)00027-4 [DOI] [PubMed] [Google Scholar]
- Wiley, J. L. , Marusich, J. A. , & Huffman, J. W. (2014). Moving around the molecule: Relationship between chemical structure and in vivo activity of synthetic cannabinoids. Life Sciences, 97, 55–63. 10.1016/j.lfs.2013.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley, J. L. , Marusich, J. A. , Lefever, T. W. , Antonazzo, K. R. , Wallgren, M. T. , Cortes, R. A. , … Thomas, B. F. (2015). AB‐CHMINACA, AB‐PINACA, and FUBIMINA: Affinity and potency of novel synthetic cannabinoids in producing Δ9‐tetrahydrocannabinol–like effects in mice. Journal of Pharmacology and Experimental Therapeutics, 354, 328–339. 10.1124/jpet.115.225326 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Structure of selected SCRAs
Figure S2. Supporting information
Table S1. Functional activity of CP55940, Δ9‐THC, and other SCRAs. Values were calculated using the four‐parameter nonlinear regression to fit the concentration response curves.